High-Resolution Respirometry Reveals MPP+ Mitochondrial Toxicity Mechanism in a Cellular Model of Parkinson’s Disease


 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
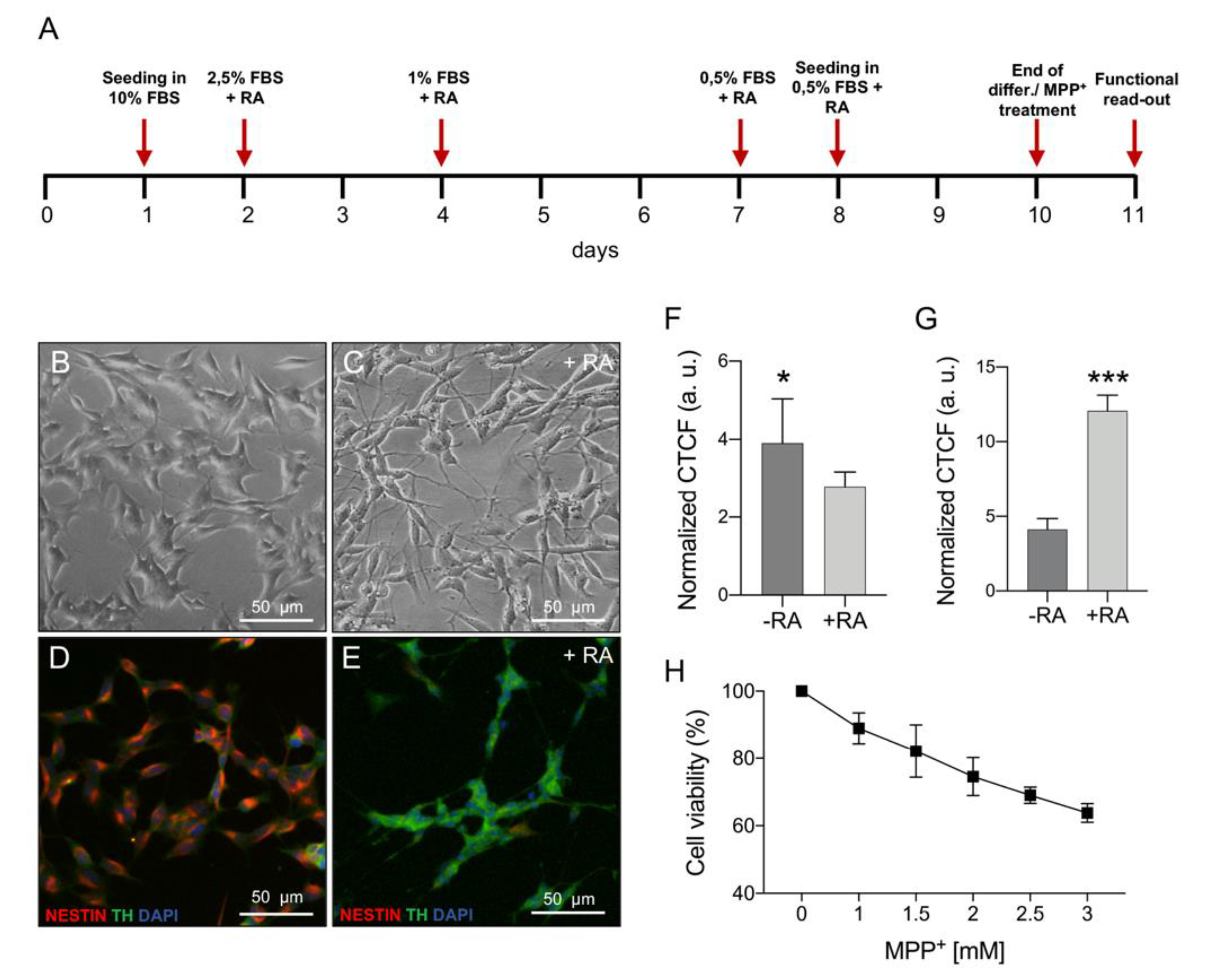
2.1. MPP+ Toxicity Assessment in Differentiated SH-SY5Y Cells
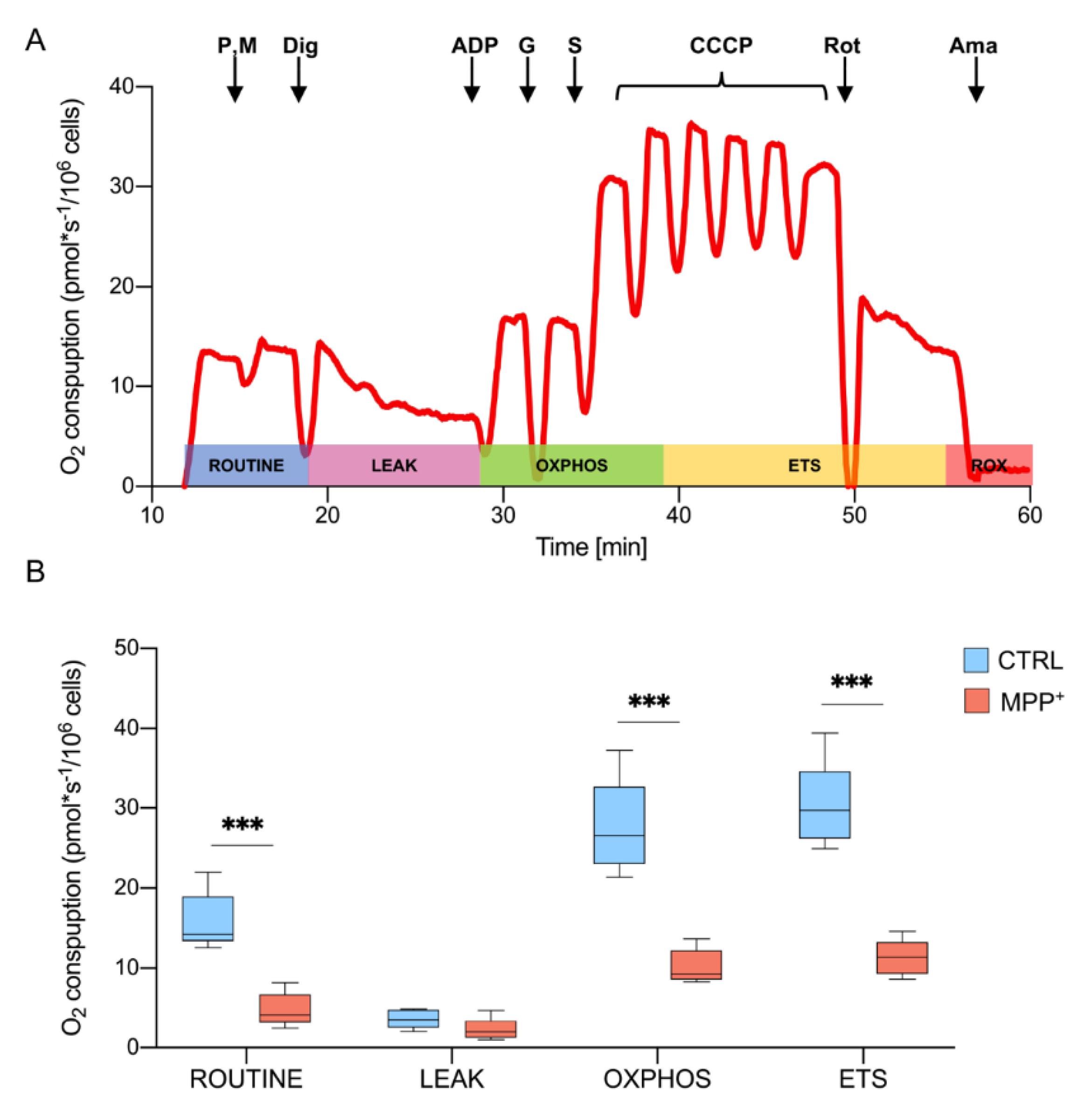
2.2. MPP+ Drastically Reduces Oxygen Consumption Associated with the Main Respiratory States
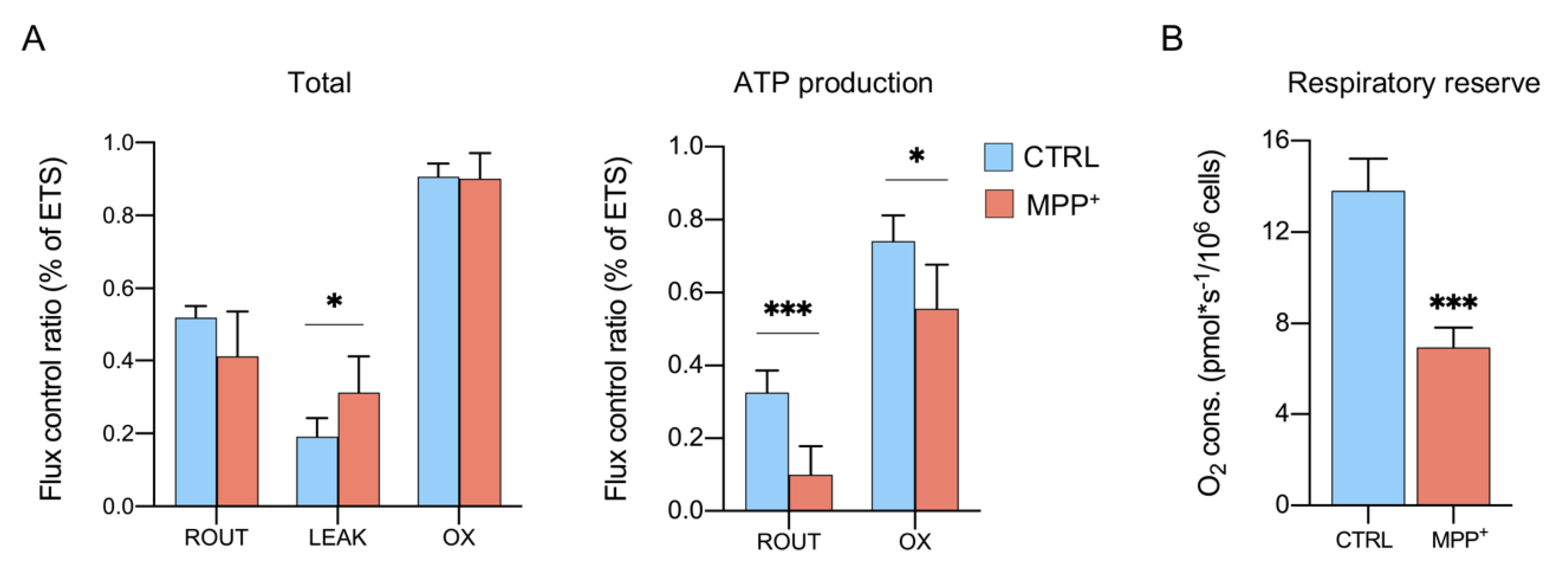
2.3. MPP+ Increases the Dissipative Ratio of Oxygen Flux
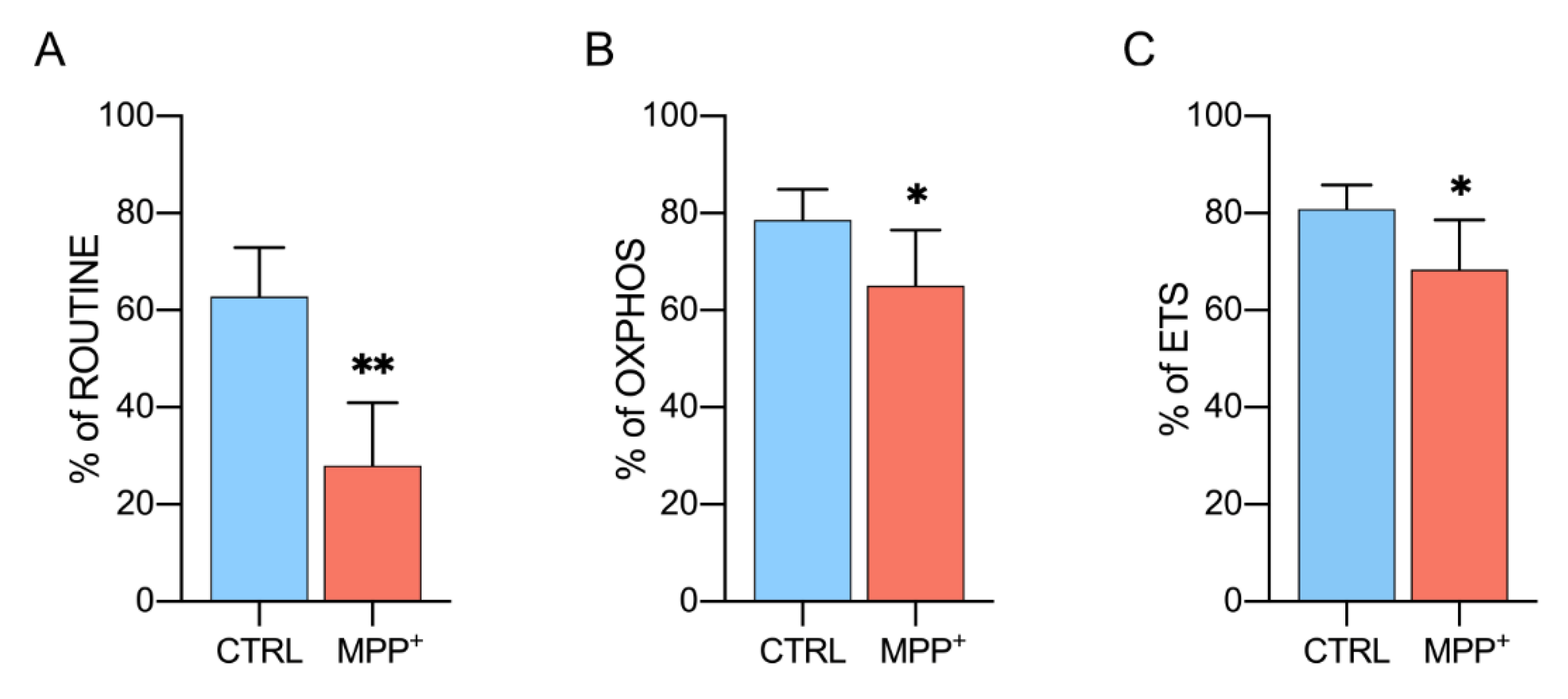
2.4. MPP+ Reduces Coupling Efficiency in Each Respiratory State
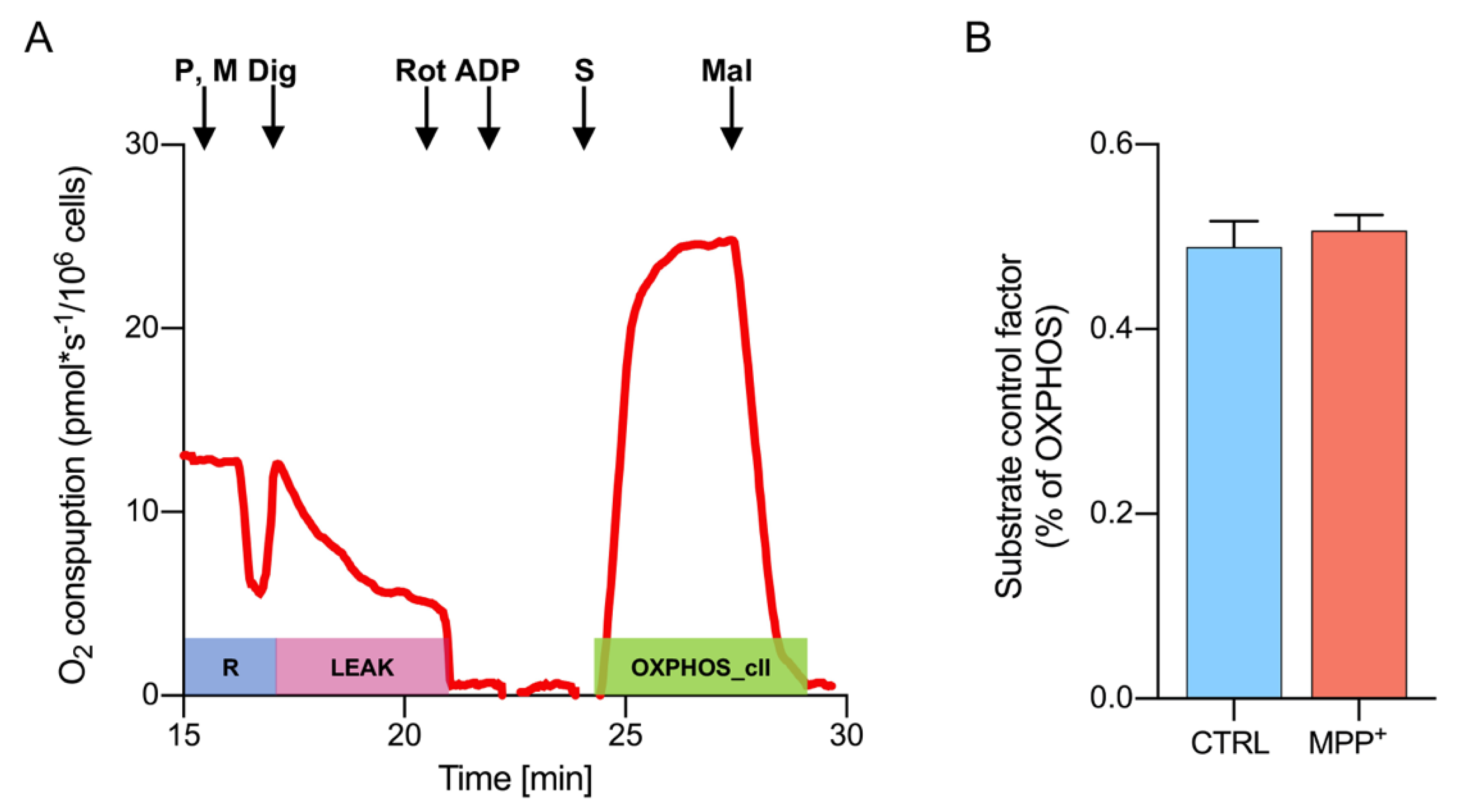
2.5. Activity of Complex II Is Increased Upon Complex I Inhibition
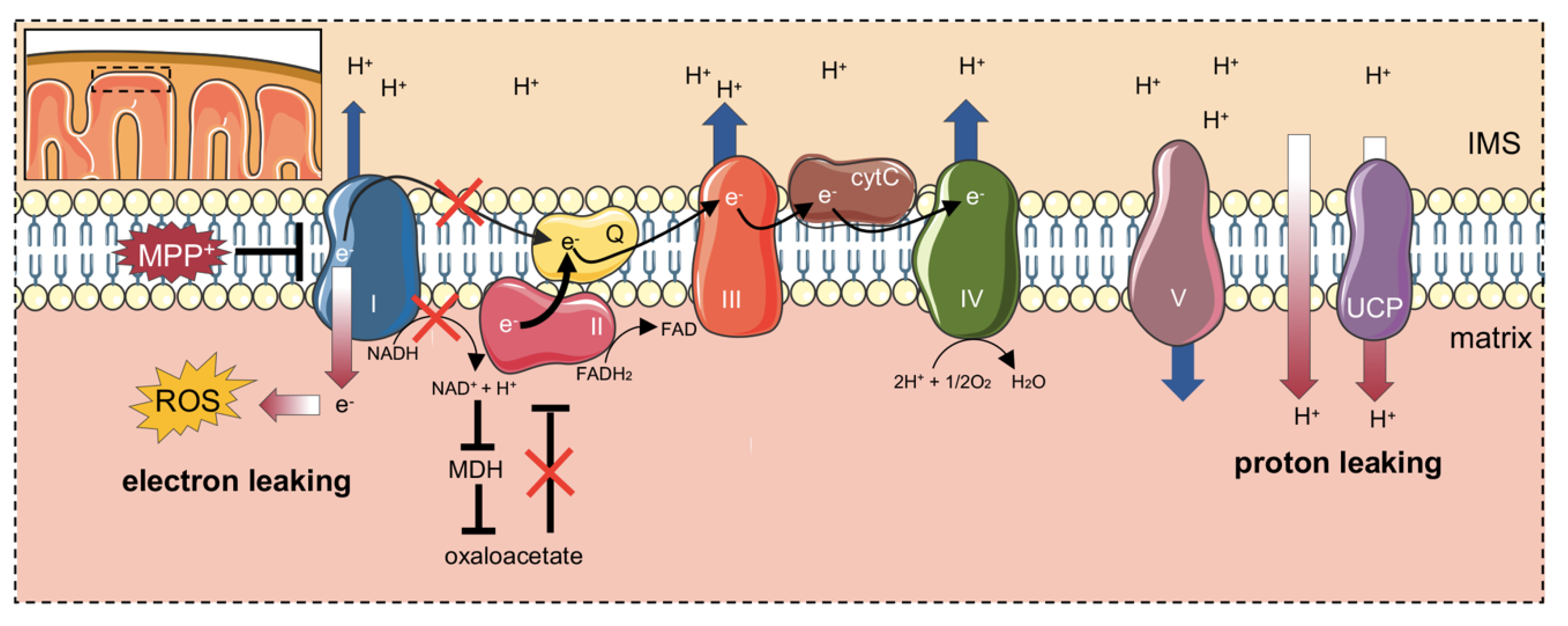
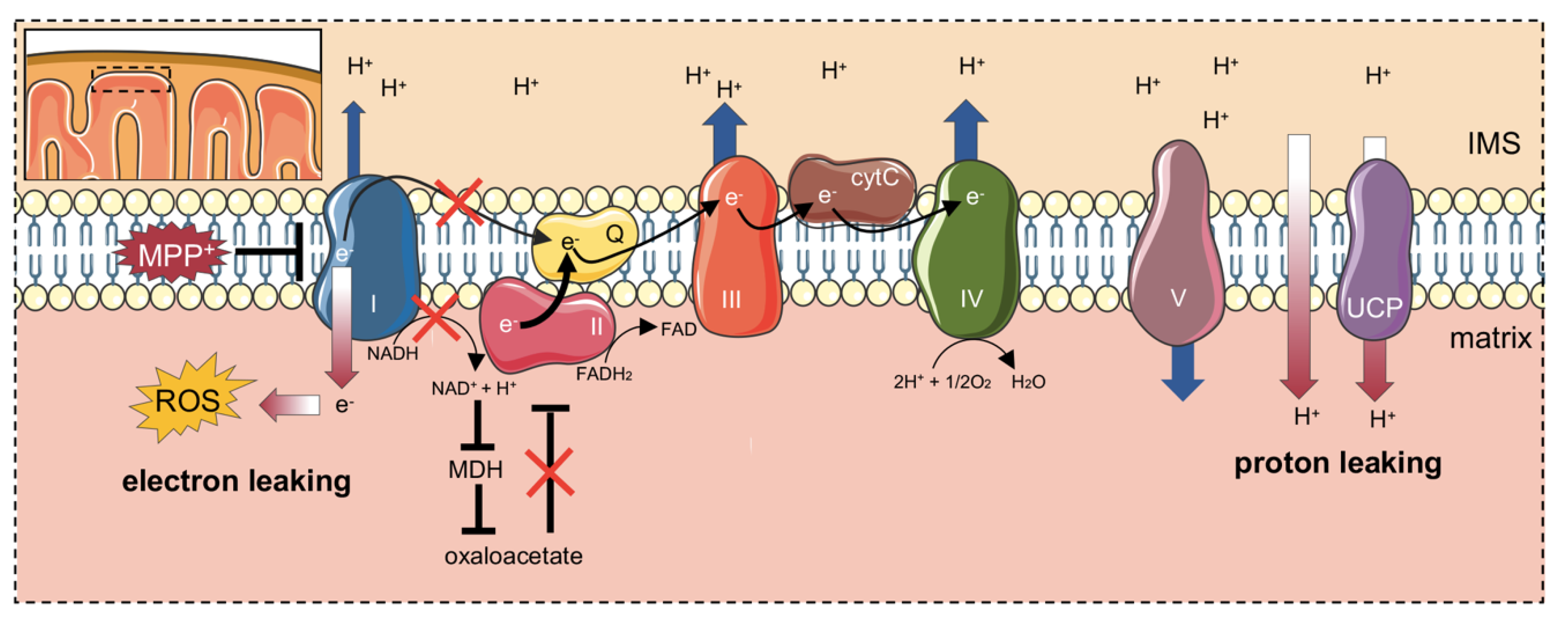
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Differentiation
4.2. Immunofluorescence
4.3. MPP+ Treatment and Cell Viability
4.4. High-Resolution Respirometry (HRR) Analysis
4.5. Complex II Activity Analysis
4.6. Data Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| ND | Neurodegenerative disease |
| DA | Dopamine |
| LBs | Lewy’s bodies |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MPP+ | 1-methyl-4-phenylpyridinium ion |
| ETS | Electron transport system |
| HRR | High-resolution respirometry |
| RA | Retinoic acid |
| TH | Tyrosine hydroxylase |
| SUIT | Substrate-uncoupler-inhibitor titration |
| FCR | Flux control ratio |
| SCF | Substrate control factor |
| IMM | Inner mitochondrial membrane |
| OMM | Outer mitochondrial membrane |
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Kim, W.S.; Kagedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, B.; Leggio, L.; L’Episcopo, F.; Vivarelli, S.; Tirolo, C.; Paternò, G.; Giachino, C.; Caniglia, S.; Serapide, M.F.; Iraci, N. Glia-Derived Extracellular Vesicles in Parkinson’s Disease. J. Clin. Med. 2020, 9, 1941. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Jenner, P.; Przedborski, S. Pathogenesis of Parkinson’s disease. Mov. Disord. 2013, 28, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Leggio, L.; Patern, G.; Vivarelli, S.; Episcopo, F.L.; Tirolo, C.; Raciti, G.; Pappalardo, F.; Giachino, C.; Caniglia, S.; Serapide, M.F.; et al. Extracellular Vesicles as Nanotherapeutics for Parkinson ’s Disease. Biomolecules 2020, 10, E1327. [Google Scholar] [CrossRef]
- Leggio, L.; Arrabito, G.; Ferrara, V.; Vivarelli, S.; Paternò, G.; Marchetti, B.; Pignataro, B.; Iraci, N. Mastering the Tools: Natural versus Artificial Vesicles in Nanomedicine. Adv. Healthc. Mater. 2020, e2000731. [Google Scholar] [CrossRef] [PubMed]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Marchetti, B. Nrf2/Wnt resilience orchestrates rejuvenation of glia-neuron dialogue in Parkinson’s disease. Redox Biol. 2020, 36, 101664. [Google Scholar] [CrossRef]
- Toffoli, M.; Vieira, S.R.L.; Schapira, A.H.V. Genetic causes of PD: A pathway to disease modification. Neuropharmacology 2020, 170, 108022. [Google Scholar] [CrossRef]
- Marchetti, B. Wnt/β-catenin signaling pathway governs a full program for dopaminergic neuron survival, neurorescue and regeneration in the MPTP mouse model of Parkinson’s disease. Int. J. Mol. Sci. 2018, 19, 3743. [Google Scholar] [CrossRef] [Green Version]
- William Langston, J.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef]
- Langston, J.W. The MPTP story. J. Parkinsons. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltazar, M.T.; Dinis-Oliveira, R.J.; de Lourdes Bastos, M.; Tsatsakis, A.M.; Duarte, J.A.; Carvalho, F. Pesticides exposure as etiological factors of Parkinson’s disease and other neurodegenerative diseases-A mechanistic approach. Toxicol. Lett. 2014, 230, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, C.; El Dib, R.; de Camargo, J.L.V. Paraquat and Parkinson’s disease: A systematic review protocol according to the OHAT approach for hazard identification. Syst. Rev. 2017, 6, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, P.; Manna, J.; Dunbar, G.L.; Maiti, P.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Di Monte, D.A.; Wu, E.Y.; Irwin, I.; Delanney, L.E.; Langston, J.W. Biotransformation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in primary cultures of mouse astrocytes. J. Pharmacol. Exp. Ther. 1991, 258, 594–600. [Google Scholar]
- Heikkila, R.E.; Hess, A.; Duvoisin, R.C. Dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine in mice. Science 1984, 224, 1451–1453. [Google Scholar] [CrossRef]
- Javitch, J.A.; D’Amato, R.J.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Himeda, T.; Araki, T. Mechanisms of MPTP toxicity and their implications for therapy of Parkinson’s disease. Med. Sci. Monit. 2005, 11, RA17–RA23. [Google Scholar] [PubMed]
- Nicklas, W.J.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985, 36, 2503–2508. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Mapa, M.S.T.; Le, V.Q.; Wimalasena, K. Characteristics of the mitochondrial and cellular uptake of MPP+, as probed by the fluorescent mimic, 4’I-MPP+. PLoS ONE 2018, 13, e0197946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.H.; Gusdon, A.M.; Cimen, H.; Van Houten, B.; Koc, E.; Chu, C.T. Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: Dual roles for ERK1/2. Cell Death Dis. 2012, 3, e312. [Google Scholar] [CrossRef]
- Choi, W.S.; Kruse, S.E.; Palmiter, R.D.; Xia, Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc. Natl. Acad. Sci. USA 2008, 105, 15136–15141. [Google Scholar] [CrossRef] [Green Version]
- Biedler, J.L.; Schachner, M. Multiple Neurotransmitter Synthesis by Human Neuroblastoma Cell Lines and Clones. Cancer Res. 1978, 38, 3751–3757. [Google Scholar]
- Ross, R.A.; Biedler, J.L. Presence and Regulation of Tyrosinase Activity in Human Neuroblastoma Cell Variants in Vitro. Cancer Res. 1985, 45, 1628–1632. [Google Scholar]
- Påhlman, S.; Ruusala, A.I.; Abrahamsson, L.; Mattsson, M.E.K.; Esscher, T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: A comparison with phorbolester-induced differentiation. Cell. Differ. 1984, 14, 135–144. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Lopes, F.M.; Schröder, R.; Júnior, M.L.C. da F.; Zanotto-Filho, A.; Müller, C.B.; Pires, A.S.; Meurer, R.T.; Colpo, G.D.; Gelain, D.P.; Kapczinski, F.; et al. Comparison between proliferative and neuron-like SH-SY5Y cells as an in vitro model for Parkinson disease studies. Brain Res. 2010, 1337, 85–94. [Google Scholar] [CrossRef]
- Cheung, Y.T.; Lau, W.K.W.; Yu, M.S.; Lai, C.S.W.; Yeung, S.C.; So, K.F.; Chang, R.C.C. Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. Neurotoxicology 2009, 30, 127–135. [Google Scholar] [CrossRef]
- Teppola, H.; Sarkanen, J.R.; Jalonen, T.O.; Linne, M.L. Morphological Differentiation Towards Neuronal Phenotype of SH-SY5Y Neuroblastoma Cells by Estradiol, Retinoic Acid and Cholesterol. Neurochem. Res. 2016, 41, 731–747. [Google Scholar] [CrossRef] [Green Version]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar]
- Gnaiger, E.; MitoEAGLE Task Group. Mitochondrial physiology. Bioenerg. Commun. 2020, 1. [Google Scholar] [CrossRef]
- Zhu, J.H.; Horbinski, C.; Guo, F.; Watkins, S.; Uchiyama, Y.; Chu, C.T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol. 2007, 170, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Zilocchi, M.; Finzi, G.; Lualdi, M.; Sessa, F.; Fasano, M.; Alberio, T. Mitochondrial alterations in Parkinson’s disease human samples and cellular models. Neurochem. Int. 2018, 118, 61–72. [Google Scholar] [CrossRef]
- Dukes, A.A.; Bai, Q.; Van Laar, V.S.; Zhou, Y.; Ilin, V.; David, C.N.; Agim, Z.S.; Bonkowsky, J.L.; Cannon, J.R.; Watkins, S.C.; et al. Live imaging of mitochondrial dynamics in CNS dopaminergic neurons in vivo demonstrates early reversal of mitochondrial transport following MPP+ exposure. Neurobiol. Dis. 2016, 95, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnaiger, E. Capacity of oxidative phosphorylation in human skeletal muscle. New perspectives of mitochondrial physiology. Int. J. Biochem. Cell Biol. 2009, 41, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Djafarzadeh, S.; Jakob, S.M. High-resolution respirometry to assess mitochondrial function in permeabilized and intact cells. J. Vis. Exp. 2017, 120, 54985. [Google Scholar] [CrossRef]
- Pfleger, J.; He, M.; Abdellatif, M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015, 6, e1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evinova, A.; Cizmarova, B.; Hatokova, Z.; Racay, P. High-Resolution Respirometry in Assessment of Mitochondrial Function in Neuroblastoma SH-SY5Y Intact Cells. J. Membr. Biol. 2020, 253, 12–136. [Google Scholar]
- Stepanova, A.; Shurubor, Y.; Valsecchi, F.; Manfredi, G.; Galkin, A. Differential susceptibility of mitochondrial complex II to inhibition by oxaloacetate in brain and heart. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1561–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The voltage-dependent anion channel 1 mediates amyloid β toxicity and represents a potential target for Alzheimer disease therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef] [Green Version]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrì, A.; Belfiore, R.; Reina, S.; Tomasello, M.F.; Di Rosa, M.C.; Guarino, F.; Leggio, L.; De Pinto, V.; Messina, A. Hexokinase i N-terminal based peptide prevents the VDAC1-SOD1 G93A interaction and re-establishes ALS cell viability. Sci. Rep. 2016, 6, 34802. [Google Scholar] [CrossRef]
- Shteinfer-Kuzmine, A.; Argueti, S.; Gupta, R.; Shvil, N.; Abu-Hamad, S.; Gropper, Y.; Hoeber, J.; Magrì, A.; Messina, A.; Kozlova, E.N.; et al. A VDAC1-Derived N-Terminal Peptide Inhibits Mutant SOD1-VDAC1 Interactions and Toxicity in the SOD1 Model of ALS. Front. Cell. Neurosci. 2019, 13, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. α-synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef] [Green Version]
- Magri, A.; Messina, A. Interactions of VDAC with proteins involved in neurodegenerative aggregation: An opportunity for advancement on therapeutic molecules. Curr. Med. Chem. 2017, 24, 4470–4487. [Google Scholar] [CrossRef]
- Leggio, L.; Guarino, F.; Magrì, A.; Accardi-Gheit, R.; Reina, S.; Specchia, V.; Damiano, F.; Tomasello, M.F.; Tommasino, M.; Messina, A. Mechanism of translation control of the alternative Drosophila melanogaster Voltage Dependent Anion-selective Channel 1 mRNAs. Sci. Rep. 2018, 8, 5347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrì, A.; Di Rosa, M.C.; Tomasello, M.F.; Guarino, F.; Reina, S.; Messina, A.; De Pinto, V. Overexpression of human SOD1 in VDAC1-less yeast restores mitochondrial functionality modulating beta-barrel outer membrane protein genes. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1466–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrì, A.; Di Rosa, M.C.; Orlandi, I.; Guarino, F.; Reina, S.; Guarnaccia, M.; Morello, G.; Spampinato, A.; Cavallaro, S.; Messina, A.; et al. Deletion of Voltage-Dependent Anion Channel 1 knocks mitochondria down triggering metabolic rewiring in yeast. Cell. Mol. Life Sci. 2020, 77, 3195–3213. [Google Scholar] [CrossRef] [PubMed]
- Hoogerheide, D.P.; Gurnev, P.A.; Rostovtseva, T.K.; Bezrukov, S.M. Mechanism of α-synuclein translocation through a VDAC nanopore revealed by energy landscape modeling of escape time distributions. Nanoscale 2017, 9, 183–192. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Elkon, H.; Don, J.; Melamed, E.; Ziv, I.; Shirvan, A.; Offen, D. Mutant and wild-type α-synuclein interact with mitochondrial cytochrome C oxidase. J. Mol. Neurosci. 2002, 18, 229–238. [Google Scholar] [CrossRef]
- Fritz, R.R.; Abell, C.W.; Patel, N.T.; Gessner, W.; Brossi, A. Metabolism of the neurotoxin in MPTP by human liver monoamine oxidase B. FEBS Lett. 1985, 186, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar]
- Ramonet, D.; Perier, C.; Recasens, A.; Dehay, B.; Bové, J.; Costa, V.; Scorrano, L.; Vila, M. Optic atrophy 1 mediates mitochondria remodeling and dopaminergic neurodegeneration linked to complex i deficiency. Cell Death Differ. 2013, 20, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Calabria, E.; Scambi, I.; Bonafede, R.; Schiaffino, L.; Peroni, D.; Potrich, V.; Capelli, C.; Schena, F.; Mariotti, R. Ascs-exosomes recover coupling efficiency and mitochondrial membrane potential in an in vitro model of als. Front. Neurosci. 2019, 13, 1070. [Google Scholar] [CrossRef] [Green Version]
- Ricquier, D.; Bouillaud, F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem. J. 2000, 345, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.K. Mitochondrial proton leak: A role for uncoupling proteins 2 and 3? Biochim. Biophys. Acta Bioenerg. 2001, 1504, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Cannon, B.; Shabalina, I.G.; Kramarova, T.V.; Petrovic, N.; Nedergaard, J. Uncoupling proteins: A role in protection against reactive oxygen species-or not? Biochim. Biophys. Acta Bioenerg. 2006, 1757, 449–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.W.L.; Chan, D.Y.L.; Kwok, K.H.H.; Chu, A.C.Y.; Ho, J.W.M.; Kung, M.H.W.; Ramsden, D.B.; Ho, S.L. Methyl-4-phenylpyridinium ion modulates expression of mitochondrial uncoupling proteins 2, 4, and 5 in catecholaminergic (SK-N-SH) cells. J. Neurosci. Res. 2005, 81, 261–268. [Google Scholar] [CrossRef]
- Chu, A.C.Y.; Ho, P.W.L.; Kwok, K.H.H.; Ho, J.W.M.; Chan, K.H.; Liu, H.F.; Kung, M.H.W.; Ramsden, D.B.; Ho, S.L. Mitochondrial UCP4 attenuates MPP+- and dopamine-induced oxidative stress, mitochondrial depolarization, and ATP deficiency in neurons and is interlinked with UCP2 expression. Free Radic. Biol. Med. 2009, 46, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Kwok, K.H.H.; Ho, P.W.L.; Chu, A.C.Y.; Ho, J.W.M.; Liu, H.F.; Yiu, D.C.W.; Chan, K.H.; Kung, M.H.W.; Ramsden, D.B.; Ho, S.L. Mitochondrial UCP5 is neuroprotective by preserving mitochondrial membrane potential, ATP levels, and reducing oxidative stress in MPP+ and dopamine toxicity. Free Radic. Biol. Med. 2010, 49, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.L.; Ho, J.W.M.; Tse, H.M.; So, D.H.F.; Yiu, D.C.W.; Liu, H.F.; Chan, K.H.; Kung, M.H.W.; Ramsden, D.B.; Ho, S.L. Uncoupling protein-4 (UCP4) increases ATP supply by interacting with mitochondrial complex II in neuroblastoma cells. PLoS ONE 2012, 7, e32810. [Google Scholar] [CrossRef] [Green Version]
- L’Episcopo, F.; Serapide, M.F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Pluchino, S.; Marchetti, B. A Wnt1 regulated Frizzled-1/β-catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: Therapeutical relevance for neuron survival and neuroprotection. Mol. Neurodegener. 2011, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y human neuroblastoma cell line. J. Vis. Exp. 2016, 108, 53193. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Risiglione, P.; Leggio, L.; Cubisino, S.A.M.; Reina, S.; Paternò, G.; Marchetti, B.; Magrì, A.; Iraci, N.; Messina, A. High-Resolution Respirometry Reveals MPP+ Mitochondrial Toxicity Mechanism in a Cellular Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 7809. https://doi.org/10.3390/ijms21217809
Risiglione P, Leggio L, Cubisino SAM, Reina S, Paternò G, Marchetti B, Magrì A, Iraci N, Messina A. High-Resolution Respirometry Reveals MPP+ Mitochondrial Toxicity Mechanism in a Cellular Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2020; 21(21):7809. https://doi.org/10.3390/ijms21217809
Chicago/Turabian StyleRisiglione, Pierpaolo, Loredana Leggio, Salvatore A. M. Cubisino, Simona Reina, Greta Paternò, Bianca Marchetti, Andrea Magrì, Nunzio Iraci, and Angela Messina. 2020. "High-Resolution Respirometry Reveals MPP+ Mitochondrial Toxicity Mechanism in a Cellular Model of Parkinson’s Disease" International Journal of Molecular Sciences 21, no. 21: 7809. https://doi.org/10.3390/ijms21217809
APA StyleRisiglione, P., Leggio, L., Cubisino, S. A. M., Reina, S., Paternò, G., Marchetti, B., Magrì, A., Iraci, N., & Messina, A. (2020). High-Resolution Respirometry Reveals MPP+ Mitochondrial Toxicity Mechanism in a Cellular Model of Parkinson’s Disease. International Journal of Molecular Sciences, 21(21), 7809. https://doi.org/10.3390/ijms21217809