RAGE Up-Regulation Differently Affects Cell Proliferation and Migration in Pancreatic Cancer Cells

 ,
,
Abstract
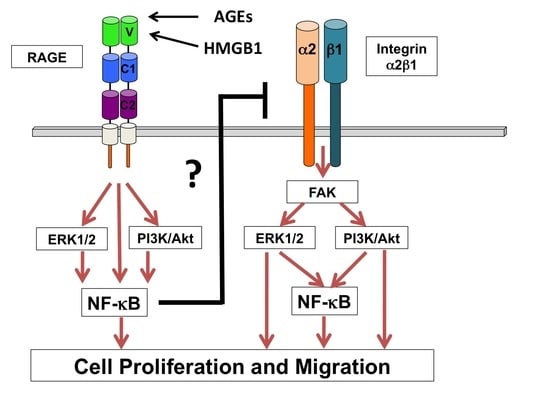
1. Introduction
2. Results and Discussion
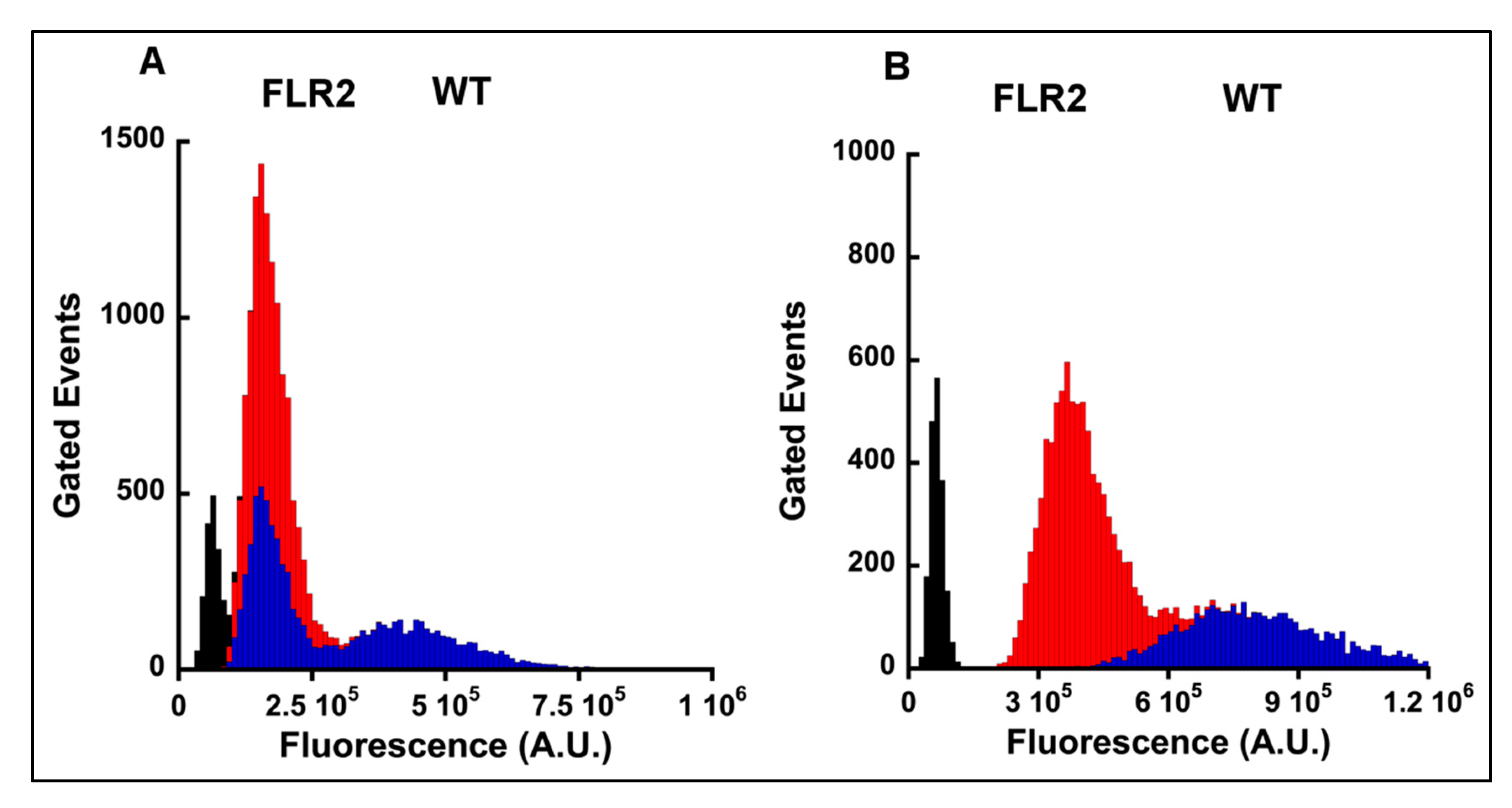
2.1. Generation and Characterization of RAGE Overexpressing Panc-1 Cells
2.2. Effect of RAGE Up-Regulation on Cell Proliferation
2.3. Effect of RAGE Up-Regulation on Cell Migration
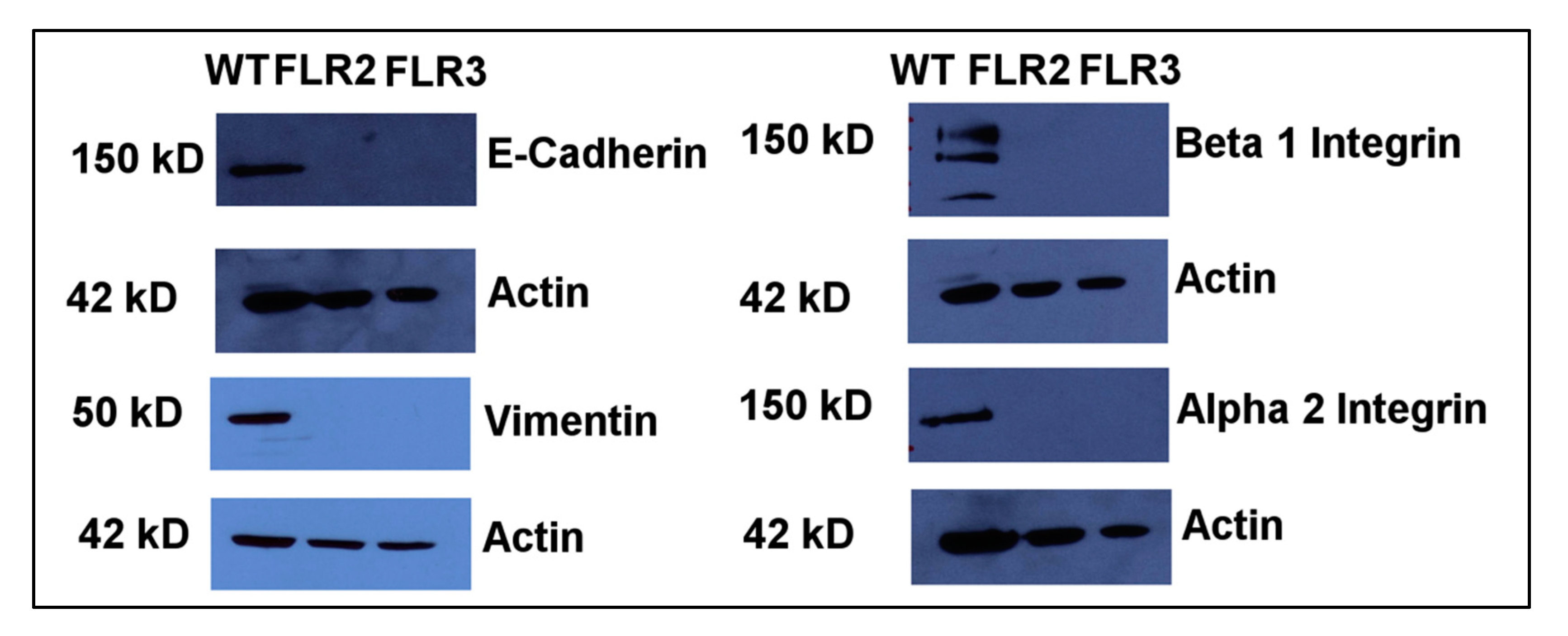
2.4. The Levels of α2 and β1 Integrins Are Significantly Decreased in RAGE Overexpressing Cells
2.5. RAGE Overexpression Is Associated with Changes in Expression Levels of Vimentin and E-Cadherin
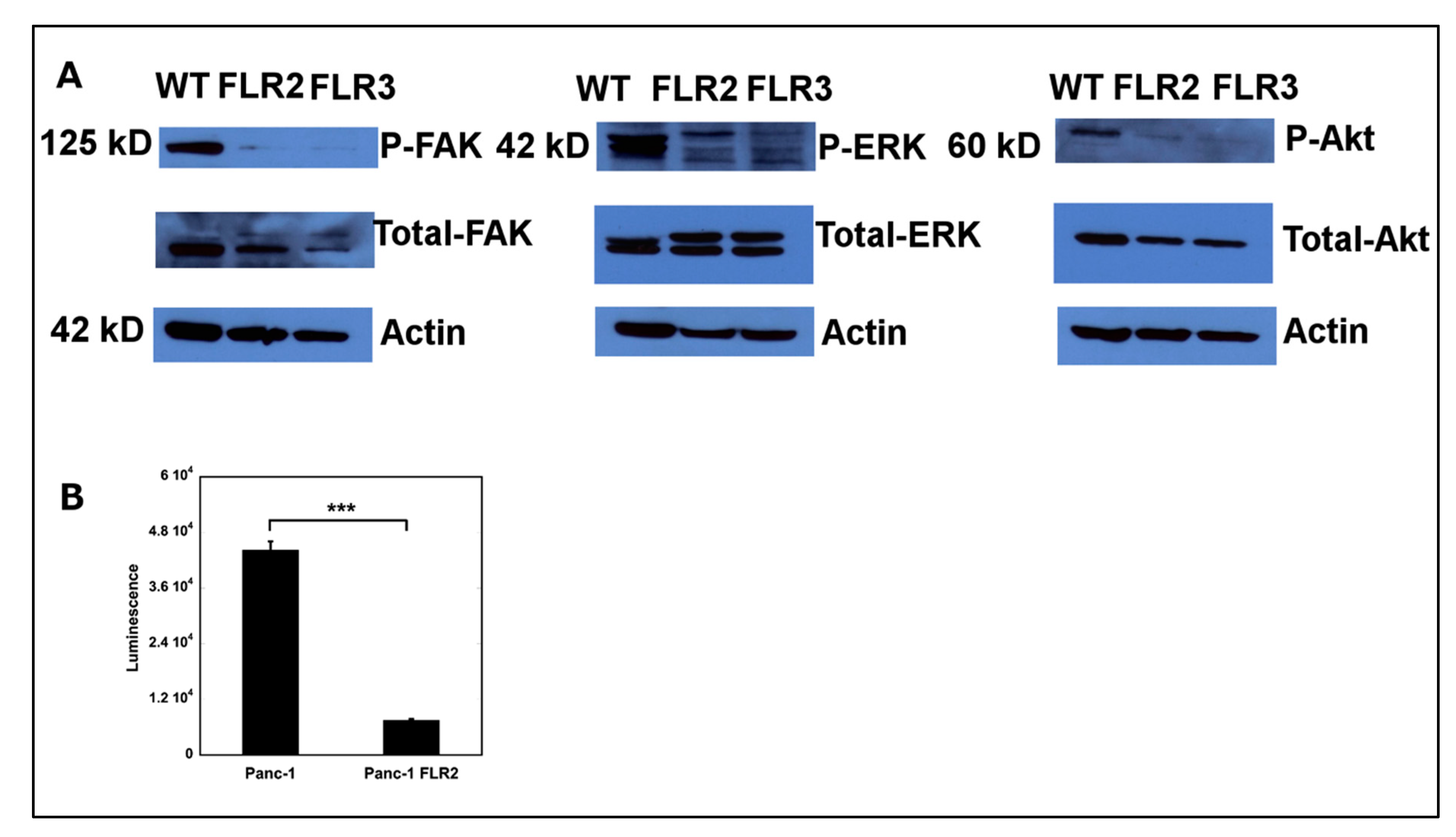
2.6. Overexpression of RAGE Results in Down-Regulation of FAK, ERK1/2, Akt, and NF-κB
3. Materials and Methods
3.1. Cell Culture
3.2. Generation of Stably Transfected FLR2 and FLR3 Panc-1 Cells
3.3. Determination of RAGE Transcript and Protein Levels in FLR2 and FLR3 Panc-1 Cells
3.4. Western Blot Detection of Akt/P-Akt, ERK/P-ERK, FAK/P-FAK, Vimentin, E-Cadherin, N-Cadherin, β-Catenin, Snail, Slug, ZEB1, ZO-1, α2 and β1 Integrins, and Actin
3.5. Flow Cytometry Studies
3.6. Quantitative ELISA
3.7. Cell Proliferation Assays
3.8. Boyden Chamber Assay
signal without insert − Resazurin background signal)
3.9. Wound Healing Assay
3.10. NF-κB Transcription Factor Activation Assay
3.11. Transfection with RAGE siRNAs
3.12. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RAGE | Receptor for advanced glycation endproducts |
| FLR2 | Full-length RAGE Panc-1 clone 2 |
| FLR3 | Full-length RAGE Panc-1 clone 3 |
| WT | Wild-type |
References
- Yan, S.F.; Yan, S.D.; Ramasamy, R.; Schmidt, A.M. Tempering the wrath of RAGE: An emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann. Med. 2009, 41, 408–422. [Google Scholar] [CrossRef]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Munch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Logsdon, C.D.; Fuentes, M.K.; Huang, E.H.; Arumugam, T. RAGE and RAGE ligands in cancer. Curr. Mol. Med. 2007, 7, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Foell, D.; Wittkowski, H.; Vogl, T.; Roth, J. S100 proteins expressed in phagocytes: A novel group of damage-associated molecular pattern molecules. J. Leukoc. Biol. 2007, 81, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, D.; Foell, D.; Kessel, C. The role of S100 proteins in the pathogenesis and monitoring of autoinflammatory diseases. Mol. Cell. Pediatr. 2018, 5, 7. [Google Scholar] [CrossRef]
- Kessel, C.; Holzinger, D.; Foell, D. Phagocyte-derived S100 proteins in autoinflammation: Putative role in pathogenesis and usefulness as biomarkers. Clin. Immunol. 2013, 147, 229–241. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Hofmann, M.; Taguchi, A.; Yan, S.D.; Stern, D.M. RAGE: A multiligand receptor contributing to the cellular response in diabetic vasculopathy and inflammation. Semin. Thromb. Hemost. 2000, 26, 485–493. [Google Scholar] [CrossRef]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar]
- Bierhaus, A.; Stern, D.M.; Nawroth, P.P. RAGE in inflammation: A new therapeutic target? Curr. Opin. Investig. Drugs 2006, 7, 985–991. [Google Scholar]
- Sakatani, S.; Yamada, K.; Homma, C.; Munesue, S.; Yamamoto, Y.; Yamamoto, H.; Hirase, H. Deletion of RAGE causes hyperactivity and increased sensitivity to auditory stimuli in mice. PLoS ONE 2009, 4, e8309. [Google Scholar] [CrossRef]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. RAGE: Therapeutic target and biomarker of the inflammatory response--the evidence mounts. J. Leukoc. Biol. 2009, 86, 505–512. [Google Scholar] [CrossRef]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Grone, H.J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef]
- Lutterloh, E.C.; Opal, S.M.; Pittman, D.D.; Keith, J.C., Jr.; Tan, X.Y.; Clancy, B.M.; Palmer, H.; Milarski, K.; Sun, Y.; Palardy, J.E.; et al. Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit. Care 2007, 11, R122. [Google Scholar] [CrossRef]
- DiNorcia, J.; Lee, M.K.; Moroziewicz, D.N.; Winner, M.; Suman, P.; Bao, F.; Remotti, H.E.; Zou, Y.S.; Yan, S.F.; Qiu, W.; et al. RAGE gene deletion inhibits the development and progression of ductal neoplasia and prolongs survival in a murine model of pancreatic cancer. J. Gastrointest. Surg. 2012, 16, 104–112. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J., 3rd. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef]
- Yamamoto, K.; Murata, H.; Putranto, E.W.; Kataoka, K.; Motoyama, A.; Hibino, T.; Inoue, Y.; Sakaguchi, M.; Huh, N.H. DOCK7 is a critical regulator of the RAGE-Cdc42 signaling axis that induces formation of dendritic pseudopodia in human cancer cells. Oncol. Rep. 2013, 29, 1073–1079. [Google Scholar] [CrossRef]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Nemeth, J.; Furstenberger, G.; Muller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef]
- Donato, R. RAGE: A single receptor for several ligands and different cellular responses: The case of certain S100 proteins. Curr. Mol. Med. 2007, 7, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Nawroth, P.P. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009, 52, 2251–2263. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, X.; Schmidt, A.M. Sp1-binding elements in the promoter of RAGE are essential for amphoterin-mediated gene expression in cultured neuroblastoma cells. J. Biol. Chem. 1998, 273, 30870–30878. [Google Scholar] [CrossRef]
- Riehl, A.; Nemeth, J.; Angel, P.; Hess, J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Bartling, B.; Demling, N.; Silber, R.E.; Simm, A. Proliferative stimulus of lung fibroblasts on lung cancer cells is impaired by the receptor for advanced glycation end-products. Am. J. Respir. Cell Mol. Biol. 2006, 34, 83–91. [Google Scholar] [CrossRef]
- Riuzzi, F.; Sorci, G.; Donato, R. The amphoterin (HMGB1)/receptor for advanced glycation end products (RAGE) pair modulates myoblast proliferation, apoptosis, adhesiveness, migration, and invasiveness. Functional inactivation of RAGE in L6 myoblasts results in tumor formation in vivo. J. Biol. Chem. 2006, 281, 8242–8253. [Google Scholar] [CrossRef]
- Riuzzi, F.; Sorci, G.; Donato, R. RAGE expression in rhabdomyosarcoma cells results in myogenic differentiation and reduced proliferation, migration, invasiveness, and tumor growth. Am. J. Pathol. 2007, 171, 947–961. [Google Scholar] [CrossRef]
- Arumugam, T.; Simeone, D.M.; Van Golen, K.; Logsdon, C.D. S100P promotes pancreatic cancer growth, survival, and invasion. Clin. Cancer Res. 2005, 11, 5356–5364. [Google Scholar] [CrossRef]
- Arumugam, T.; Logsdon, C.D. S100P: A novel therapeutic target for cancer. Amino Acids 2011, 41, 893–899. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Gomez, S.B.; Schmidt, A.M.; Logsdon, C.D. S100P-Derived RAGE Antagonistic Peptide Reduces Tumor Growth and Metastasis. Clin. Cancer Res. 2012, 18, 4356–4364. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Logsdon, C.D. Effect of cromolyn on S100P interactions with RAGE and pancreatic cancer growth and invasion in mouse models. J. Natl. Cancer Inst. 2006, 98, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Sun, D.; Peng, Z.; Pal, A.; Maxwell, D.S.; Bornmann, W.G.; Logsdon, C.D. Designing and developing S100P inhibitor 5-methyl cromolyn for pancreatic cancer therapy. Mol. Cancer Ther. 2013, 12, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Simeone, D.M.; Schmidt, A.M.; Logsdon, C.D. S100P stimulates cell proliferation and survival via receptor for activated glycation end products (RAGE). J. Biol. Chem. 2004, 279, 5059–5065. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Schapiro, N.E.; Loux, T.; Livesey, K.M.; Billiar, T.R.; Wang, H.; Van Houten, B.; Lotze, M.T.; Zeh, H.J. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 2014, 33, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D. Autophagy in pancreatic cancer pathogenesis and treatment. Am. J. Cancer Res. 2012, 2, 383–396. [Google Scholar] [PubMed]
- Kang, R.; Tang, D.; Livesey, K.M.; Schapiro, N.E.; Lotze, M.T.; Zeh, H.J., 3rd. The Receptor for Advanced Glycation End-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid. Redox Signal. 2011, 15, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Lotze, M.T.; Zeh, H.J., 3rd. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy 2012, 8, 989–991. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D.; Schapiro, N.E.; Livesey, K.M.; Farkas, A.; Loughran, P.; Bierhaus, A.; Lotze, M.T.; Zeh, H.J. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010, 17, 666–676. [Google Scholar] [CrossRef]
- Li, D.; Lei, C.; Zhang, S.; Zhang, S.; Liu, M.; Wu, B. Blockade of high mobility group box-1 signaling via the receptor for advanced glycation end-products ameliorates inflammatory damage after acute intracerebral hemorrhage. Neurosci. Lett. 2015, 609, 109–119. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Yatime, L.; Andersen, G.R. Structural insights into the oligomerization mode of the human receptor for advanced glycation end-products. FEBS J. 2013, 280, 6556–6568. [Google Scholar] [CrossRef] [PubMed]
- Prica, F.; Radon, T.; Cheng, Y.; Crnogorac-Jurcevic, T. The life and works of S100P—From conception to cancer. Am. J. Cancer Res. 2016, 6, 562–576. [Google Scholar]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Grzesiak, J.J.; Bouvet, M. The alpha2beta1 integrin mediates the malignant phenotype on type I collagen in pancreatic cancer cell lines. Br. J. Cancer 2006, 94, 1311–1319. [Google Scholar] [CrossRef]
- Grzesiak, J.J.; Tran Cao, H.S.; Burton, D.W.; Kaushal, S.; Vargas, F.; Clopton, P.; Snyder, C.S.; Deftos, L.J.; Hoffman, R.M.; Bouvet, M. Knockdown of the beta(1) integrin subunit reduces primary tumor growth and inhibits pancreatic cancer metastasis. Int. J. Cancer 2011, 129, 2905–2915. [Google Scholar] [CrossRef]
- Bassaganas, S.; Carvalho, S.; Dias, A.M.; Perez-Garay, M.; Ortiz, M.R.; Figueras, J.; Reis, C.A.; Pinho, S.S.; Peracaula, R. Pancreatic cancer cell glycosylation regulates cell adhesion and invasion through the modulation of alpha2beta1 integrin and E-cadherin function. PLoS ONE 2014, 9, e98595. [Google Scholar] [CrossRef]
- Kreis, S.; Schonfeld, H.J.; Melchior, C.; Steiner, B.; Kieffer, N. The intermediate filament protein vimentin binds specifically to a recombinant integrin alpha2/beta1 cytoplasmic tail complex and co-localizes with native alpha2/beta1 in endothelial cell focal adhesions. Exp. Cell Res. 2005, 305, 110–121. [Google Scholar] [CrossRef]
- Gilles, C.; Polette, M.; Zahm, J.M.; Tournier, J.M.; Volders, L.; Foidart, J.M.; Birembaut, P. Vimentin contributes to human mammary epithelial cell migration. J. Cell Sci. 1999, 112 Pt 24, 4615–4625. [Google Scholar]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Birchmeier, W.; Behrens, J. Cadherin expression in carcinomas: Role in the formation of cell junctions and the prevention of invasiveness. Biochim. Biophys. Acta 1994, 1198, 11–26. [Google Scholar] [CrossRef]
- St Croix, B.; Sheehan, C.; Rak, J.W.; Florenes, V.A.; Slingerland, J.M.; Kerbel, R.S. E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1). J. Cell Biol. 1998, 142, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Caveda, L.; Martin-Padura, I.; Navarro, P.; Breviario, F.; Corada, M.; Gulino, D.; Lampugnani, M.G.; Dejana, E. Inhibition of cultured cell growth by vascular endothelial cadherin (cadherin-5/VE-cadherin). J. Clin. Investig. 1996, 98, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Perrais, M.; Chen, X.; Perez-Moreno, M.; Gumbiner, B.M. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol. Biol. Cell 2007, 18, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef]
- Stockinger, A.; Eger, A.; Wolf, J.; Beug, H.; Foisner, R. E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J. Cell Biol. 2001, 154, 1185–1196. [Google Scholar] [CrossRef]
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006, 66, 4662–4671. [Google Scholar] [CrossRef]
- Zhou, W.; Lv, R.; Qi, W.; Wu, D.; Xu, Y.; Liu, W.; Mou, Y.; Wang, L. Snail contributes to the maintenance of stem cell-like phenotype cells in human pancreatic cancer. PLoS ONE 2014, 9, e87409. [Google Scholar] [CrossRef]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta (BBA)-Biomembr. 2008, 1778, 660–669. [Google Scholar] [CrossRef]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adhes. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. Embo J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Delaney, M.K.; Du, X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr. Opin. Cell Biol. 2012, 24, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Downey-Biechler, C.; Craig, D.H.; More, S.K.; Basson, M.D. Inside-out signaling through FAK-integrin axis may regulate circulating cancer cell metastatic adhesion. Proc. Natl. Acad. Sci. USA 2019, 116, 19795–19796. [Google Scholar] [CrossRef]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- De Heer, P.; Koudijs, M.M.; van de Velde, C.J.; Aalbers, R.I.; Tollenaar, R.A.; Putter, H.; Morreau, J.; van de Water, B.; Kuppen, P.J. Combined expression of the non-receptor protein tyrosine kinases FAK and Src in primary colorectal cancer is associated with tumor recurrence and metastasis formation. Eur. J. Surg. Oncol. 2008, 34, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Tabellini, G.; Bressanin, D.; Ognibene, A.; Goto, K.; Cocco, L.; Evangelisti, C. The emerging multiple roles of nuclear Akt. Biochim. Biophys. Acta 2012, 1823, 2168–2178. [Google Scholar] [CrossRef]
- Toker, A.; Marmiroli, S. Signaling specificity in the Akt pathway in biology and disease. Adv. Biol. Regul. 2014, 55, 28–38. [Google Scholar] [CrossRef]
- Chin, Y.R.; Toker, A. The actin-bundling protein palladin is an Akt1-specific substrate that regulates breast cancer cell migration. Mol. Cell 2010, 38, 333–344. [Google Scholar] [CrossRef]
- Dillon, R.L.; Marcotte, R.; Hennessy, B.T.; Woodgett, J.R.; Mills, G.B.; Muller, W.J. Akt1 and akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res. 2009, 69, 5057–5064. [Google Scholar] [CrossRef]
- Hutchinson, J.N.; Jin, J.; Cardiff, R.D.; Woodgett, J.R.; Muller, W.J. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res. 2004, 64, 3171–3178. [Google Scholar] [CrossRef]
- Irie, H.Y.; Pearline, R.V.; Grueneberg, D.; Hsia, M.; Ravichandran, P.; Kothari, N.; Natesan, S.; Brugge, J.S. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J. Cell Biol. 2005, 171, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Radisky, D.C.; Nelson, C.M.; Zhang, H.; Fata, J.E.; Roth, R.A.; Bissell, M.J. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc. Natl. Acad. Sci. USA 2006, 103, 4134–4139. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-kappaB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Oue, N.; Wakikawa, A.; Shigeishi, H.; Matsutani, N.; Kuraoka, K.; Ito, R.; Yokozaki, H.; Yasui, W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J. Pathol. 2002, 196, 163–170. [Google Scholar] [CrossRef]
- Meghnani, V.; Vetter, S.W.; Leclerc, E. RAGE overexpression confers a metastatic phenotype to the WM115 human primary melanoma cell line. Biochim. Biophys. Acta 2014, 1842, 1017–1027. [Google Scholar] [CrossRef]
- Bartling, B.; Hofmann, H.S.; Weigle, B.; Silber, R.E.; Simm, A. Down-regulation of the receptor for advanced glycation end-products (RAGE) supports non-small cell lung carcinoma. Carcinogenesis 2005, 26, 293–301. [Google Scholar] [CrossRef]
- Bhawal, U.K.; Ozaki, Y.; Nishimura, M.; Sugiyama, M.; Sasahira, T.; Nomura, Y.; Sato, F.; Fujimoto, K.; Sasaki, N.; Ikeda, M.A.; et al. Association of expression of receptor for advanced glycation end products and invasive activity of oral squamous cell carcinoma. Oncology 2005, 69, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Kwak, T.; Drews-Elger, K.; Ergonul, A.; Miller, P.C.; Braley, A.; Hwang, G.H.; Zhao, D.; Besser, A.; Yamamoto, Y.; Yamamoto, H.; et al. Targeting of RAGE-ligand signaling impairs breast cancer cell invasion and metastasis. Oncogene 2017, 36, 1559–1572. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Chihara, Y.; Kondo, H. Differential effects between amphoterin and advanced glycation end products on colon cancer cells. Int. J. Cancer 2003, 104, 722–727. [Google Scholar] [CrossRef]
- Bao, J.M.; He, M.Y.; Liu, Y.W.; Lu, Y.J.; Hong, Y.Q.; Luo, H.H.; Ren, Z.L.; Zhao, S.C.; Jiang, Y. AGE/RAGE/Akt pathway contributes to prostate cancer cell proliferation by promoting Rb phosphorylation and degradation. Am. J. Cancer Res. 2015, 5, 1741–1750. [Google Scholar] [CrossRef]
- Hudson, B.I.; Kalea, A.Z.; Del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Murata, H.; Yamamoto, K.; Ono, T.; Sakaguchi, Y.; Motoyama, A.; Hibino, T.; Kataoka, K.; Huh, N.H. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS ONE 2011, 6, e23132. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence |
|---|---|
| β-Actin fwd | CATGTACGTTGCTATCCAGGC |
| β-Actin rev | CTCCTTAATGTCACGCACGAT |
| RAGE fwd | TGTGTGGCCACCCATTCCAG |
| RAGE rev | GCCCTCCAGTACTACTCTCG |
| α2 Integrin fwd | CCTACAATGTTGGTCTCCCAGA |
| α2 Integrin rev | AGTAACCAGTTGCCTTTTGGATT |
| β1 Integrin fwd | CCTACTTCTGCACGATGTGATG |
| β1 Integrin rev | CCTTTGCTACGGTTGGTTACATT |
| Antigen Name (Poly- or Mono-Clonal) | Method (Dilution Used) | Cat. Number | Provider | Species |
|---|---|---|---|---|
| Akt (mono) | WB (1/1000) | 4685 | Cell Signaling | Rabbit |
| P-Akt (mono) | WB (1/2000) | 4060 | Cell Signaling | Rabbit |
| ERK1/2 (mono) | WB (1/1000) | 4695 | Cell Signaling | Rabbit |
| P-ERK1/2 (poly) | WB (1/1000) | 9101 | Cell Signaling | Rabbit |
| RAGE N-16 (poly) | WB (1/500) | sc-8230 | Santa Cruz Biotechnologies | Goat |
| β-Actin (poly) | WB (1/500) | sc-1616 | Santa Cruz Biotechnologies | Goat |
| FAK (mono) | WB (1/500) | sc-271126 | Santa Cruz Biotechnologies | Mouse |
| P-FAK (mono) | WB (1/500) | sc-81493 | Santa Cruz Biotechnologies | Mouse |
| Vimentin (mono) | WB (1/1000) | sc-6260 | Santa Cruz Biotechnologies | Mouse |
| E-Cadherin (mono) | WB (1/10000) | ab40772 | Abcam | Rabbit |
| N-Cadherin (mono) | WB (1/1000) | 13116 | Cell signaling | Rabbit |
| β-Catenin (mono) | WB (1/1000) | 8480 | Cell Signaling | Rabbit |
| ZEB1 (mono) | WB (1/1000) | 3396 | Cell Signaling | Rabbit |
| ZO-1 (mono) | WB (1/1000) | 8193 | Cell Signaling | Rabbit |
| Snail (mono) | WB (1/1000) | 3879 | Cell Signaling | Rabbit |
| Slug (mono) | WB (1/1000) | 9585 | Cell Signaling | Rabbit |
| β1 Integrin (mono) | WB (1/10000) | ab52971 | Abcam | Rabbit |
| β1 Integrin (mono) | FC (1/100) | MA5-31981 | Invitrogen | Rabbit |
| α2 Integrin (mono) | WB (1/10000) | ab133557 | Abcam | Rabbit |
| α2 Integrin (mono) | FC (1/500) | EPR5788 | Abcam | Rabbit |
| AlexaFluor 488 conjugated secondary antibody (poly) | FC (1/1000) | 4412 | Cell Signaling | Goat |
| HRP-conjugated donkey anti-goat antibody (poly) | WB (1/50000) | 711-035-152 | Jackson ImunoResearch | Donkey |
| HRP-conjugated goat anti-mouse antibody (poly) | WB (1/50000) | 115-035-062 | Jackson ImmunoResearch | Goat |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swami, P.; Thiyagarajan, S.; Vidger, A.; Indurthi, V.S.K.; Vetter, S.W.; Leclerc, E. RAGE Up-Regulation Differently Affects Cell Proliferation and Migration in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2020, 21, 7723. https://doi.org/10.3390/ijms21207723
Swami P, Thiyagarajan S, Vidger A, Indurthi VSK, Vetter SW, Leclerc E. RAGE Up-Regulation Differently Affects Cell Proliferation and Migration in Pancreatic Cancer Cells. International Journal of Molecular Sciences. 2020; 21(20):7723. https://doi.org/10.3390/ijms21207723
Chicago/Turabian StyleSwami, Priyanka, Swetha Thiyagarajan, Arianna Vidger, Venkata S. K. Indurthi, Stefan W. Vetter, and Estelle Leclerc. 2020. "RAGE Up-Regulation Differently Affects Cell Proliferation and Migration in Pancreatic Cancer Cells" International Journal of Molecular Sciences 21, no. 20: 7723. https://doi.org/10.3390/ijms21207723
APA StyleSwami, P., Thiyagarajan, S., Vidger, A., Indurthi, V. S. K., Vetter, S. W., & Leclerc, E. (2020). RAGE Up-Regulation Differently Affects Cell Proliferation and Migration in Pancreatic Cancer Cells. International Journal of Molecular Sciences, 21(20), 7723. https://doi.org/10.3390/ijms21207723