Setup and Validation of a Reliable Docking Protocol for the Development of Neuroprotective Agents by Targeting the Sigma-1 Receptor (S1R)

 , , , ,
, , , ,  ,
,  , ,
, , 

Abstract
1. Introduction
2. Results
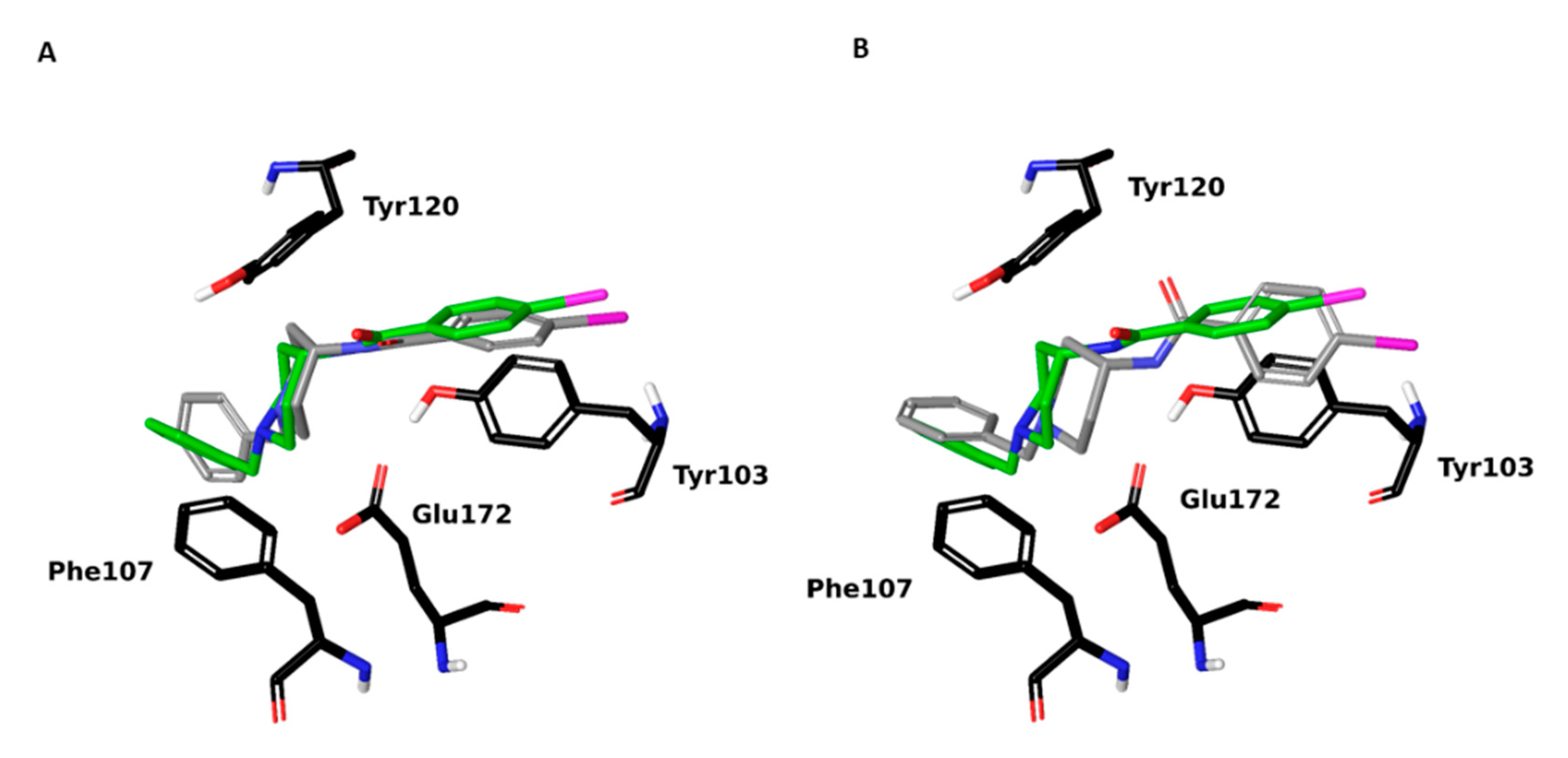
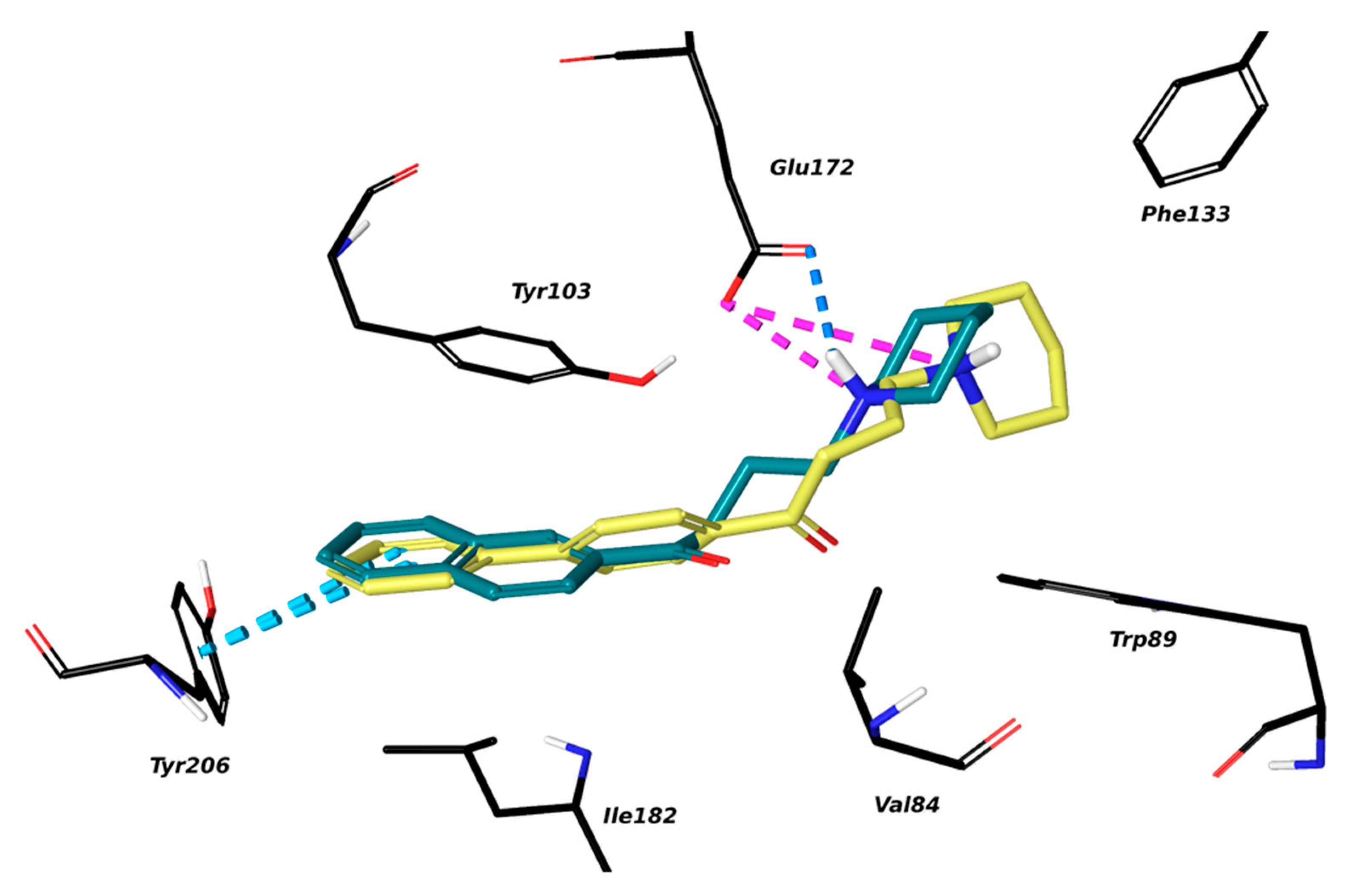
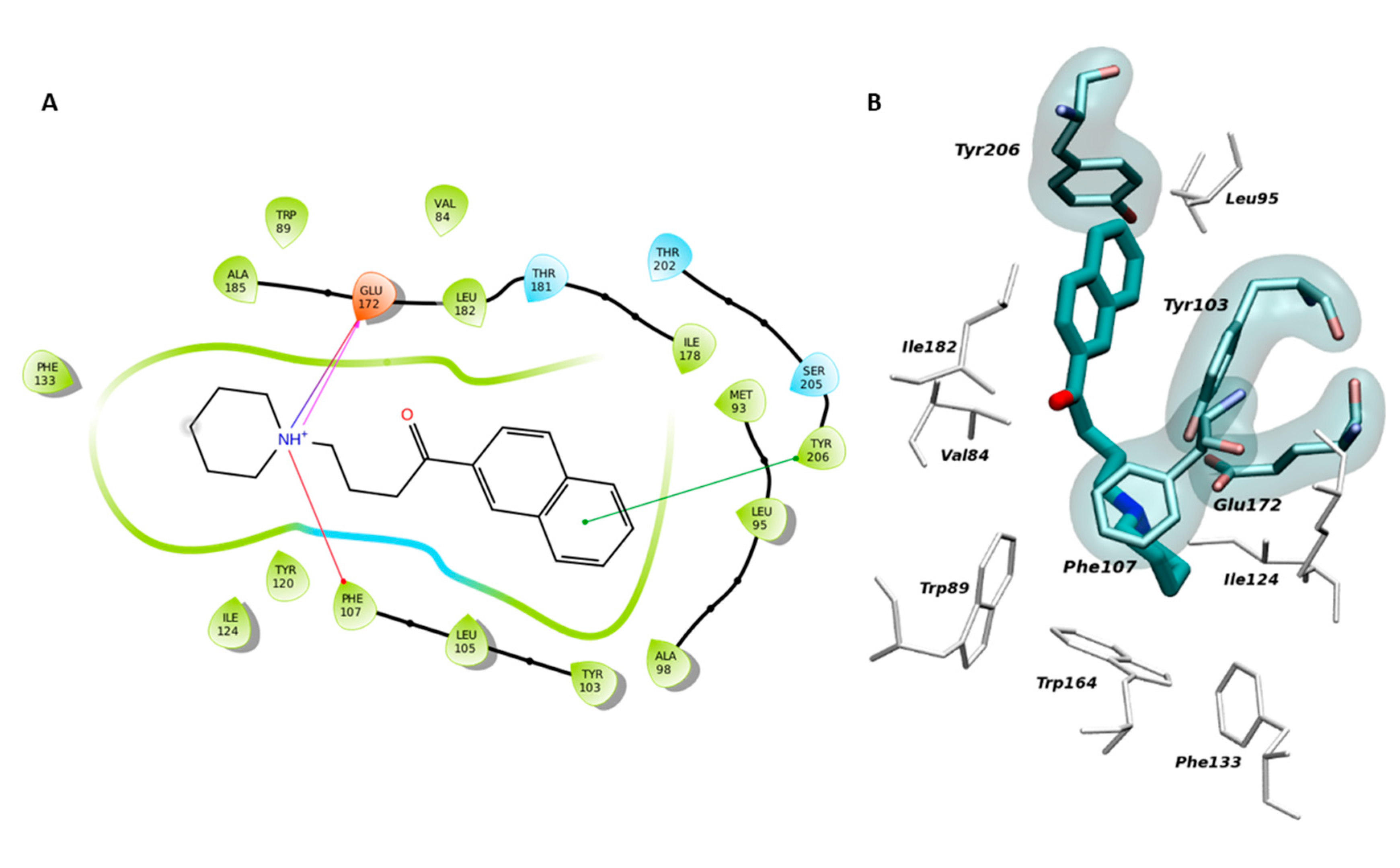
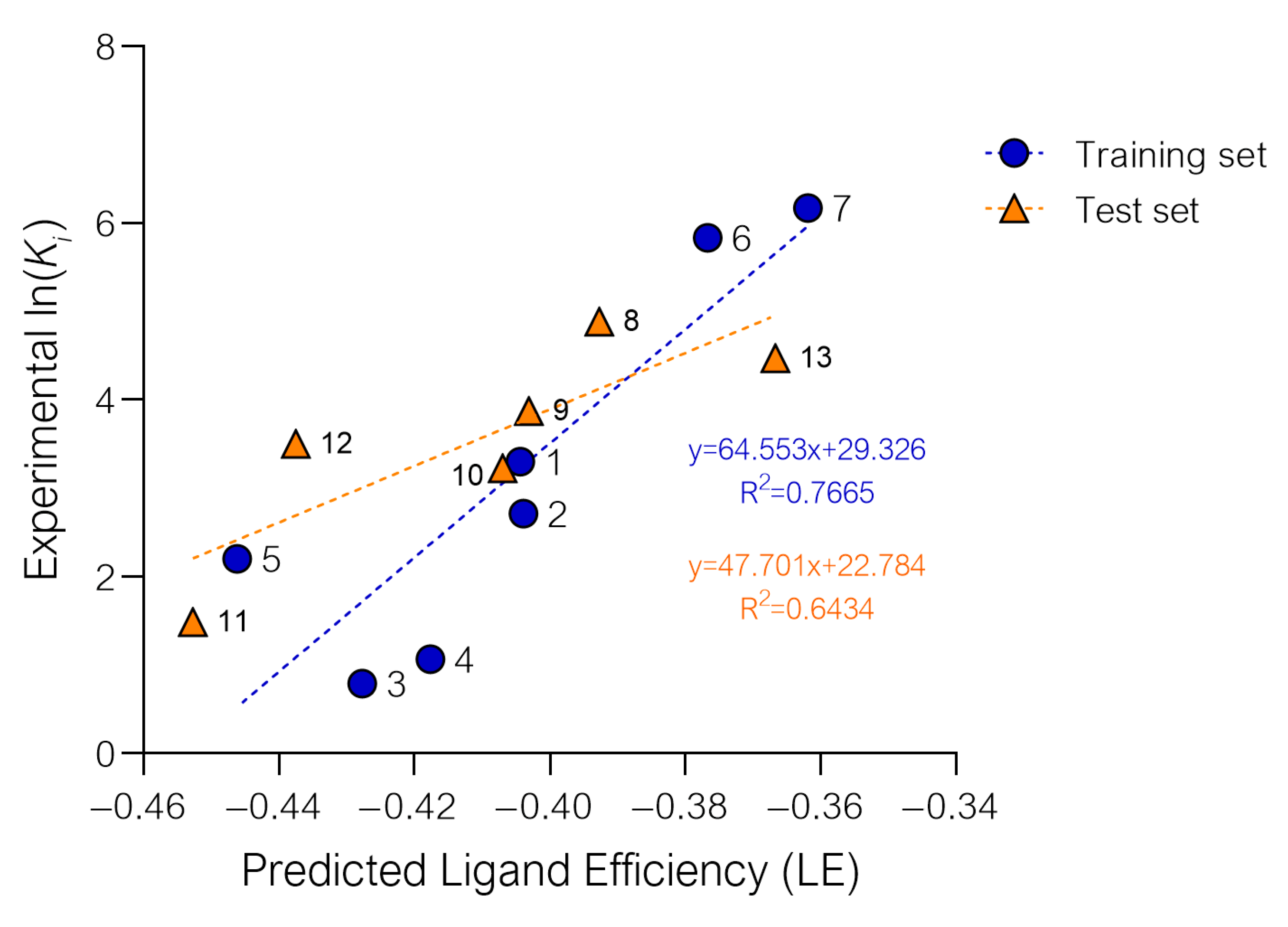
2.1. Setup and Validation of a Docking Model to Predict Affinity toward S1R
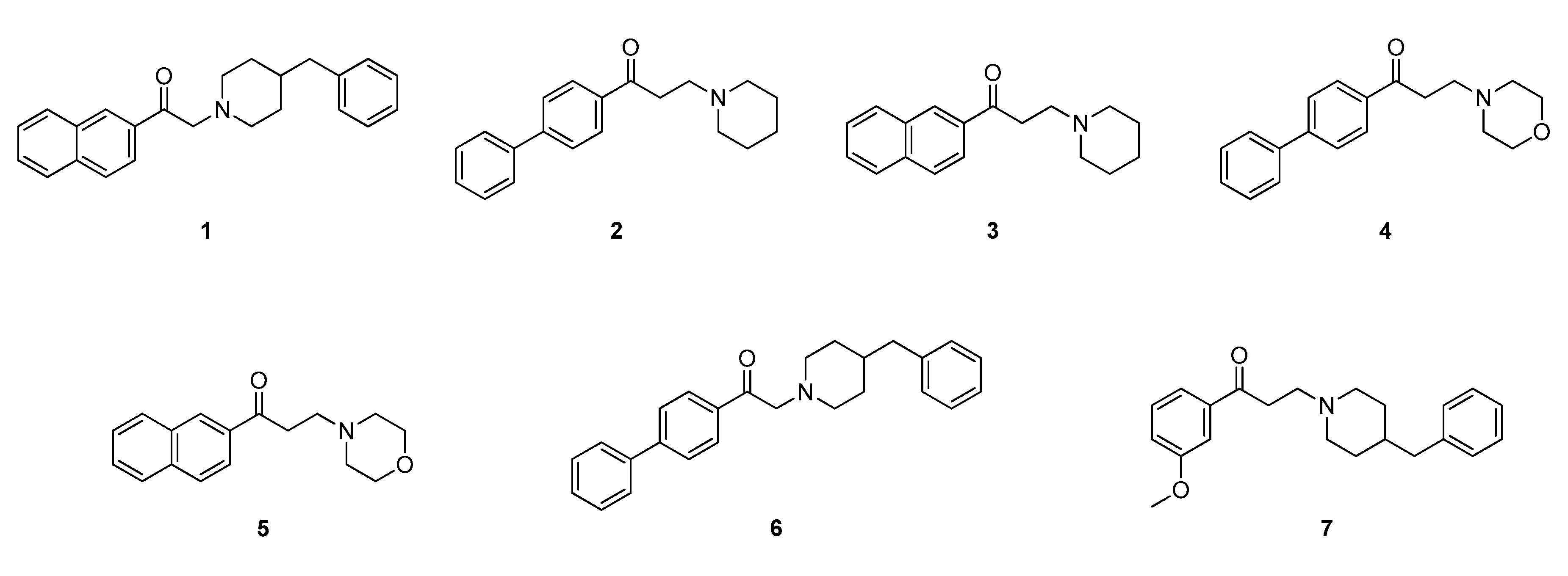
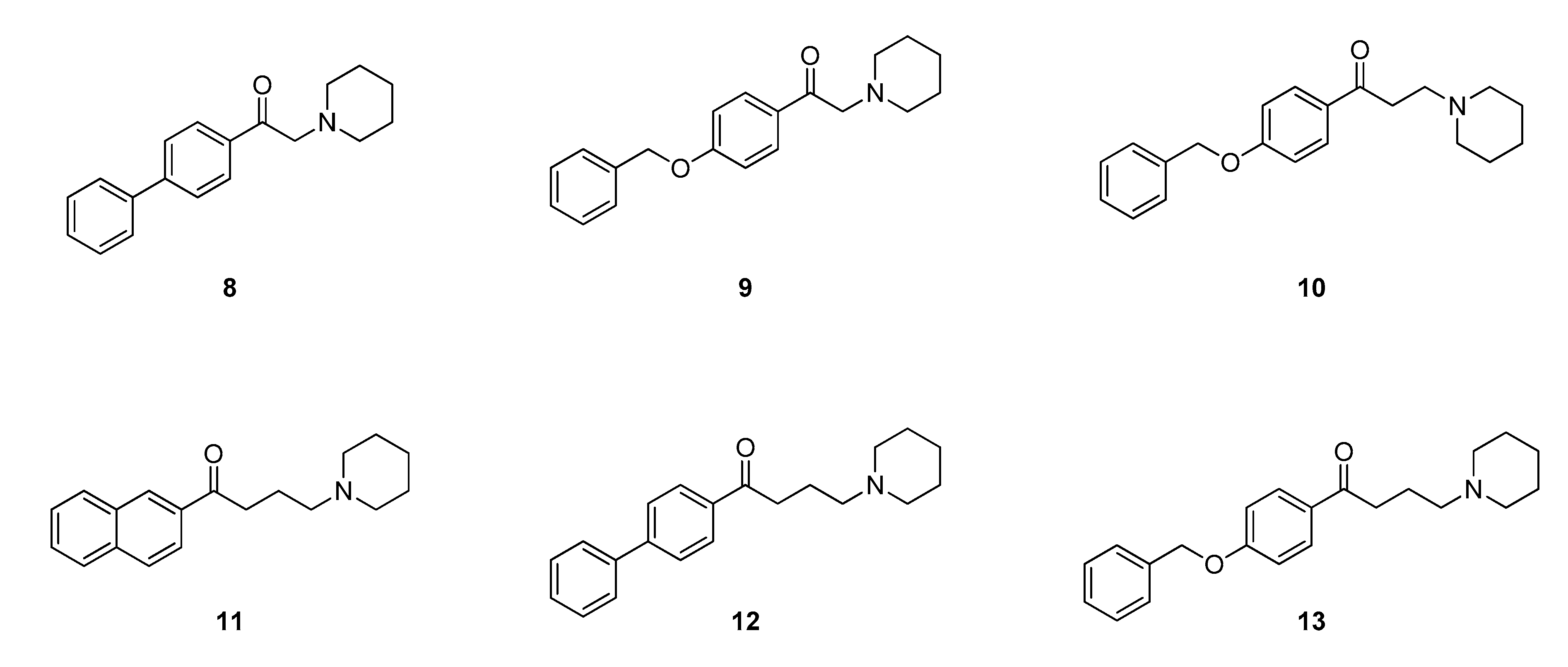
2.2. Design of New Ligands
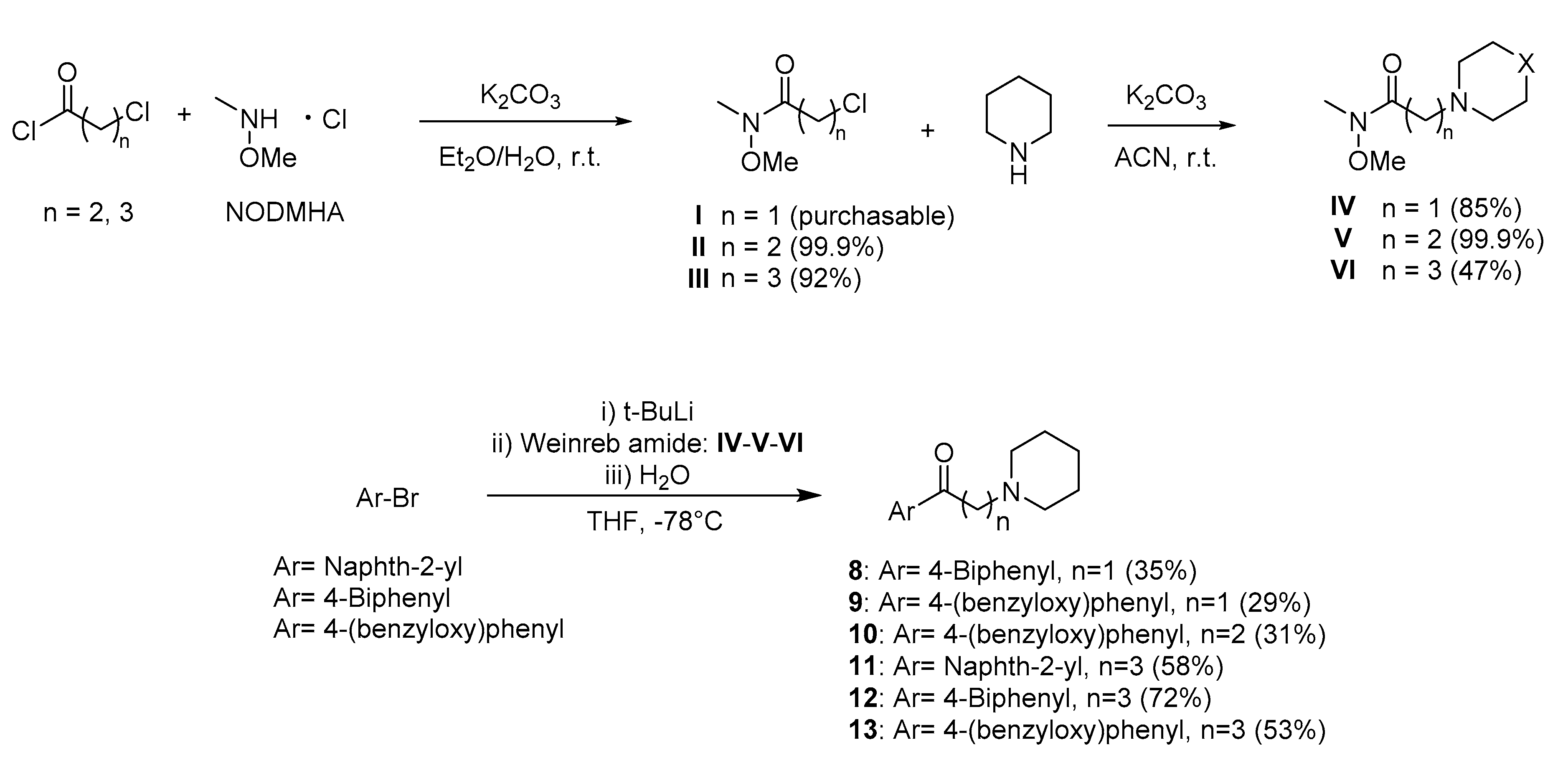
2.3. Synthesis
2.4. S1R Binding Profile
2.5. In Depth Pharmacological Profile
3. Discussion
4. Materials and Methods
4.1. Laboratory Materials and Equipment
4.2. General Experimental Details
4.3. Synthetic Procedures
4.3.1. General Procedure for the Preparation of Compounds II–III
4.3.2. General Procedure for the Preparation of Compounds IV–V–VI
4.3.3. General Procedure for the Preparation of Compounds 8–13
4.4. Molecular Modelling on S1R
4.4.1. Ligand Preparation
4.4.2. Protein Preparation
4.4.3. Grid Generation
4.4.4. Ligand Docking
4.4.5. Induced-Fit Docking
4.5. General Protocol for Binding Assays
4.5.1. S1R Binding Assay
4.5.2. S2R Binding Assay
4.5.3. GluN2 Binding Assay
4.6. Inhibition of AChE
4.7. Water Permeability Measurements
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| ADME | Absorption, distribution, metabolism and excretion |
| AQP | Aquaporin |
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| ER | Endoplasmic reticulum |
| IFD | Induced fit docking |
| LE | Ligand efficiency |
| MAM | Mitochondria-associated ER membrane |
| NMDA | N-methyl-d-aspartate |
| NMDAR | N-methyl-d-aspartate receptor |
| RMSD | Root-mean-square deviation |
| ROS | Reactive oxygen species |
| S1R | Sigma-1 receptor |
| S2R | Sigma-2 receptor |
| SEM | Standard error of the mean |
References
- Golpich, M.; Amini, E.; Mohamed, Z.; Ali, R.A.; Ibrahim, N.M.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- De Felice, A.; Ricceri, L.; Venerosi, A.; Chiarotti, F.; Calamandrei, G. Multifactorial Origin of Neurodevelopmental Disorders: Approaches to Understanding Complex Etiologies. Toxics 2015, 3, 89–129. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Models Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef]
- Fontanilla, D.; Johannessen, M.; Hajipour, A.R.; Cozzi, N.V.; Jackson, M.B.; Ruoho, A.E. The Hallucinogen N,N-Dimethyltryptamine (DMT) Is an Endogenous Sigma-1 Receptor Regulator. Science 2009, 323, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Chakraborty, S.; Brailoiu, G.C.; Zhao, P.; Barr, J.L.; Ilies, M.A.; Unterwald, E.M.; Abood, M.E.; Taylor, C.W. Choline Is an Intracellular Messenger Linking Extracellular Stimuli to IP3-Evoked Ca2+ Signals through Sigma-1 Receptors. Cell Rep. 2019, 26, 330–337.e4. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Pohl, P.; Zhou, G.Z. Steroid binding at sigma-“opioid” receptors. Science 1989, 246, 1635–1637. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ramachandran, S.; Chu, U.B.; Mavlyutov, T.A.; Pal, A.; Pyne, S.; Ruoho, A.E. The sigma1 receptor interacts with N-alkyl amines and endogenous sphingolipids. Eur. J. Pharmacol. 2009, 609, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Tesei, A.; Cortesi, M.; Zamagni, A.; Arienti, C.; Pignatta, S.; Zanoni, M.; Paolillo, M.; Curti, D.; Rui, M.; Rossi, D.; et al. Sigma Receptors as Endoplasmic Reticulum Stress “Gatekeepers” and their Modulators as Emerging New Weapons in the Fight Against Cancer. Front. Pharm. 2018, 9. [Google Scholar] [CrossRef]
- Delprat, B.; Crouzier, L.; Su, T.-P.; Maurice, T. At the Crossing of ER Stress and MAMs: A Key Role of Sigma-1 Receptor? Adv. Exp. Med. Biol. 2020, 1131, 699–718. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Su, T.-P.; Su, T.-C.; Nakamura, Y.; Tsai, S.-Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharm. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef]
- Penke, B.; Fulop, L.; Szucs, M.; Frecska, E. The Role of Sigma-1 Receptor, an Intracellular Chaperone in Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 97–116. [Google Scholar] [CrossRef]
- Gao, H.-M.; Hong, J.-S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef]
- Rui, M.; Rossino, G.; Coniglio, S.; Monteleone, S.; Scuteri, A.; Malacrida, A.; Rossi, D.; Catenacci, L.; Sorrenti, M.; Paolillo, M.; et al. Identification of dual Sigma1 receptor modulators/acetylcholinesterase inhibitors with antioxidant and neurotrophic properties, as neuroprotective agents. Eur. J. Med. Chem. 2018, 158, 353–370. [Google Scholar] [CrossRef]
- Black, S.A.G.; Stys, P.K.; Zamponi, G.W.; Tsutsui, S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front. Cell Dev. Biol. 2014, 2, 45. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol. 2016, 53, 648–661. [Google Scholar] [CrossRef]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta (BBA) Biomembr. 2006, 1758, 994–1003. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Pellavio, G.; Rui, M.; Caliogna, L.; Martino, E.; Gastaldi, G.; Collina, S.; Laforenza, U. Regulation of Aquaporin Functional Properties Mediated by the Antioxidant Effects of Natural Compounds. Int. J. Mol. Sci. 2017, 18, 2665. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human σ 1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef]
- Cavalluzzi, M.M.; Mangiatordi, G.F.; Nicolotti, O.; Lentini, G. Ligand efficiency metrics in drug discovery: The pros and cons from a practical perspective. Expert Opin. Drug Discov. 2017, 12, 1087–1104. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Betz, R.M.; Dror, R.O.; Kruse, A.C. Structural basis for σ 1 receptor ligand recognition. Nat. Struct. Mol. Biol. 2018, 25, 981–987. [Google Scholar] [CrossRef]
- Laoui, A.; Polyakov, V.R. Web services as applications’ integration tool: QikProp case study. J. Comput. Chem. 2011, 32, 1944–1951. [Google Scholar] [CrossRef]
- QikProp 3.5 User Manual. Available online: https://www.schrodinger.com/qikprop (accessed on 16 October 2020).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid Med. Cell Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Bozzo, F.; Mirra, A.; Carrì, M.T. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef]
- Bekdash, R.A. Choline, the brain and neurodegeneration: Insights from epigenetics. Front. Biosci (Landmark Ed.) 2018, 23, 1113–1143. [Google Scholar] [CrossRef]
- Brewster, J.T.; Dell’Acqua, S.; Thach, D.Q.; Sessler, J.L. Classics in Chemical Neuroscience: Donepezil. ACS Chem. Neurosci. 2019, 10, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Borgnia, M.; Nielsen, S.; Engel, A.; Agre, P. Cellular and Molecular Biology of the Aquaporin Water Channels. Annu. Rev. Biochem. 1999, 68, 425–458. [Google Scholar] [CrossRef] [PubMed]
- Hedfalk, K.; Törnroth-Horsefield, S.; Nyblom, M.; Johanson, U.; Kjellbom, P.; Neutze, R. Aquaporin gating. Curr. Opin. Struct. Biol. 2006, 16, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Beitz, E.; Golldack, A.; Rothert, M.; von Bülow, J. Challenges and achievements in the therapeutic modulation of aquaporin functionality. Pharmacol. Ther. 2015, 155, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.J.; Tsujita, M.; Nakada, T. Aquaporins in drug discovery and pharmacotherapy. Mol. Asp. Med. 2012, 33, 691–703. [Google Scholar] [CrossRef]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | State | Glide Gscore | Average Gscore | Ligand Efficiency (LE) | Average LE | Ki S1R (nM) ± SEM a | ln(Ki) |
|---|---|---|---|---|---|---|---|
| 1 | diss | −10.560 | −10.514 | −0.4062 | −0.4044 | 27 ± 1.8 | 3.296 |
| undiss | −10.467 | −0.4026 | |||||
| 2 | diss | −8.885 | −8.885 | −0.4039 | −0.4039 | 15 ± 1.1 | 2.708 |
| 3 | diss | −8.553 | −8.553 | −0.4277 | −0.4277 | 2.2 ± 0.7 | 0.788 |
| 4 | diss | −10.295 | −9.188 | −0.4680 | −0.4176 | 2.9 ± 0.3 | 1.065 |
| undiss | −8.081 | −0.3673 | |||||
| 5 | diss | −9.101 | −8.923 | −0.4551 | −0.4462 | 9 ± 0.7 | 2.197 |
| undiss | −8.745 | −0.4373 | |||||
| 6 | diss | −11.411 | −10.548 | −0.4075 | −0.3767 | 340 | 5.829 |
| undiss | −9.685 | −0.3459 | |||||
| 7 | diss | −9.048 | −9.048 | −0.3619 | −0.3619 | 478 | 6.170 |
| Cmpd | MW | PSA | HBD | HBA | QPlogPo/w | QPlogS | QPlog HERG | QPP Caco | QPlog BB | Oral Abs b |
|---|---|---|---|---|---|---|---|---|---|---|
| 8 a | 279.38 | 30.874 | 0 | 4 | 3.51 | −3.414 | −6.564 | 1053.825 | 0.34 | 3 |
| 9 a | 309.40 | 38.211 | 0 | 4.75 | 3.924 | −3.75 | −7.077 | 1057.244 | 0.198 | 3 |
| 9 | 309.40 | 36.978 | 0 | 4.75 | 3.899 | −3.604 | −6.893 | 1049.048 | 0.202 | 3 |
| 10 | 323.43 | 38.951 | 0 | 4.75 | 4.112 | −4.229 | −7.246 | 972.122 | 0.085 | 3 |
| 11 | 281.39 | 31.865 | 0 | 4 | 3.612 | −3.565 | −6.442 | 984.507 | 0.242 | 3 |
| 12 | 307.43 | 32.094 | 0 | 4 | 4.268 | −4.372 | −6.98 | 971.725 | 0.156 | 3 |
| 13 | 337.46 | 39.309 | 0 | 4.75 | 4.28 | −4.622 | −7.356 | 899.612 | −0.023 | 3 |
| Drug-likeness c | 130–725 | 7.0–200 | 0–6 | 2–20 | −2.0–6.5 | −6.5–0.5 | <−5 | <25 poor >500 great | −3.0–1.2 | 1 low 2 med 3 high |
| cmpd | LE | Predicted ln(Ki) | Predicted Ki (nM) | Experimental Ki S1R (nM) ± SEM | Experimental ln(Ki) |
|---|---|---|---|---|---|
| 8 | −0.39271 | 3.9754 | 44.92 | 132 ± 55 | 4.8828 |
| 9 | −0.40309 | 3.3053 | 21.10 | 48 ± 12 | 3.8712 |
| 10 | −0.40696 | 3.0555 | 15.92 | 25 ± 7 | 3.2189 |
| 11 | −0.45271 | 0.1022 | 0.57 | 4.4 ± 2.4 | 1.4816 |
| 12 | −0.43748 | 1.0854 | 1.72 | 33 ± 14 | 3.4965 |
| 13 | −0.36672 | 5.6531 | 298.28 | 87 ± 28 | 4.4659 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossino, G.; Rui, M.; Pozzetti, L.; Schepmann, D.; Wünsch, B.; Zampieri, D.; Pellavio, G.; Laforenza, U.; Rinaldi, S.; Colombo, G.; et al. Setup and Validation of a Reliable Docking Protocol for the Development of Neuroprotective Agents by Targeting the Sigma-1 Receptor (S1R). Int. J. Mol. Sci. 2020, 21, 7708. https://doi.org/10.3390/ijms21207708
Rossino G, Rui M, Pozzetti L, Schepmann D, Wünsch B, Zampieri D, Pellavio G, Laforenza U, Rinaldi S, Colombo G, et al. Setup and Validation of a Reliable Docking Protocol for the Development of Neuroprotective Agents by Targeting the Sigma-1 Receptor (S1R). International Journal of Molecular Sciences. 2020; 21(20):7708. https://doi.org/10.3390/ijms21207708
Chicago/Turabian StyleRossino, Giacomo, Marta Rui, Luca Pozzetti, Dirk Schepmann, Bernhard Wünsch, Daniele Zampieri, Giorgia Pellavio, Umberto Laforenza, Silvia Rinaldi, Giorgio Colombo, and et al. 2020. "Setup and Validation of a Reliable Docking Protocol for the Development of Neuroprotective Agents by Targeting the Sigma-1 Receptor (S1R)" International Journal of Molecular Sciences 21, no. 20: 7708. https://doi.org/10.3390/ijms21207708
APA StyleRossino, G., Rui, M., Pozzetti, L., Schepmann, D., Wünsch, B., Zampieri, D., Pellavio, G., Laforenza, U., Rinaldi, S., Colombo, G., Morelli, L., Linciano, P., Rossi, D., & Collina, S. (2020). Setup and Validation of a Reliable Docking Protocol for the Development of Neuroprotective Agents by Targeting the Sigma-1 Receptor (S1R). International Journal of Molecular Sciences, 21(20), 7708. https://doi.org/10.3390/ijms21207708