Impact of Mitophagy and Mitochondrial Unfolded Protein Response as New Adaptive Mechanisms Underlying Old Pathologies: Sarcopenia and Non-Alcoholic Fatty Liver Disease


Abstract
1. Introduction
1.1. Mitochondrial Proteostasis
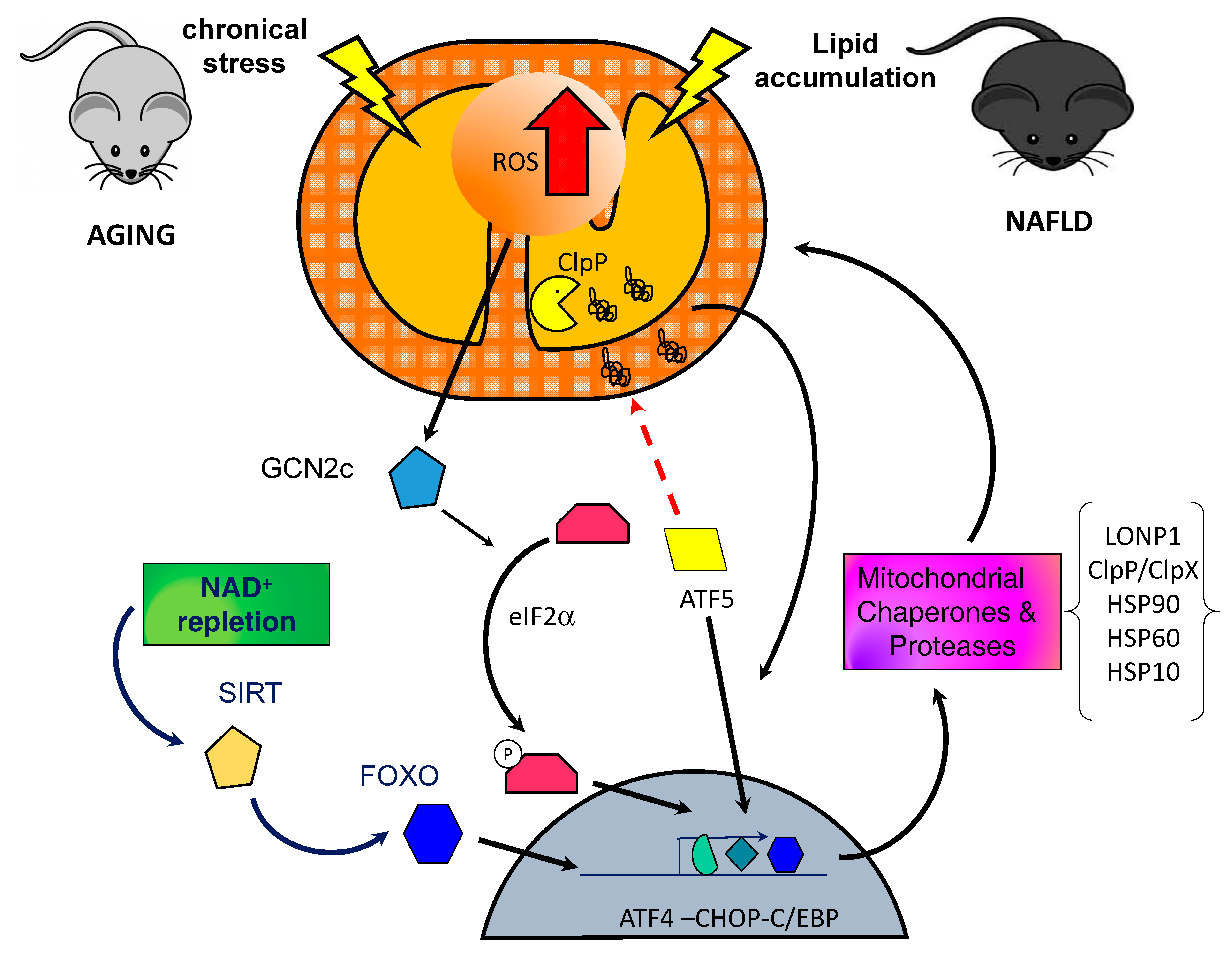
1.2. Mitochondrial Unfolded Protein Response Signaling
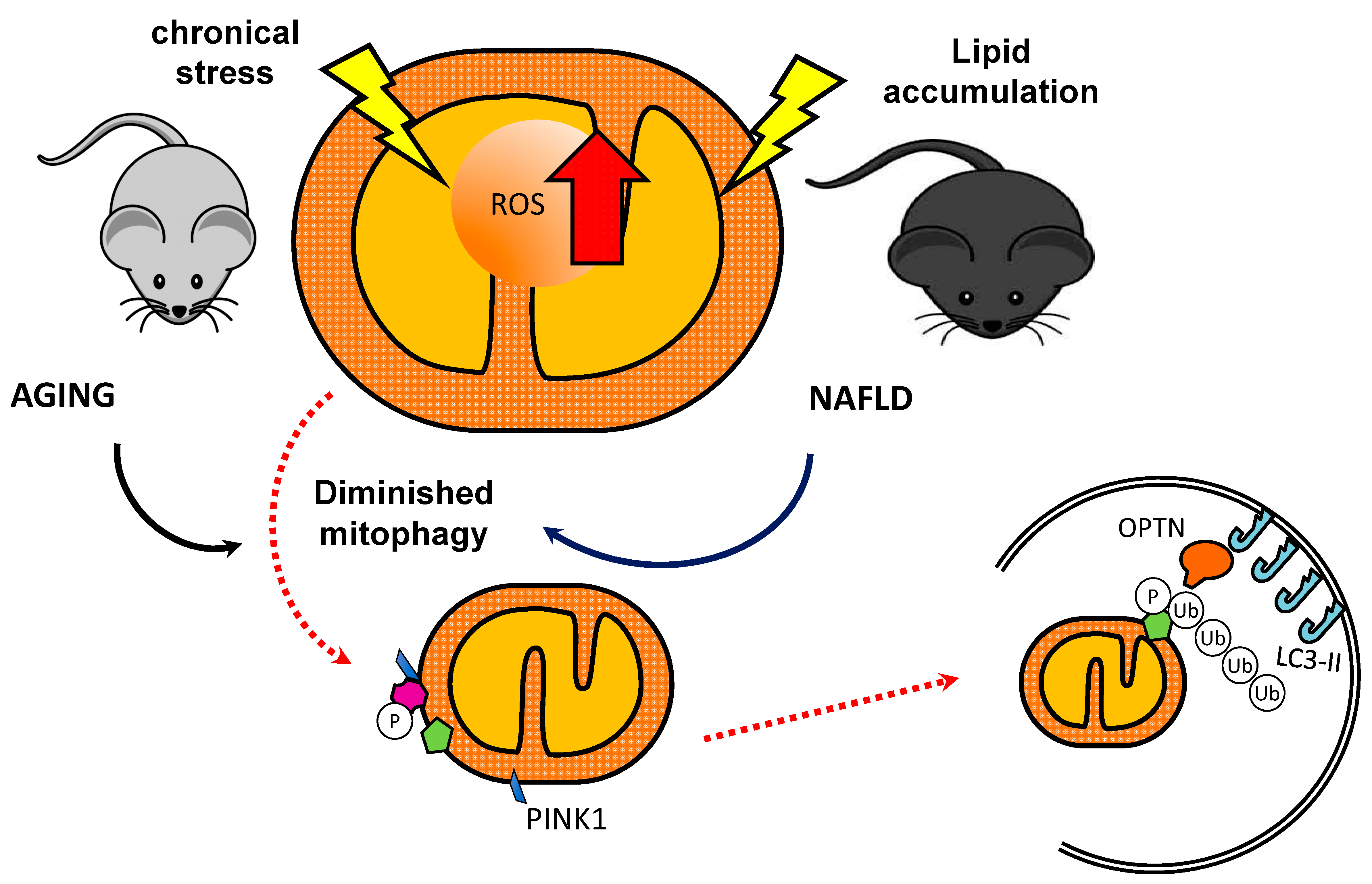
1.3. Mitophagy
1.3.1. The PINK1/Parkin Pathway
1.3.2. BNIP3/NIX Pathway
2. Mitophagy and Associated Disorders in Skeletal Muscle
3. UPRmt in Sarcopenia
4. Mitophagy and Non-Alcoholic Fatty Liver Disease (NAFLD)
5. UPRmt and NAFLD
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| AMPK | AMP-activated protein kinase |
| Atg7 | Autophagy related protein 7 |
| Atg12 | Autophagy related protein 12 |
| ATP5α | ATP synthase subunit alpha |
| ATF-4 | Activating transcription factor 4 |
| ATF-5 | Activating transcription factor 5 |
| ATFS-1 | Activating transcription factor associated with stress |
| BAK | BCL2 homologous antagonist/killer |
| BAX | BCL2-like protein 4 |
| BNIP3 | BCL2 and adenovirus E1B 19-kDa-interacting protein 3 |
| BNIP3L | BNIP3-like |
| bZIP | Basic leucine zipper |
| C26 | Colon-26 carcinoma |
| CEBPβ | CCAAT/enhancer binding protein β |
| CFCM | Cross-fiber connection mitochondria |
| CHF | Congestive heart failure |
| CHOP | CEBP Homologous Protein |
| ClpP | Caseinolytic protease proteolytic subunit |
| ClpXP | ATP-dependent Clp protease ATP-binding subunit ClpX-like |
| COPD | Chronic obstructive pulmonary disease |
| Crif1 | CR6-interacting factor 1 |
| DHA | Docosahexaenoic acid |
| DNA | Deoxyribonucleic acid |
| Dve-1 | Homeobox domain-containing protein |
| eIF2α | Eukaryotic translation initiation factor 2 alpha |
| FA | Fatty acid |
| FGF21 | Fibroblast growth factor 21 |
| FPM | Fiber parallel mitochondria |
| GCN2 | General control non-derepressible 2 kinase |
| GDF15 | Growth differentiation factor 15 |
| Haf-1 | Matrix peptide exporter |
| HFD | High-fat diet |
| HFHS | High-fat and high-sucrose |
| HRI | Heme-regulated inhibitor kinase |
| Hsp10 | Heat shock protein 10 |
| Hsp60 | Heat shock protein 60 |
| Hsp90 | Heat shock protein 90 |
| HT | Hydroxytyrosol |
| IMM | Inner mitochondrial membrane |
| IMS | Intermembrane space |
| IP3R1 | Inositol 1,4,5-triphosphate receptor type 1 |
| IP3R2 | Inositol 1,4,5-triphosphate receptor type 2 |
| IRE1 | Inositol-requiring enzyme-1 |
| ISR | Integrated stress response |
| Jmjd-3.1 | Lysine-specific demethylase (JuMonJi Domain protein) |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| LONP1 | Lon protease 1 |
| MAM | Mitochondrial associated ER membrane |
| Mfn-2 | Mitofusin 2 |
| MPP | Mitochondrial processing peptidase |
| MRC | Mitochondrial respiratory chain |
| MTCO1 | Mitochondrially encoded cytochrome c oxidase 1 |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| mtHsp70 | Mitochondrial heat shock protein 70 |
| MTS | Mitochondrial targeting sequence |
| LCPUFA | Long-chain polyunsaturated FA |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NRF-1 | Nuclear respiratory factor 1 |
| OMM | Outer mitochondrial membrane |
| OPTN | Optineurin protein |
| p62 | Classical receptor of autophagy |
| PACS-2 | Phosphofurin acidic cluster sorting protein 2 |
| PERK | Protein kinase RNA-like ER kinase |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PINK1 | PTEN-induced putative kinase 1 |
| PKR | Protein kinase RNA-activated |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PUFA | Polyunsaturated fatty acid |
| SIRT1 | Sirtuin 1 |
| SIRT3 | Sirtuin 3 |
| SREBP-1c | Sterol regulatory element-binding protein-1c |
| TIM | Translocase of the inner membrane |
| TNF-α | Tumor necrosis factor-alpha |
| TOM | Translocase of the outer membrane |
| VDAC | Voltage-dependent anion channel |
| XBP1 | X-box-binding protein-1 |
| Yme1L1 | Yme1-like 1 ATPase |
References
- Bereiter-Hahn, J.; Vöth, M.; Mai, S.; Jendrach, M. Structural implications of mitochondrial dynamics. Biotechnol. J. 2008, 3, 765–780. [Google Scholar] [CrossRef]
- Benard, G.; Rossignol, R. Ultrastructure of the mitochondrion and its bearing on function and bioenergetics. Antioxid. Redox Signal. 2008, 10, 1313–1342. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med. 2004, 25, 365–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, H. Translational regulation of mitochondrial biogenesis. Biochem. Soc. Trans. 2016, 44, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W.; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Münch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef]
- Stadtman, E.R. Metal ion-catalyzed oxidation of proteins: Biochemical mechanism and biological consequences. Free. Radic. Biol. Med. 1990, 9, 315–325. [Google Scholar] [CrossRef]
- Reznick, A.Z.; Packer, L. Oxidative damage to proteins: Spectrophotometric method for carbonyl assay. Methods Enzymol. 1994, 233, 357–363. [Google Scholar] [CrossRef]
- Videla, L.A.; Fernández, V.; Cornejo, P.; Vargas, R.; Carrasco, J.; Fernández, J.; Varela, N. Causal role of oxidative stress in unfolded protein response development in the hyperthyroid state. Free. Radic. Biol. Med. 2015, 89, 401–408. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef]
- Yi, H.-S.; Chang, J.Y.; Shong, M. The mitochondrial unfolded protein response and mitohormesis: A perspective on metabolic diseases. J. Mol. Endocrinol. 2018, 61, R91–R105. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Naresh, N.U.; Haynes, C.M. Signaling and Regulation of the Mitochondrial Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033944. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.C.; Yan, Z. Mitophagy in maintaining skeletal muscle mitochondrial proteostasis and metabolic health with ageing. J. Physiol. 2017, 595, 6391–6399. [Google Scholar] [CrossRef]
- Fiesel, F.C.; James, E.D.; Hudec, R.; Springer, W. Mitochondrial targeted HSP90 inhibitor Gamitrinib-TPP (G-TPP) induces PINK1/Parkin-dependent mitophagy. Oncotarget 2017, 8, 106233–106248. [Google Scholar] [CrossRef] [PubMed]
- Quiles, J.M.; Gustafsson, Å.B. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front. Physiol. 2020, 11, 515. [Google Scholar] [CrossRef]
- Ngo, J.K.; Pomatto, L.C.D.; Davies, K.J.A. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef]
- Cordeiro, A.V.; Brícola, R.S.; Braga, R.R.; Lenhare, L.; Silva, V.R.R.; Anaruma, C.P.; Katashima, C.K.; Crisol, B.M.; Simabuco, F.M.; Silva, A.S.R.; et al. Aerobic Exercise Training Induces the Mitonuclear Imbalance and UPRmt in the Skeletal Muscle of Aged Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2020. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Matsunaga, Y.; Kitaoka, Y.; Hatta, H. Effects of Heat Stress Treatment on Age-dependent Unfolded Protein Response in Different Types of Skeletal Muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Memme, J.M.; Oliveira, A.N.; Hood, D.A. Chronology of UPR activation in skeletal muscle adaptations to chronic contractile activity. Am. J. Physiol.-Cell Physiol. 2016, 310, C1024–C1036. [Google Scholar] [CrossRef] [PubMed]
- Al-Furoukh, N.; Ianni, A.; Nolte, H.; Hölper, S.; Krüger, M.; Wanrooij, S.; Braun, T. ClpX stimulates the mitochondrial unfolded protein response (UPRmt) in mammalian cells. BBA — Mol. Cell Res. 2015, 10, 2580–2591. [Google Scholar] [CrossRef]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatol. Baltim. Md 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. CB 2016, 26, 2037–2043. [Google Scholar] [CrossRef]
- Bennett, C.F.; Vander Wende, H.; Simko, M.; Klum, S.; Barfield, S.; Choi, H.; Pineda, V.V.; Kaeberlein, M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 2014, 5, 3483. [Google Scholar] [CrossRef]
- Michel, S.; Canonne, M.; Arnould, T.; Renard, P. Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion. 2015, 21, 58–68. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial speci®c stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Chen, P.B.; Yang, J.S.; Park, Y. Adaptations of Skeletal Muscle Mitochondria to Obesity, Exercise, and Polyunsaturated Fatty Acids. Lipids 2018, 53, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Chomentowski, P.; Coen, P.M.; Radiková, Z.; Goodpaster, B.H.; Toledo, F.G.S. Skeletal muscle mitochondria in insulin resistance: Differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J. Clin. Endocrinol. Metab. 2011, 96, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Manoli, I.; Alesci, S.; Blackman, M.R.; Su, Y.A.; Rennert, O.M.; Chrousos, G.P. Mitochondria as key components of the stress response. Trends Endocrinol. Metab. TEM 2007, 18, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Melber, A.; Haynes, C.M. UPRmt regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef]
- Ting, S.-Y.; Yan, N.L.; Schilke, B.A.; Craig, E.A. Dual interaction of scaffold protein Tim44 of mitochondrial import motor with channel-forming translocase subunit Tim23. eLife 2017, 6, e23609. [Google Scholar] [CrossRef]
- Mapa, K.; Sikor, M.; Kudryavtsev, V.; Waegemann, K.; Kalinin, S.; Seidel, C.A.M.; Neupert, W.; Lamb, D.C.; Mokranjac, D. The conformational dynamics of the mitochondrial Hsp70 chaperone. Mol. Cell 2010, 38, 89–100. [Google Scholar] [CrossRef]
- Mokranjac, D. How to get to the other side of the mitochondrial inner membrane—The protein import motor. Biol. Chem. 2020, 401, 723–736. [Google Scholar] [CrossRef]
- Caumont-Sarcos, A.; Moulin, C.; Poinot, L.; Guiard, B.; van der Laan, M.; Ieva, R. Transmembrane Coordination of Preprotein Recognition and Motor Coupling by the Mitochondrial Presequence Receptor Tim50. Cell Rep. 2020, 30, 3092–3104. [Google Scholar] [CrossRef]
- Fischer, M.; Riemer, J. The Mitochondrial Disulfide Relay System: Roles in Oxidative Protein Folding and Beyond. Int. J. Cell Biol. 2013, 2013, 1–12. [Google Scholar] [CrossRef]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef]
- Baker, B.M.; Nargund, A.M.; Sun, T.; Haynes, C.M. Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 2012, 8, e1002760. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Peixeiro, I.; Romão, L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013, 9, e1003529. [Google Scholar] [CrossRef]
- Zhou, D.; Palam, L.R.; Jiang, L.; Narasimhan, J.; Staschke, K.A.; Wek, R.C. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J. Biol. Chem. 2008, 283, 7064–7073. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef] [PubMed]
- Münch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 2011, 3, a007559. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coupling mitogenesis and mitophagy for longevity. Autophagy 2015, 11, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2015, 1853, 2784–2790. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.K.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449. [Google Scholar] [CrossRef]
- Kubli, D.A.; Ycaza, J.E.; Gustafsson, A.B. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem. J. 2007, 405, 407–415. [Google Scholar] [CrossRef]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef]
- Gan, Z.; Fu, T.; Kelly, D.P.; Vega, R.B. Skeletal muscle mitochondrial remodeling in exercise and diseases. Cell Res. 2018, 28, 969–980. [Google Scholar] [CrossRef]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C.; Boncompagni, S. The Evolution of the Mitochondria-to-Calcium Release Units Relationship in Vertebrate Skeletal Muscles. Available online: https://www.hindawi.com/journals/bmri/2011/830573/ (accessed on 14 August 2020).
- Glancy, B.; Hartnell, L.M.; Malide, D.; Yu, Z.-X.; Combs, C.A.; Connelly, P.S.; Subramaniam, S.; Balaban, R.S. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 2015, 523, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Riva, A.; Tandler, B.; Loffredo, F.; Vazquez, E.; Hoppel, C. Structural differences in two biochemically defined populations of cardiac mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H868–H872. [Google Scholar] [CrossRef]
- Koves, T.R.; Noland, R.C.; Bates, A.L.; Henes, S.T.; Muoio, D.M.; Cortright, R.N. Subsarcolemmal and intermyofibrillar mitochondria play distinct roles in regulating skeletal muscle fatty acid metabolism. Am. J. Physiol. Cell Physiol. 2005, 288, C1074–C1082. [Google Scholar] [CrossRef]
- Ferreira, R.; Vitorino, R.; Alves, R.M.P.; Appell, H.J.; Powers, S.K.; Duarte, J.A.; Amado, F. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 2010, 10, 3142–3154. [Google Scholar] [CrossRef]
- Rantanen, T.; Harris, T.; Leveille, S.G.; Visser, M.; Foley, D.; Masaki, K.; Guralnik, J.M. Muscle strength and body mass index as long-term predictors of mortality in initially healthy men. J. Gerontol. A Biol. Sci. Med. Sci. 2000, 55, M168–M173. [Google Scholar] [CrossRef]
- Metter, E.J.; Talbot, L.A.; Schrager, M.; Conwit, R. Skeletal muscle strength as a predictor of all-cause mortality in healthy men. J. Gerontol. A Biol. Sci. Med. Sci. 2002, 57, B359–B365. [Google Scholar] [CrossRef]
- Nair, K.S. Aging muscle. Am. J. Clin. Nutr. 2005, 81, 953–963. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Park, S.W.; Harris, T.B.; Kritchevsky, S.B.; Nevitt, M.; Schwartz, A.V.; Simonsick, E.M.; Tylavsky, F.A.; Visser, M.; Newman, A.B. The loss of skeletal muscle strength, mass, and quality in older adults: The health, aging and body composition study. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 1059–1064. [Google Scholar] [CrossRef]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef]
- Joseph, A.-M.; Adhihetty, P.J.; Buford, T.W.; Wohlgemuth, S.E.; Lees, H.A.; Nguyen, L.M.-D.; Aranda, J.M.; Sandesara, B.D.; Pahor, M.; Manini, T.M.; et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012, 11, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Kruse, S.E.; Karunadharma, P.P.; Basisty, N.; Johnson, R.; Beyer, R.P.; MacCoss, M.J.; Rabinovitch, P.S.; Marcinek, D.J. Age modifies respiratory complex I and protein homeostasis in a muscle type-specific manner. Aging Cell 2016, 15, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Marcinek, D.J.; Schenkman, K.A.; Ciesielski, W.A.; Lee, D.; Conley, K.E. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J. Physiol. 2005, 569, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, I.H. Sarcopenia: Origins and clinical relevance. Clin. Geriatr. Med. 2011, 27, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Hughes, V.A.; Frontera, W.R.; Wood, M.; Evans, W.J.; Dallal, G.E.; Roubenoff, R.; Fiatarone Singh, M.A. Longitudinal muscle strength changes in older adults: Influence of muscle mass, physical activity, and health. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B209–B217. [Google Scholar] [CrossRef] [PubMed]
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B. Role of Age-Related Mitochondrial Dysfunction in Sarcopenia. Int. J. Mol. Sci. 2020, 21, 5236. [Google Scholar] [CrossRef] [PubMed]
- Sayed, R.K.A.; de Leonardis, E.C.; Guerrero-Martínez, J.A.; Rahim, I.; Mokhtar, D.M.; Saleh, A.M.; Abdalla, K.E.H.; Pozo, M.J.; Escames, G.; López, L.C.; et al. Identification of morphological markers of sarcopenia at early stage of aging in skeletal muscle of mice. Exp. Gerontol. 2016, 83, 22–30. [Google Scholar] [CrossRef]
- Del Campo, A.; Contreras-Hernández, I.; Castro-Sepúlveda, M.; Campos, C.A.; Figueroa, R.; Tevy, M.F.; Eisner, V.; Casas, M.; Jaimovich, E. Muscle function decline and mitochondria changes in middle age precede sarcopenia in mice. Aging 2018, 10, 34–55. [Google Scholar] [CrossRef]
- Huang, D.-D.; Fan, S.-D.; Chen, X.-Y.; Yan, X.-L.; Zhang, X.-Z.; Ma, B.-W.; Yu, D.-Y.; Xiao, W.-Y.; Zhuang, C.-L.; Yu, Z. Nrf2 deficiency exacerbates frailty and sarcopenia by impairing skeletal muscle mitochondrial biogenesis and dynamics in an age-dependent manner. Exp. Gerontol. 2019, 119, 61–73. [Google Scholar] [CrossRef]
- Kitaoka, Y.; Tamura, Y.; Takahashi, K.; Takeda, K.; Takemasa, T.; Hatta, H. Effects of Nrf2 deficiency on mitochondrial oxidative stress in aged skeletal muscle. Physiol. Rep. 2019, 7, e13998. [Google Scholar] [CrossRef]
- Dillon, L.M.; Williams, S.L.; Hida, A.; Peacock, J.D.; Prolla, T.A.; Lincoln, J.; Moraes, C.T. Increased mitochondrial biogenesis in muscle improves aging phenotypes in the mtDNA mutator mouse. Hum. Mol. Genet. 2012, 21, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Ritchie, D.; Thomas, M.M.; Wright, K.J.; Hepple, R.T. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell 2011, 10, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Gouspillou, G.; Sgarioto, N.; Kapchinsky, S.; Purves-Smith, F.; Norris, B.; Pion, C.H.; Barbat-Artigas, S.; Lemieux, F.; Taivassalo, T.; Morais, J.A.; et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014, 28, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Tamilselvan, J.; Jayaraman, G.; Sivarajan, K.; Panneerselvam, C. Age-dependent upregulation of p53 and cytochrome c release and susceptibility to apoptosis in skeletal muscle fiber of aged rats: Role of carnitine and lipoic acid. Free Radic. Biol. Med. 2007, 43, 1656–1669. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yun Chong, S.; Lim, A.; Singh, B.K.; Sinha, R.A.; Salmon, A.B.; Yen, P.M. Changes in macroautophagy, chaperone-mediated autophagy, and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging 2017, 9, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yao, Z.; Klionsky, D.J. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015, 25, 354–363. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Baehr, L.M.; West, D.W.D.; Marcotte, G.; Marshall, A.G.; De Sousa, L.G.; Baar, K.; Bodine, S.C. Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging 2016, 8, 127–146. [Google Scholar] [CrossRef]
- Joseph, A.-M.; Adhihetty, P.J.; Wawrzyniak, N.R.; Wohlgemuth, S.E.; Picca, A.; Kujoth, G.C.; Prolla, T.A.; Leeuwenburgh, C. Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS ONE 2013, 8, e69327. [Google Scholar] [CrossRef]
- Bujak, A.L.; Crane, J.D.; Lally, J.S.; Ford, R.J.; Kang, S.J.; Rebalka, I.A.; Green, A.E.; Kemp, B.E.; Hawke, T.J.; Schertzer, J.D.; et al. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab. 2015, 21, 883–890. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Grumati, P.; Sandri, M.; Bonaldo, P. Skeletal muscle, autophagy, and physical activity: The ménage à trois of metabolic regulation in health and disease. J. Mol. Med. Berl. Ger. 2014, 92, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef] [PubMed]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; LaStayo, P.C.; Marcus, R.L. Downregulation of E3 Ubiquitin Ligases and Mitophagy-Related Genes in Skeletal Muscle of Physically Inactive, Frail Older Women: A Cross-Sectional Comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Triolo, M.; Hood, D.A. Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle. Oxidative Med. Cell. Longev. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Pietrangelo, L.; Loefler, S.; Fruhmann, H.; Vogelauer, M.; Burggraf, S.; Pond, A.; Grim-Stieger, M.; Cvecka, J.; Sedliak, M.; et al. Lifelong physical exercise delays age-associated skeletal muscle decline. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 163–173. [Google Scholar] [CrossRef]
- Stanford, K.I.; Goodyear, L.J. Exercise and type 2 diabetes: Molecular mechanisms regulating glucose uptake in skeletal muscle. Adv. Physiol. Educ. 2014, 38, 308–314. [Google Scholar] [CrossRef]
- Maltais, F.; Decramer, M.; Casaburi, R.; Barreiro, E.; Burelle, Y.; Debigaré, R.; Dekhuijzen, P.N.R.; Franssen, F.; Gayan-Ramirez, G.; Gea, J.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update on Limb Muscle Dysfunction in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2014, 189, e15–e62. [Google Scholar] [CrossRef]
- Kinugawa, S.; Takada, S.; Matsushima, S.; Okita, K.; Tsutsui, H. Skeletal Muscle Abnormalities in Heart Failure. Int. Heart. J. 2015, 56, 475–484. [Google Scholar] [CrossRef]
- van de Bool, C.; Gosker, H.R.; van den Borst, B.; Op den Kamp, C.M.; Slot, I.G.M.; Schols, A.M.W.J. Muscle Quality is More Impaired in Sarcopenic Patients With Chronic Obstructive Pulmonary Disease. J. Am. Med. Dir. Assoc. 2016, 17, 415–420. [Google Scholar] [CrossRef]
- Huffman, K.M.; Koves, T.R.; Hubal, M.J.; Abouassi, H.; Beri, N.; Bateman, L.A.; Stevens, R.D.; Ilkayeva, O.R.; Hoffman, E.P.; Muoio, D.M.; et al. Metabolite signatures of exercise training in human skeletal muscle relate to mitochondrial remodelling and cardiometabolic fitness. Diabetologia 2014, 57, 2282–2295. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Gosker, H.R.; Schols, A.M.W.J.; Kapchinsky, S.; Bourbeau, J.; Sandri, M.; Jagoe, R.T.; Debigaré, R.; Maltais, F.; Taivassalo, T.; et al. Autophagy in locomotor muscles of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2013, 188, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Kruse, R.; Vind, B.F.; Petersson, S.J.; Kristensen, J.M.; Højlund, K. Markers of autophagy are adapted to hyperglycaemia in skeletal muscle in type 2 diabetes. Diabetologia 2015, 58, 2087–2095. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Pearson, T.; Lightfoot, A.P.; Nye, G.A.; Wells, N.; Giakoumaki, I.I.; Vasilaki, A.; Griffiths, R.D.; Jackson, M.J.; McArdle, A. Mitochondrial ROS regulate oxidative damage and mitophagy but not age-related muscle fiber atrophy. Sci. Rep. 2016, 6, 33944. [Google Scholar] [CrossRef] [PubMed]
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function: Parkin is essential for optimal muscle and mitochondrial functions. J. Physiol. 2018, 596, 2565–2579. [Google Scholar] [CrossRef] [PubMed]
- Peker, N.; Donipadi, V.; Sharma, M.; McFarlane, C.; Kambadur, R. Loss of Parkin impairs mitochondrial function and leads to muscle atrophy. Am. J. Physiol.-Cell Physiol. 2018, 315, C164–C185. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Guzmán Mentesana, G.; Báez, A.L.; Lo Presti, M.S.; Domínguez, R.; Córdoba, R.; Bazán, C.; Strauss, M.; Fretes, R.; Rivarola, H.W.; Paglini-Oliva, P. Functional and Structural Alterations of Cardiac and Skeletal Muscle Mitochondria in Heart Failure Patients. Arch. Med. Res. 2014, 45, 237–246. [Google Scholar] [CrossRef]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef]
- Bach, D.; Naon, D.; Pich, S.; Soriano, F.X.; Vega, N.; Rieusset, J.; Laville, M.; Guillet, C.; Boirie, Y.; Wallberg-Henriksson, H.; et al. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: Effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 2005, 54, 2685–2693. [Google Scholar] [CrossRef]
- Garnier, A.; Fortin, D.; Zoll, J.; N’Guessan, B.; Mettauer, B.; Lampert, E.; Veksler, V.; Ventura-Clapier, R. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Molina, A.J.A.; Bharadwaj, M.S.; Van Horn, C.; Nicklas, B.J.; Lyles, M.F.; Eggebeen, J.; Haykowsky, M.J.; Brubaker, P.H.; Kitzman, D.W. Skeletal Muscle Mitochondrial Content, Oxidative Capacity, and Mfn2 Expression Are Reduced in Older Patients with Heart Failure and Preserved Ejection Fraction and Are Related to Exercise Intolerance. JACC Heart Fail. 2016, 4, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef]
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and Proteomic Profiling Reveals Reduced Mitochondrial Function and Disruption of the Neuromuscular Junction Driving Rat Sarcopenia. Mol. Cell. Biol. 2013, 33, 194–212. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.-P.; Picard, M.; St-Jean Pelletier, F.; Sgarioto, N.; Auger, M.-J.; Vallée, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef]
- Leermakers, P.A.; Gosker, H.R. Skeletal muscle mitophagy in chronic disease: Implications for muscle oxidative capacity? Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 427–433. [Google Scholar] [CrossRef]
- Schulz, A.M.; Haynes, C.M. UPR(mt)-mediated cytoprotection and organismal aging. Biochim. Biophys. Acta 2015, 1847, 1448–1456. [Google Scholar] [CrossRef]
- Ben-Meir, A.; Yahalomi, S.; Moshe, B.; Shufaro, Y.; Reubinoff, B.; Saada, A. Coenzyme Q-dependent mitochondrial respiratory chain activity in granulosa cells is reduced with aging. Fertil. Steril. 2015, 104, 724–727. [Google Scholar] [CrossRef]
- Duarte, J.M.N.; Schuck, P.F.; Wenk, G.L.; Ferreira, G.C. Metabolic disturbances in diseases with neurological involvement. Aging Dis. 2014, 5, 238–255. [Google Scholar] [CrossRef]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.K.; Ryu, D.; Kim, K.S.; Chang, J.Y.; Kim, Y.K.; Yi, H.-S.; Kang, S.G.; Choi, M.J.; Lee, S.E.; Jung, S.-B.; et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 2017, 216, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Copeland, J.M.; Cho, J.; Lo, T.; Hur, J.H.; Bahadorani, S.; Arabyan, T.; Rabie, J.; Soh, J.; Walker, D.W. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. CB 2009, 19, 1591–1598. [Google Scholar] [CrossRef]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt). Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533, 416–419. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef]
- Gitschlag, B.L.; Kirby, C.S.; Samuels, D.C.; Gangula, R.D.; Mallal, S.A.; Patel, M.R. Homeostatic Responses Regulate Selfish Mitochondrial Genome Dynamics in C. elegans. Cell Metab. 2016, 24, 91–103. [Google Scholar] [CrossRef]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivelä, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 2017, 26, 419–428. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Mjosund, K.P.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Spelbrink, J.N.; Paetau, A.; Suomalainen, A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W.; De Vries, R.L.A.; Lebot, P.; Wikstrom, J.D.; Torgyekes, E.; Shirihai, O.S.; Przedborski, S.; Schon, E.A. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 2012, 21, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Ogborn, D.I.; McKay, B.R.; Crane, J.D.; Parise, G.; Tarnopolsky, M.A. The unfolded protein response is triggered following a single, unaccustomed resistance-exercise bout. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2014, 307, R664–R669. [Google Scholar] [CrossRef]
- Kim, K.; Kim, Y.-H.; Lee, S.-H.; Jeon, M.-J.; Park, S.-Y.; Doh, K.-O. Effect of exercise intensity on unfolded protein response in skeletal muscle of rat. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2014, 18, 211–216. [Google Scholar] [CrossRef]
- Passos, E.; Ascensão, A.; Martins, M.J.; Magalhães, J. Endoplasmic Reticulum Stress Response in Non-alcoholic Steatohepatitis: The Possible Role of Physical Exercise. Metabolism 2015, 64, 780–792. [Google Scholar] [CrossRef]
- Pereira, B.C.; da Rocha, A.L.; Pinto, A.P.; Pauli, J.R.; de Souza, C.T.; Cintra, D.E.; Ropelle, E.R.; de Freitas, E.C.; Zagatto, A.M.; da Silva, A.S.R. Excessive eccentric exercise-induced overtraining model leads to endoplasmic reticulum stress in mice skeletal muscles. Life Sci. 2016, 145, 144–151. [Google Scholar] [CrossRef]
- Wu, J.; Ruas, J.L.; Estall, J.L.; Rasbach, K.A.; Choi, J.H.; Ye, L.; Boström, P.; Tyra, H.M.; Crawford, R.W.; Campbell, K.P.; et al. The Unfolded Protein Response Mediates Adaptation to Exercise in Skeletal Muscle through a PGC-1α/ATF6α Complex. Cell Metab. 2011, 13, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; DAmico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Thoma, A.; Akter-Miah, T.; Reade, R.L.; Lightfoot, A.P. Targeting reactive oxygen species (ROS) to combat the age-related loss of muscle mass and function. Biogerontology 2020, 21, 475–484. [Google Scholar] [CrossRef]
- Morley, J.E. Pharmacologic Options for the Treatment of Sarcopenia. Calcif. Tissue Int. 2016, 98, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.-N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef]
- Li, Y.; Ruan, D.-Y.; Jia, C.-C.; Zheng, J.; Wang, G.-Y.; Zhao, H.; Yang, Q.; Liu, W.; Yi, S.-H.; Li, H.; et al. Aging aggravates hepatic ischemia-reperfusion injury in mice by impairing mitophagy with the involvement of the EIF2α-parkin pathway. Aging 2018, 10, 1902–1920. [Google Scholar] [CrossRef]
- Zhou, H.; Du, W.; Li, Y.; Shi, C.; Hu, N.; Ma, S.; Wang, W.; Ren, J. Effects of melatonin on fatty liver disease: The role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J. Pineal Res. 2018, 64, e12450. [Google Scholar] [CrossRef]
- Lin, C.-W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.-M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef]
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019, 864, 172715. [Google Scholar] [CrossRef]
- Li, X.; Shi, Z.; Zhu, Y.; Shen, T.; Wang, H.; Shui, G.; Loor, J.J.; Fang, Z.; Chen, M.; Wang, X.; et al. Cyanidin-3-O-glucoside improves non-alcoholic fatty liver disease by promoting PINK1-mediated mitophagy in mice. Br. J. Pharmacol. 2020, 177, 3591–3607. [Google Scholar] [CrossRef]
- Li, R.; Xin, T.; Li, D.; Wang, C.; Zhu, H.; Zhou, H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018, 18, 229–243. [Google Scholar] [CrossRef]
- Gao, A.; Jiang, J.; Xie, F.; Chen, L. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 506, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Sädevirta, S.; Zhou, Y.; Kayser, B.; Ali, A.; Ahonen, L.; Lallukka, S.; Pelloux, V.; Gaggini, M.; Jian, C.; et al. Saturated Fat Is More Metabolically Harmful for the Human Liver Than Unsaturated Fat or Simple Sugars. Diabetes Care 2018, 41, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Oteng, A.-B.; Loregger, A.; van Weeghel, M.; Zelcer, N.; Kersten, S. Industrial Trans Fatty Acids Stimulate SREBP2-Mediated Cholesterogenesis and Promote Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900385. [Google Scholar] [CrossRef]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef]
- Valenzuela, R.; Videla, L.A. The importance of the long-chain polyunsaturated fatty acid n-6/n-3 ratio in development of non-alcoholic fatty liver associated with obesity. Food Funct. 2011, 2, 644–648. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quiñones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. Lond. Engl. 1979 2004, 106, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhu, H.; Li, Y.; Lin, H.; Gabrielson, K.; Trush, M.A.; Diehl, A.M. Mitochondrial adaptations to obesity-related oxidant stress. Arch. Biochem. Biophys. 2000, 378, 259–268. [Google Scholar] [CrossRef]
- Aronis, A.; Madar, Z.; Tirosh, O. Mechanism underlying oxidative stress-mediated lipotoxicity: Exposure of J774.2 macrophages to triacylglycerols facilitates mitochondrial reactive oxygen species production and cellular necrosis. Free. Radic. Biol. Med. 2005, 38, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Videla, L.A.; Rodrigo, R.; Araya, J.; Poniachik, J. Insulin resistance and oxidative stress interdependency in non-alcoholic fatty liver disease. Trends Mol. Med. 2006, 12, 555–558. [Google Scholar] [CrossRef]
- Tsukamoto, H. Redox regulation of cytokine expression in Kupffer cells. Antioxid. Redox Signal. 2002, 4, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, L.; Rosa, M.D.; Zambito, A.M.; dell’Ombra, N.; Nicoletti, F.; Malaguarnera, M. Chitotriosidase gene expression in Kupffer cells from patients with non-alcoholic fatty liver disease. Gut 2006, 55, 1313–1320. [Google Scholar] [CrossRef]
- Orellana, M.; Rodrigo, R.; Varela, N.; Araya, J.; Poniachik, J.; Csendes, A.; Smok, G.; Videla, L.A. Relationship between in vivo chlorzoxazone hydroxylation, hepatic cytochrome P450 2E1 content and liver injury in obese non-alcoholic fatty liver disease patients. Hepatol. Res. 2006, 34, 57–63. [Google Scholar] [CrossRef]
- Weltman, M.D.; Farrell, G.C.; Liddle, C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 1996, 111, 1645–1653. [Google Scholar] [CrossRef]
- Caro, A.A.; Cederbaum, A.I. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Valenzuela, R.; Espinosa, A.; González-Mañán, D.; D’Espessailles, A.; Fernández, V.; Videla, L.A.; Tapia, G. N-3 long-chain polyunsaturated fatty acid supplementation significantly reduces liver oxidative stress in high fat induced steatosis. PLoS ONE 2012, 7, e46400. [Google Scholar] [CrossRef]
- Gentile, C.L.; Frye, M.A.; Pagliassotti, M.J. Fatty acids and the endoplasmic reticulum in nonalcoholic fatty liver disease. BioFactors Oxf. Engl. 2011, 37, 8–16. [Google Scholar] [CrossRef]
- Zhang, X.-Q.; Xu, C.-F.; Yu, C.-H.; Chen, W.-X.; Li, Y.-M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef]
- Yi, H.S. Implications of Mitochondrial Unfolded Protein Response and Mitokines: A Perspective on Fatty Liver Diseases. Endocrinol. Metab. Seoul Korea 2019, 34, 39–46. [Google Scholar] [CrossRef]
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193–199. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.; Soto-Alarcón, S.A.; Orellana, P.; Espinosa, A.; Campos, C.; López-Arana, S.; Rincón, M.A.; Illesca, P.; Valenzuela, R.; Videla, L.A. Suppression of high-fat diet-induced obesity-associated liver mitochondrial dysfunction by docosahexaenoic acid and hydroxytyrosol co-administration. Dig. Liver Dis. 2020, 52, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.S.; Duarte, F.V.; Gomes, A.P.; Varela, A.T.; Peixoto, F.M.; Rolo, A.P.; Palmeira, C.M. Berberine reverts hepatic mitochondrial dysfunction in high-fat fed rats: A possible role for SirT3 activation. Mitochondrion 2013, 13, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Lohr, K.; Pachl, F.; Moghaddas Gholami, A.; Geillinger, K.E.; Daniel, H.; Kuster, B.; Klingenspor, M. Reduced mitochondrial mass and function add to age-related susceptibility toward diet-induced fatty liver in C57BL/6J mice. Physiol. Rep. 2016, 4, e12988. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Sugden, M.C.; Caton, P.W.; Holness, M.J. PPAR control: It’s SIRTainly as easy as PGC. J. Endocrinol. 2010, 204, 93–104. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef]
- Okada, L.S.D.R.R.; Oliveira, C.P.; Stefano, J.T.; Nogueira, M.A.; da Silva, I.D.C.G.; Cordeiro, F.B.; Alves, V.A.F.; Torrinhas, R.S.; Carrilho, F.J.; Puri, P.; et al. Omega-3 PUFA modulate lipogenesis, ER stress, and mitochondrial dysfunction markers in NASH—Proteomic and lipidomic insight. Clin. Nutr. 2018, 37, 1474–1484. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Wang, J.; He, W.; Tsai, P.-J.; Chen, P.-H.; Ye, M.; Guo, J.; Su, Z. Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Health Dis. 2020, 19, 72. [Google Scholar] [CrossRef]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-Erkkilä, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjøsund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 19, 3948–3958. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Model | Main Findings |
|---|---|---|
| Cordeiro et al., 2020 [21] | Young and old mice underwent 4 weeks of aerobic exercise training | Increased UPRmt markers in gastrocnemius muscle of aged mice |
| Tamura et al., 2017 [22] | Young and aged mice received heat stress treatment | Remarkable improvements in age-related changes in soleus, but minor effects in gastrocnemius and plantaris |
| Memme et al., 2016 [23] | Rats subjected to chronic contractile activity (CCA) for 1 to 7 days | UPRmt-specific markers were induced 10 to 80% between days 1 and 7 |
| Al-Furoukh et al., 2015 [24] | ClpX overexpression in C2C12 mouse myoblasts and HEK293T cells | Upregulation of markers of the UPRmt |
| Gariani et al., 2016 [25] | Mice fed with HFHS diet plus nicotinamide riboside | Induction of a SIRT1- and SIRT3-dependent UPRmt, preventing/reverting NAFLD |
| Quirós et al., 2017 [26] | Multiomics approach in mammalian cells treated with 4 mitochondrial stressors | Identification of ATF4 as the main regulator of the stress response |
| Fiorese et al., 2016 [27] | C. elegans lacking ATFS-1 and expressing a reporter of UPRmt | Rescue of UPRmt activation under stress conditions by ATF5 |
| Bennett et al., 2014 [28] | Genome-wide RNAi screen for negative regulators of the UPRmt | UPRmt is neither necessary nor sufficient for lifespan extension |
| Michel et al., 2015 [29] | Impairment of mtDNA expression | Triggering of ISR, but not of UPRmt |
| Zhao et al., 2002 [30] | Mutated OTC to provoke mitochondrial protein accumulation | Induction of nuclear genes encoding for Hsp60, Hsp10 and ClpP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbina-Varela, R.; Castillo, N.; Videla, L.A.; del Campo, A. Impact of Mitophagy and Mitochondrial Unfolded Protein Response as New Adaptive Mechanisms Underlying Old Pathologies: Sarcopenia and Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 7704. https://doi.org/10.3390/ijms21207704
Urbina-Varela R, Castillo N, Videla LA, del Campo A. Impact of Mitophagy and Mitochondrial Unfolded Protein Response as New Adaptive Mechanisms Underlying Old Pathologies: Sarcopenia and Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2020; 21(20):7704. https://doi.org/10.3390/ijms21207704
Chicago/Turabian StyleUrbina-Varela, Rodrigo, Nataly Castillo, Luis A. Videla, and Andrea del Campo. 2020. "Impact of Mitophagy and Mitochondrial Unfolded Protein Response as New Adaptive Mechanisms Underlying Old Pathologies: Sarcopenia and Non-Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 21, no. 20: 7704. https://doi.org/10.3390/ijms21207704
APA StyleUrbina-Varela, R., Castillo, N., Videla, L. A., & del Campo, A. (2020). Impact of Mitophagy and Mitochondrial Unfolded Protein Response as New Adaptive Mechanisms Underlying Old Pathologies: Sarcopenia and Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences, 21(20), 7704. https://doi.org/10.3390/ijms21207704