Facts and Challenges in Immunotherapy for T-Cell Acute Lymphoblastic Leukemia


Abstract
1. Introduction
2. Immunotherapy of T-ALL with Monoclonal Antibodies
2.1. Mechanisms Underlying Monoclonal Antibody Therapy
2.2. CD38
2.3. CD52
3. Immunotherapy of T-ALL with CAR T Cells
3.1. CD5
3.2. CD7
3.3. CD3
3.4. CD4
3.5. CD1a
3.6. TCRβ Constant Region
4. Potential New Targets for T-ALL Immunotherapy
4.1. CXCR4
4.2. IL-7R
4.3. CD44
4.4. Other Potential Targets: CD30 and CD99
5. Conclusions and Remarks
Funding
Conflicts of Interest
References
- Borella, L.; Sen, L. T cell surface markers on lymphoblasts from acute lymphocytic leukemia. J. Immunol. 1973, 111, 1257–1260. [Google Scholar]
- Pui, C.H.; Relling, M.V.; Downing, J.R. Mechanisms of disease: Acute lymphoblastic leukemia. N. Engl. J. Med. 2004, 350, 1535–1548. [Google Scholar] [CrossRef]
- Goldberg, J.M.; Silverman, L.B.; Levy, D.E.; Dalton, V.K.; Gelber, R.D.; Lehmann, L.; Cohen, H.J.; Sallan, S.E.; Asselin, B.L. Childhood T-cell acute lymphoblastic leukemia: The Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium experience. J. Clin. Oncol. 2003, 21, 3616–3622. [Google Scholar] [CrossRef]
- Pui, C.H.; Howard, S.C. Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol. 2008, 9, 257–268. [Google Scholar] [CrossRef]
- Bene, M.C.; Castoldi, G.; Knapp, W.; Ludwig, W.D.; Matutes, E.; Orfao, A.; Van’t Veer, M.B. Proposals for the immunological classification of acute leukemias. Leukemia 1995, 9, 1783–1786. [Google Scholar]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef]
- Chen, B.; Jiang, L.; Zhong, M.L.; Li, J.F.; Li, B.S.; Peng, L.J.; Dai, Y.T.; Cui, B.W.; Yan, T.Q.; Zhang, W.N.; et al. Identification of fusion genes and characterization of transcriptome features in T-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2017, 115, 373–378. [Google Scholar] [CrossRef]
- De Bie, J.; Demeyer, S.; Alberti-Servera, L.; Geerdens, E.; Segers, H.; Broux, M.; De Keersmaecker, K.; Michaux, L.; Vandenberghe, P.; Voet, T.; et al. Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 1358–1369. [Google Scholar] [CrossRef]
- Mirji, G.; Bhat, J.; Kode, J.; Banavali, S.; Sengar, M.; Khadke, P.; Sait, O.; Chiplunkar, S. Risk stratification of T-cell Acute Lymphoblastic Leukemia patients based on gene expression, mutations and copy number variation. Leuk. Res. 2016, 45, 33–39. [Google Scholar] [CrossRef]
- Kataoka, K.; Iwanaga, M.; Yasunaga, J.I.; Nagata, Y.; Kitanaka, A.; Kameda, T.; Yoshimitsu, M.; Shiraishi, Y.; Sato-Otsubo, A.; Sanada, M.; et al. Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma. Blood 2018, 131, 215–225. [Google Scholar] [CrossRef]
- Petit, A.; Trinquand, A.; Chevret, S.; Ballerini, P.; Cayuela, J.M.; Grardel, N.; Touzart, A.; Brethon, B.; Lapillonne, H.; Schmitt, C.; et al. Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 2018, 131, 289–300. [Google Scholar] [CrossRef]
- Begley, C.G.; Aplan, P.D.; Davey, M.P.; Nakahara, K.; Tchorz, K.; Kurtzberg, J.; Hershfield, M.S.; Haynes, B.F.; Cohen, D.I.; Waldmann, T.A.; et al. Chromosomal translocation in a human leukemic stem-cell line disrupts the T-cell antigen receptor δ-chain diversity region and results in a previously unreported fusion transcript. Proc. Natl. Acad. Sci. USA 1989, 86, 2031–2035. [Google Scholar] [CrossRef]
- Mellentin, J.D.; Smith, S.D.; Cleary, M.L. lyl-1, a novel gene altered by chromosomal translocation in T cell leukemia, codes for a protein with a helix-loop-helix DNA binding motif. Cell 1989, 58, 77–83. [Google Scholar] [CrossRef]
- Dube, I.D.; Kamel-Reid, S.; Yuan, C.C.; Lu, M.; Wu, X.; Corpus, G.; Raimondi, S.C.; Crist, W.M.; Carroll, A.J.; Minowada, J.; et al. A novel human homeobox gene lies at the chromosome 10 breakpoint in lymphoid neoplasias with chromosomal translocation t(10;14). Blood 1991, 78, 2996–3003. [Google Scholar] [CrossRef]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Pieters, R.; Beverloo, H.B.; Meijerink, J.P.P. Molecular-genetic insights in paediatric T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2008, 143, 153–168. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris IV, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef]
- Van der Zwet, J.C.G.; Cordo’, V.; Canté-Barrett, K.; Meijerink, J.P.P. Multi-omic approaches to improve outcome for T-cell acute lymphoblastic leukemia patients. Adv. Biol. Regul. 2019, 74. [Google Scholar] [CrossRef]
- Noronha, E.P.; Codeço Marques, L.V.; Andrade, F.G.; Santos Thuler, L.C.; Terra-Granado, E.; Pombo-De-Oliveira, M.S. The profile of immunophenotype and genotype aberrations in subsets of pediatric T-cell acute lymphoblastic leukemia. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Girardi, T.; Vicente, C.; Cools, J.; De Keersmaecker, K. The genetics and molecular biology of T-ALL. Blood 2017, 129, 1113–1123. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3398–3406. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; WHO: Lyon, France, 2017; Volume 4, p. 326. [Google Scholar] [CrossRef]
- Wenzinger, C.; Williams, E.; Gru, A.A. Updates in the Pathology of Precursor Lymphoid Neoplasms in the Revised Fourth Edition of the WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. Curr. Hematol. Malig. Rep. 2018, 13, 275–288. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Mullighan, C.G.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.E.; Basso, G.; et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009, 10, 147–156. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Haydu, J.E.; Rigo, I.; Hadler, M.; Tosello, V.; Della Gatta, G.; Paietta, E.; Racevskis, J.; et al. ETV6 mutations in early immature human T cell leukemias. J. Exp. Med. 2011, 208, 2571–2579. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Ambesi-Impiombato, A.; De Keersmaecker, K.; Hadler, M.; Paietta, E.; Tallman, M.S.; Rowe, J.M.; Forne, C.; Rue, M.; Ferrando, A.A. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood 2013, 122, 74–82. [Google Scholar] [CrossRef]
- Jain, N.; Lamb, A.V.; O’Brien, S.; Ravandi, F.; Konopleva, M.; Jabbour, E.; Zuo, Z.; Jorgensen, J.; Lin, P.; Pierce, S.; et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: A high-risk subtype. Blood 2016, 127, 1863–1869. [Google Scholar] [CrossRef]
- Jain, N.; Lamb, A.; O’Brien, S.M.; Ravandi, F.; Konopleva, M.; Jabbour, E.; Zuo, Z.; Jorgensen, J.L.; Lin, P.; Pierce, S.; et al. Early T-Cell Precursor Acute Lymphoblastic Leukemia (ETP-ALL) Is a High-Risk Subtype in Adults. Blood 2015, 126, 1418. [Google Scholar] [CrossRef]
- Patrick, K.; Wade, R.; Goulden, N.; Mitchell, C.; Moorman, A.V.; Rowntree, C.; Jenkinson, S.; Hough, R.; Vora, A. Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br. J. Haematol. 2014, 166, 421–424. [Google Scholar] [CrossRef]
- Asnafi, V.; Bond, J.; Graux, C.; Lhermitte, L.; Lara, D.; Cluzeau, T.; Leguay, T.; Cieslak, A.; Trinquand, A.; Pastoret, C.; et al. Early response–based therapy stratification improves survival in adult early thymic precursor acute lymphoblastic leukemia: A Group for research on adult acute lymphoblastic leukemia study. J. Clin. Oncol. 2017, 35, 2683–2691. [Google Scholar] [CrossRef]
- Hefazi, M.; Litzow, M.R. Recent Advances in the Biology and Treatment of T Cell Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2018, 13, 265–274. [Google Scholar] [CrossRef]
- Patel, A.A.; Thomas, J.; Rojek, A.E.; Stock, W. Biology and Treatment Paradigms in T Cell Acute Lymphoblastic Leukemia in Older Adolescents and Adults. Curr. Treat. Options Oncol. 2020, 21. [Google Scholar] [CrossRef]
- Teachey, D.T.; O’Connor, D. How i treat newly diagnosed T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma in children. Blood 2020, 135, 159–166. [Google Scholar] [CrossRef]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 1663–1669. [Google Scholar] [CrossRef]
- Winter, S.S.; Dunsmore, K.P.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Improved survival for children and young adults with t-lineage acute lymphoblastic leukemia: Results from the children’s oncology Group AALL0434 methotrexate randomization. J. Clin. Oncol. 2018, 36, 2926–2934. [Google Scholar] [CrossRef]
- Marks, D.I.; Paietta, E.M.; Moorman, A.V.; Richards, S.M.; Buck, G.; DeWald, G.; Ferrando, A.; Fielding, A.K.; Goldstone, A.H.; Ketterling, R.P.; et al. T-cell acute lymphoblastic leukemia in adults: Clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood 2009, 114, 5136–5145. [Google Scholar] [CrossRef]
- Marks, D.I.; Rowntree, C. Management of adults with T-cell lymphoblastic leukemia. Blood 2017, 129, 1134–1142. [Google Scholar] [CrossRef]
- Abaza, Y.; Kantarjian, H.M.; Faderl, S.; Jabbour, E.; Jain, N.; Thomas, D.; Kadia, T.; Borthakur, G.; Khoury, J.D.; Burger, J.; et al. Hyper-CVAD plus nelarabine in newly diagnosed adult T-cell acute lymphoblastic leukemia and T-lymphoblastic lymphoma. Am. J. Hematol. 2018, 93, 91–99. [Google Scholar] [CrossRef]
- Schrappe, M.; Valsecchi, M.G.; Bartram, C.R.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Parasole, R.; Zimmermann, M.; Dworzak, M.; Buldini, B.; et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: Results of the AIEOP-BFM-ALL 2000 study. Blood 2011, 118, 2077–2084. [Google Scholar] [CrossRef]
- Modvig, S.; Madsen, H.O.; Siitonen, S.M.; Rosthøj, S.; Tierens, A.; Juvonen, V.; Osnes, L.T.N.; Vålerhaugen, H.; Hultdin, M.; Thörn, I.; et al. Minimal residual disease quantification by flow cytometry provides reliable risk stratification in T-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 1324–1336. [Google Scholar] [CrossRef]
- Brüggemann, M.; Raff, T.; Flohr, T.; Gökbuget, N.; Nakao, M.; Droese, J.; Lüschen, S.; Pott, C.; Ritgen, M.; Scheuring, U.; et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood 2006, 107, 1116–1123. [Google Scholar] [CrossRef]
- Beldjord, K.; Chevret, S.; Asnafi, V.; Huguet, F.; Boulland, M.L.; Leguay, T.; Thomas, X.; Cayuela, J.M.; Grardel, N.; Chalandon, Y.; et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood 2014, 123, 3739–3749. [Google Scholar] [CrossRef]
- Berg, S.L.; Blaney, S.M.; Devidas, M.; Lampkin, T.A.; Murgo, A.; Bernstein, M.; Billett, A.; Kurtzberg, J.; Reaman, G.; Gaynon, P.; et al. Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: A report from the children’s oncology group. J. Clin. Oncol. 2005, 23, 3376–3382. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Yu, D.; Johnson, J.L.; Coutre, S.E.; Stone, R.M.; Stopeck, A.T.; Gockerman, J.P.; Mitchell, B.S.; Appelbaum, F.R.; Larson, R.A. Nelarabine induces complete remissions in adults with relapsed or refractory T-lineage acute lymphoblastic leukemia or lymphoblastic lymphoma: Cancer and Leukemia Group B study 19801. Blood 2007, 109, 5136–5142. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.; Ravandi, F.; Thomas, D.; O’Brien, S.; Kadia, T.; Burger, J.; Borthakur, G.; Daver, N.; Jabbour, E.; et al. The combination of hyper-CVAD plus nelarabine as frontline therapy in adult T-cell acute lymphoblastic leukemia and T-lymphoblastic lymphoma: MD Anderson Cancer Center experience. Leukemia 2014, 28, 973–975. [Google Scholar] [CrossRef]
- Zwaan, C.M.; Kowalczyk, J.; Schmitt, C.; Bielorai, B.; Russo, M.W.; Woessner, M.; Ranganathan, S.; Leverger, G. Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: Results of a phase 4 study. Br. J. Haematol. 2017, 179, 284–293. [Google Scholar] [CrossRef]
- Kuhlen, M.; Bleckmann, K.; Möricke, A.; Schrappe, M.; Vieth, S.; Escherich, G.; Bronsema, A.; Vonalt, A.; Queudeville, M.; Zwaan, C.M.; et al. Neurotoxic side effects in children with refractory or relapsed T-cell malignancies treated with nelarabine based therapy. Br. J. Haematol. 2017, 179, 272–283. [Google Scholar] [CrossRef]
- Alberti, P.; Parma, M.; Pioltelli, P.; Pogliani, E.M.; Terruzzi, E.; Stasia, A.; Doni, E.; Cecchetti, C.; Cavaletti, G. Severe, reversible nelarabine-induced neuropathy and myelopathy. J. Peripher. Nerv. Syst. 2016, 21, 154–156. [Google Scholar] [CrossRef]
- Ewins, K.; Malone, A.; Phelan, E.; Webb, D.; McHugh, J.C.; Smith, O. Nelarabine-induced peripheral and central neurotoxicity: Can sequential MRI brain imaging help to define its natural history? Br. J. Haematol. 2017, 179, 294–297. [Google Scholar] [CrossRef]
- Clappier, E.; Gerby, B.; Sigaux, F.; Delord, M.; Touzri, F.; Hernandez, L.; Ballerini, P.; Baruchel, A.; Pflumio, F.; Soulier, J. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J. Exp. Med. 2011, 208, 653–661. [Google Scholar] [CrossRef]
- Immunotherapy Clinical Trials. Available online: https://www.clinicaltrials.gov (accessed on 28 August 2020).
- Simpson, A.; Caballero, O. Monoclonal antibodies for the therapy of cancer. BMC Proc. 2014, 8. [Google Scholar] [CrossRef]
- Bakema, J.E.; Van Egmond, M. Fc receptor-dependent mechanisms of monoclonal antibody therapy of cancer. Curr. Top. Microbiol. Immunol. 2014, 382, 373–392. [Google Scholar] [CrossRef]
- Muntasell, A.; Ochoa, M.C.; Cordeiro, L.; Berraondo, P.; López-Díaz de Cerio, A.; Cabo, M.; López-Botet, M.; Melero, I. Targeting NK-cell checkpoints for cancer immunotherapy. Curr. Opin. Immunol. 2017, 45, 73–81. [Google Scholar] [CrossRef]
- Chung, S.; Lin, Y.L.; Reed, C.; Ng, C.; Cheng, Z.J.; Malavasi, F.; Yang, J.; Quarmby, V.; Song, A. Characterization of in vitro antibody-dependent cell-mediated cytotoxicity activity of therapeutic antibodies—Impact of effector cells. J. Immunol. Methods 2014, 407, 63–75. [Google Scholar] [CrossRef]
- Taylor, R.P.; Lindorfer, M.A. Cytotoxic mechanisms of immunotherapy: Harnessing complement in the action of anti-tumor monoclonal antibodies. Semin. Immunol. 2016, 28, 309–316. [Google Scholar] [CrossRef]
- Weiner, L.M.; Dhodapkar, M.V.; Ferrone, S. Monoclonal antibodies for cancer immunotherapy. Lancet 2009, 373, 1033–1040. [Google Scholar] [CrossRef]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. MAbs 2020, 12. [Google Scholar] [CrossRef]
- Jackson, D.G.; Bell, J.I. Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. J. Immunol. 1990, 144, 2811–2815. [Google Scholar]
- Ferrero, E.; Malavasi, F. Human CD38, a Leukocyte Receptor and Ectoenzyme, Is a Member of a Novel Eukaryotic Gene Family of Nicotinamide Adenine Dinucleotide’Xonverting Enzymes. J. Immunol. 1997, 159, 3858–3865. [Google Scholar]
- Hara-Yokoyama, M.; Kukimoto-Niino, M.; Terasawa, K.; Harumiya, S.; Podyma-Inoue, K.A.; Hino, N.; Sakamoto, K.; Itoh, S.; Hashii, N.; Hiruta, Y.; et al. Tetrameric interaction of the ectoenzyme CD38 on the cell surface enables its catalytic and raft-association activities. Structure 2012, 20, 1585–1595. [Google Scholar] [CrossRef]
- Franco, L.; Guida, L.; Bruzzone, S.; Zocchi, E.; Usai, C.; De Flora, A. The transmembrane glycoprotein CD38 is a catalytically active transporter responsible for generation and influx of the second messenger cyclic ADP-ribose across membranes. FASEB J. 1998, 12, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar] [PubMed]
- Guse, A.H.; Berg, I.; Da Silva, C.P.; Potter, B.V.L.; Mayr, G.W. Ca2+ entry induced by cyclic ADP-ribose in intact T-lymphocytes. J. Biol. Chem. 1997, 272, 8546–8550. [Google Scholar] [CrossRef] [PubMed]
- Reinherz, E.L.; Kung, P.C.; Goldstein, G.; Levey, R.H.; Schlossman, S.F. Discrete stages of human intrathymic differentiation: Analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc. Natl. Acad. Sci. USA 1980, 77, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Rousset, F. Human B Lymphocytes: Phenotype, Proliferation, and Differentiation. Adv. Immunol. 1992, 52, 125–262. [Google Scholar] [CrossRef]
- Hercend, T.; Ritz, J.; Schlossman, S.F.; Reinherz, E.L. Comparative expression of T9, T10, and Ia antigens on activated human T cell subsets. Hum. Immunol. 1981, 3, 247–259. [Google Scholar] [CrossRef]
- Stashenko, P.; Nadler, L.M.; Hardy, R.; Schlossman, S.F. Expression of cell surface markers after human B lymphocyte activation. Proc. Natl. Acad. Sci. USA 1981, 78, 3848–3852. [Google Scholar] [CrossRef]
- Prince, H.E.; York, J.; Jensen, E.R. Phenotypic comparison of the three populations of human lymphocytes defined by CD45RO and CD45RA expression. Cell. Immunol. 1992, 145, 254–262. [Google Scholar] [CrossRef]
- Zubiaur, M.; Izquierdo, M.; Terhorst, C.; Malavasi, F.; Sancho, J. CD38 ligation results in activation of the Raf-1/mitogen-activated protein kinase and the CD3-zeta/zeta-associated protein-70 signaling pathways in Jurkat T lymphocytes. J. Immunol. 1997, 159, 193–205. [Google Scholar]
- Zubiaur, M.; Guirado, M.; Terhorst, C.; Malavasi, F.; Sancho, J. The CD3-γδε transducing module mediates CD38-induced protein-tyrosine kinase and mitogen-activated protein kinase activation in Jurkat T cells. J. Biol. Chem. 1999, 274, 20633–20642. [Google Scholar] [CrossRef] [PubMed]
- Zubiaur, M.; Fernández, O.; Ferrero, E.; Salmerón, J.; Malissen, B.; Malavasi, F.; Sancho, J. CD38 is associated with lipid rafts and upon receptor stimulation leads to Akt/protein kinase B and Erk activation in the absence of the CD3-ζ immune receptor tyrosine-based activation motifs. J. Biol. Chem. 2002, 277, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Tenca, C.; Merlo, A.; Zarcone, D.; Saverino, D.; Bruno, S.; De Santanna, A.; Ramarli, D.; Fabbi, M.; Pesce, C.; Deaglio, S.; et al. Death of T cell precursors in the human thymus: A role for CD38. Int. Immunol. 2003, 15, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 1998, 160, 395–402. [Google Scholar] [PubMed]
- Cesano, A.; Visonneau, S.; Deaglio, S.; Malavasi, F.; Santoli, D. Role of CD38 and its ligand in the regulation of MHC-nonrestricted cytotoxic T cells. J. Immunol. 1998, 160, 1106–1115. [Google Scholar]
- Calabretta, E.; Carlo-Stella, C. The Many Facets of CD38 in Lymphoma: From Tumor–Microenvironment Cell Interactions to Acquired Resistance to Immunotherapy. Cells 2020, 9, 802. [Google Scholar] [CrossRef]
- Matas-Céspedes, A.; Vidal-Crespo, A.; Rodriguez, V.; Villamor, N.; Delgado, J.; Giné, E.; Roca-Ho, H.; Menéndez, P.; Campo, E.; López-Guillermo, A.; et al. The human CD38 monoclonal antibody daratumumab shows antitumor activity and hampers leukemia-microenvironment interactions in chronic lymphocytic leukemia. Clin. Cancer Res. 2017, 23, 1493–1505. [Google Scholar] [CrossRef]
- De Weers, M.; Tai, Y.-T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Deckert, J.; Wetzel, M.C.; Bartle, L.M.; Skaletskaya, A.; Goldmacher, V.S.; Vallée, F.; Zhou-Liu, Q.; Ferrari, P.; Pouzieux, S.; Lahoute, C.; et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin. Cancer Res. 2014, 20, 4574–4583. [Google Scholar] [CrossRef]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef]
- Plesner, T.; Arkenau, H.-T.; Lokhorst, H.M.; Gimsing, P.; Krejcik, J.; Lemech, C.; Minnema, M.C.; Lassen, U.; Laubach, J.P.; Ahmadi, T.; et al. Safety and Efficacy of Daratumumab with Lenalidomide and Dexamethasone in Relapsed or Relapsed, Refractory Multiple Myeloma. Blood 2014, 124, 84. [Google Scholar] [CrossRef]
- Tembhare, P.R.; Sriram, H.; Khanka, T.; Chatterjee, G.; Panda, D.; Ghogale, S.; Badrinath, Y.; Deshpande, N.; Patkar, N.V.; Narula, G.; et al. Flow cytometric evaluation of CD38 expression levels in the newly diagnosed T-cell acute lymphoblastic leukemia and the effect of chemotherapy on its expression in measurable residual disease, refractory disease and relapsed disease: An implication for an. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Mihara, K.; Yoshida, T.; Ishida, S.; Takei, Y.; Kitanaka, A.; Shimoda, K.; Morishita, K.; Takihara, Y.; Ichinohe, T. All-Trans Retinoic Acid and Interferon-Alpha Increase CD38 Expression on Adult T-Cell Leukemia Cells and Sensitize Them to T Cells Bearing Anti-CD38 Chimeric Antigen Receptors. Blood 2015, 126, 591. [Google Scholar] [CrossRef]
- Bride, K.L.; Vincent, T.L.; Im, S.Y.; Aplenc, R.; Barrett, D.M.; Carroll, W.L.; Carson, R.; Dai, Y.; Devidas, M.; Dunsmore, K.P.; et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood 2018, 131, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, F.; Winterberg, D.; Lenk, L.; Buchmann, S.; Cario, G.; Schrappe, M.; Peipp, M.; Richter-Pechanska, P.; Kulozik, A.E.; Lentes, J.; et al. Daratumumab eradicates minimal residual disease in a preclinical model of pediatric T-cell acute lymphoblastic leukemia. Blood 2019, 134, 713–716. [Google Scholar] [CrossRef]
- Gurunathan, A.; Emberesh, M.; Norris, R. A case report of using daratumumab in refractory T-cell acute lymphoblastic leukemia. Pediatr. Blood Cancer 2019, 66, S38–S39. [Google Scholar] [CrossRef]
- Ofran, Y.; Ringelstein-Harlev, S.; Slouzkey, I.; Zuckerman, T.; Yehudai-Ofir, D.; Henig, I.; Beyar-Katz, O.; Hayun, M.; Frisch, A. Daratumumab for eradication of minimal residual disease in high-risk advanced relapse of T-cell/CD19/CD22-negative acute lymphoblastic leukemia. Leukemia 2020, 34, 293–295. [Google Scholar] [CrossRef]
- Xia, M.Q.; Hale, G.; Lifely, M.R.; Ferguson, M.A.J.; Campbell, D.; Packman, L.; Waldmann, H. Structure of the CAMPATH-1 antigen, a glycosylphosphatidylinositol-anchored glycoprotein which is an exceptionally good target for complement lysis. Biochem. J. 1993, 293, 633–640. [Google Scholar] [CrossRef]
- Zhao, Y.; Su, H.; Shen, X.; Du, J.; Zhang, X.; Zhao, Y. The immunological function of CD52 and its targeting in organ transplantation. Inflamm. Res. 2017, 66, 571–578. [Google Scholar] [CrossRef]
- Rowan, W.C.; Hale, G.; Tite, J.P.; Brett, S.J. Cross-linking of the CAMPATH-1 antigen (CD52) triggers activation of normal human T lymphocytes. Int. Immunol. 1995, 7, 69–77. [Google Scholar] [CrossRef]
- Watanabe, T.; Masuyama, J.I.; Sohma, Y.; Inazawa, H.; Horie, K.; Kojima, K.; Uemura, Y.; Aoki, Y.; Kaga, S.; Minota, S.; et al. CD52 is a novel costimulatory molecule for induction of CD4+ regulatory T cells. Clin. Immunol. 2006, 120, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Bandala-Sanchez, E.; Zhang, Y.; Reinwald, S.; Dromey, J.A.; Lee, B.H.; Qian, J.; Böhmer, R.M.; Harrison, L.C. T cell regulation mediated by interaction of soluble CD52 with the inhibitory receptor Siglec-10. Nat. Immunol. 2013, 14, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, M.; Bandala-Sanchez, E.; Lawlor, K.E.; Zhang, Y.; Neale, A.M.; Vijayaraj, S.L.; O’Donoghue, R.; Wentworth, J.M.; Adams, T.E.; Vince, J.E.; et al. CD52 inhibits Toll-like receptor activation of NF-κB and triggers apoptosis to suppress inflammation. Cell Death Differ. 2018, 25, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Hale, G.; Bright, S.; Chumbley, G.; Hoang, T.; Metcalf, D.; Munro, A.J.; Waldmann, H. Removal of T cells from bone marrow for transplantation: A monoclonal antilymphocyte antibody that fixes human complement. Blood 1983, 62, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Keating, M.J.; Flinn, I.; Jain, V.; Binet, J.L.; Hillmen, P.; Byrd, J.; Albitar, M.; Brettman, L.; Santabarbara, P.; Wacker, B.; et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: Results of a large international study. Blood 2002, 99, 3554–3561. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, M.; Ravetch, J.V.; Goldman, C.; Waldmann, T.A. Effective therapy for a murine model of adult T-cell leukemia with the humanized anti-CD2 monoclonal antibody, MEDI-507. Blood 2003, 102, 284–288. [Google Scholar] [CrossRef]
- Nückel, H.; Frey, U.H.; Röth, A.; Dührsen, U.; Siffert, W. Alemtuzumab induces enhanced apoptosis in vitro in B-cells from patients with chronic lymphocytic leukemia by antibody-dependent cellular cytotoxicity. Eur. J. Pharmacol. 2005, 514, 217–224. [Google Scholar] [CrossRef]
- Jiang, L.; Yuan, C.M.; Hubacheck, J.; Janik, J.E.; Wilson, W.; Morris, J.C.; Jasper, G.A.; Stetler-Stevenson, M. Variable CD52 expression in mature T cell and NK cell malignancies: Implications for alemtuzumab therapy. Br. J. Haematol. 2009, 145, 173–179. [Google Scholar] [CrossRef]
- Tibes, R.; Keating, M.J.; Ferrajoli, A.; Wierda, W.; Ravandi, F.; Garcia-Manero, G.; O’Brien, S.; Cortes, J.; Verstovsek, S.; Browning, M.L.; et al. Activity of alemtuzumab in patients with CD52-positive acute leukemia. Cancer 2006, 106, 2645–2651. [Google Scholar] [CrossRef]
- Oehler, V.G.; Walter, R.B.; Cummings, C.; Sala-Torra, O.; Stirewalt, D.L.; Fang, M.; Radich, J.P.; Wood, B. CD52 Expression In Leukemic Stem/Progenitor Cells. Blood 2010, 116, 2743. [Google Scholar] [CrossRef]
- Lozanski, G.; Sanford, B.; Yu, D.; Pearson, R.; Edwards, C.; Byrd, J.C.; Larson, R.A.; Bloomfield, C.D.; Stock, W. CD52 Expression in Adult Acute Lymphoblastic Leukemia (ALL): Quantitative Flow Cytometry Provides New Insights. Blood 2006, 108, 2293. [Google Scholar] [CrossRef]
- Dearden, C.E.; Matutes, E.; Catovsky, D. Alemtuzumab in T-cell malignancies. Med. Oncol. 2002, 19, S27–S32. [Google Scholar] [CrossRef]
- Dearden, C. Alemtuzumab in peripheral T-cell malignancies. Cancer Biother. Radiopharm. 2004, 19, 391–398. [Google Scholar] [CrossRef]
- Ravandi, F.; Aribi, A.; O’Brien, S.; Faderl, S.; Jones, D.; Ferrajoli, A.; Huang, X.; York, S.; Pierce, S.; Wierda, W.; et al. Phase II study of alemtuzumab in combination with pentostatin in patients with T-cell neoplasms. J. Clin. Oncol. 2009, 27, 5425–5430. [Google Scholar] [CrossRef]
- Stock, W.; Sanford, B.; Lozanski, G.; Vij, R.; Byrd, J.C.; Powell, B.L.; Wetzler, M.; Sher, D.; Edwards, C.; Kelly, M.; et al. Alemtuzumab can be Incorporated Into Front-Line Therapy of Adult Acute Lymphoblastic Leukemia (ALL): Final Phase I Results of a Cancer and Leukemia Group B Study (CALGB 10102). Blood 2009, 114, 838. [Google Scholar] [CrossRef]
- Angiolillo, A.L.; Yu, A.L.; Reaman, G.; Ingle, A.M.; Secola, R.; Adamson, P.C. A phase II study of Campath-1H in children with relapsed or refractory acute lymphoblastic leukemia: A Children’s Oncology Group report. Pediatr. Blood Cancer 2009, 53, 978–983. [Google Scholar] [CrossRef]
- Novitzky, N.; Thomas, V.; Hale, G.; Waldmann, H. Ex vivo depletion of T cells from bone marrow grafts with CAMPATH-1 in acute leukemia: Graft-versus-host disease and graft-versus-leukemia effect. Transplantation 1999, 67, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Philip, L.P.B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; Lebuhotel, C.; et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef]
- Qasim, W.; Amrolia, P.J.; Samarasinghe, S.; Ghorashian, S.; Zhan, H.; Stafford, S.; Butler, K.; Ahsan, G.; Gilmour, K.; Adams, S.; et al. First Clinical Application of Talen Engineered Universal CAR19 T Cells in B-ALL. Blood 2015, 126, 2046. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.D.; Carroll, M.; et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.L.; DiPersio, J.F. Chimeric antigen receptor T cells (CAR-T) for the treatment of T-cell malignancies. Best Pract. Res. Clin. Haematol. 2019, 32. [Google Scholar] [CrossRef] [PubMed]
- Mamonkin, M.; Rouce, R.H.; Tashiro, H.; Brenner, M.K. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood 2015, 126, 983–992. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Calderon, H.; Posey, A.D.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Irving, B.A.; Weiss, A. The cytoplasmic domain of the T cell receptor ζ chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 1991, 64, 891–901. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef]
- Van Schandevyl, S.; Kerre, T. Chimeric antigen receptor T-cell therapy: Design improvements and therapeutic strategies in cancer treatment. Acta Clin. Belgica Int. J. Clin. Lab. Med. 2020, 75, 26–32. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Mohty, M.; Gautier, J.; Malard, F.; Aljurf, M.; Bazarbachi, A.; Chabannon, C.; Kharfan-Dabaja, M.A.; Savani, B.N.; Huang, H.; Kenderian, S.; et al. CD19 chimeric antigen receptor-T cells in B-cell leukemia and lymphoma: Current status and perspectives. Leukemia 2019, 33, 2767–2778. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Uddin, S.; Steinhoff, M. CAR-T Cell Therapies: An Overview of Clinical Studies Supporting Their Approved Use against Acute Lymphoblastic Leukemia and Large B-Cell Lymphomas. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Behm, F.G.; Crist, W.M. Clinical and Biologic Relevance of Immunologic Marker Studies in Childhood Acute Lymphoblastic Leukemia. Blood 1993, 82, 343–362. [Google Scholar] [CrossRef] [PubMed]
- Campana, D.; Van Dongen, J.J.M.; Mehta, A.; Coustan-Smith, E.; Wolvers-Tettero, I.L.M.; Ganeshaguru, K.; Janossy, G. Stages of T-cell Receptor Protein Expression in T-cell Acute Lymphoblastic Leukemia. Blood 1991, 77, 1546–1554. [Google Scholar] [CrossRef]
- Jones, N.H.; Clabby, M.L.; Dialynas, D.P.; Huang, H.J.S.; Herzenberg, L.A.; Strominger, J.L. Isolation of complementary DNA clones encoding the human lymphocyte glycoprotein T1/Leu-1. Nature 1986, 323, 346–349. [Google Scholar] [CrossRef]
- Berland, R.; Wortis, H.H. Origins and functions of B-1 cells with notes on the role of CD5. Annu. Rev. Immunol. 2002, 20, 253–300. [Google Scholar] [CrossRef]
- Brossard, C.; Semichon, M.; Trautmann, A.; Bismuth, G. CD5 Inhibits Signaling at the Immunological Synapse Without Impairing Its Formation. J. Immunol. 2003, 170, 4623–4629. [Google Scholar] [CrossRef]
- Bamberger, M.; Santos, A.M.; Gonçalves, C.M.; Oliveira, M.I.; James, J.R.; Moreira, A.; Lozano, F.; Davis, S.J.; Carmo, A.M. A new pathway of CD5 glycoprotein-mediated T cell inhibition dependent on inhibitory phosphorylation of fyn kinase. J. Biol. Chem. 2011, 286, 30324–30336. [Google Scholar] [CrossRef]
- Dalloul, A. CD5: A safeguard against autoimmunity and a shield for cancer cells. Autoimmun. Rev. 2009, 8, 349–353. [Google Scholar] [CrossRef]
- Biancone, L.; Bowen, M.A.; Lim, A.; Aruffo, A.; Andres, G.; Stamenkovic, I. Identification of a novel inducible cell-surface ligand of CD5 on activated lymphocytes. J. Exp. Med. 1996, 184, 811–819. [Google Scholar] [CrossRef]
- Azzam, H.S.; Grinberg, A.; Lui, K.; Shen, H.; Shores, E.W.; Love, P.E. CD5 expression is developmentally regulated by T cell receptor (TCR) signals and TCR avidity. J. Exp. Med. 1998, 188, 2301–2311. [Google Scholar] [CrossRef] [PubMed]
- Kernan, N.A.; Knowles, R.W.; Burns, M.J.; Broxmeyer, H.E.; Lu, L.; Lee, H.M.; Kawahata, R.T.; Scannon, P.J.; Dupont, B. Specific inhibition of in vitro lymphocyte transformation by an anti-pan T cell (gp67) ricin A chain immunotoxin. J. Immunol. 1984, 133, 137–146. [Google Scholar]
- Bertram, J.; Gill, P.; Levine, A.; Boquiren, D.; Hoffman, F.; Meyer, P.; Mitchell, M. Monoclonal antibody T101 in T cell malignancies: A clinical, pharmacokinetic, and immunologic correlation. Blood 1986, 68, 752–761. [Google Scholar] [CrossRef] [PubMed]
- LeMaistre, C.F.; Rosen, S.; Frankel, A.; Kornfeld, S.; Saria, E.; Meneghetti, C.; Drajesk, J.; Fishwild, D.; Scannon, P.; Byers, V. Phase I trial of H65-RTA immunoconjugate in patients with cutaneous T-cell lymphoma. Blood 1991, 78, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Lin, K.W.; Jares, A.; Firor, A.E.; Shuai, X.; Salman, H.; Golightly, M.; et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia 2017, 31, 2151–2160. [Google Scholar] [CrossRef]
- Raikar, S.S.; Fleischer, L.C.; Moot, R.; Fedanov, A.; Paik, N.Y.; Knight, K.A.; Doering, C.B.; Spencer, H.T. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef]
- Wada, M.; Zhang, H.; Fang, L.; Feng, J.; Tse, C.O.; Zhang, W.; Chen, Q.; Sha, S.; Cao, Y.; Chen, K.H.; et al. Characterization of an Anti-CD5 Directed CAR T-Cell against T-Cell Malignancies. Stem Cell Rev. Rep. 2020. [Google Scholar] [CrossRef]
- Jones, B.S.; Lamb, L.S.; Goldman, F.; Di Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef]
- Gorczyca, W. Atlas of Differential Diagnosis in Neoplastic Hematopathology, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Sempowski, G.D.; Lee, D.; Kaufman, R.E.; Haynes, B.F. Structure and function of the CD7 molecule. Crit. Rev. Immunol. 1999, 19, 331–348. [Google Scholar]
- Bonilla, F.A.; Kokron, C.M.; Swinton, P.; Geha, R.S. Targeted gene disruption of murine CD7. Int. Immunol. 1997, 9, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.M.; Staats, H.F.; Sundy, J.S.; Patel, D.D.; Sempowski, G.D.; Scearce, R.M.; Jones, D.M.; Haynes, B.F. Immunologic Characterization of CD7-Deficient Mice. J. Immunol. 1998, 160, 5749–5756. [Google Scholar]
- Frankel, A.E.; Laver, J.H.; Willingham, M.C.; Burns, L.J.; Kersey, J.H.; Vallera, D.A. Therapy of patients with T-cell lymphomas and leukemias using an anti-CD7 monoclonal antibody-ricin A chain immunotoxin. Leuk. Lymphoma 1997, 26, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Silva, D.; Srinivasan, M.; Sharma, S.; Lee, C.M.; Wagner, D.L.; Davis, T.H.; Rouce, R.H.; Bao, G.; Brenner, M.K.; Mamonkin, M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017, 130, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef] [PubMed]
- Png, Y.T.; Vinanica, N.; Kamiya, T.; Shimasaki, N.; Coustan-Smith, E.; Campana, D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017, 1, 2348–2360. [Google Scholar] [CrossRef]
- Sabattini, E.; Bacci, F.; Sagramoso, C.; Pileri, S.A. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: An overview. Pathologica 2010, 102, 83–87. [Google Scholar]
- Kuhn, C.; Weiner, H.L. Therapeutic anti-CD3 monoclonal antibodies: From bench to bedside. Immunotherapy 2016, 8, 889–906. [Google Scholar] [CrossRef]
- Trinquand, A.; Dos Santos, N.R.; Quang, C.T.; Rocchetti, F.; Zaniboni, B.; Belhocine, M.; De Jesus, C.D.C.; Lhermitte, L.; Tesio, M.; Dussiot, M.; et al. Triggering the TCR developmental checkpoint activates a therapeutically targetable tumor suppressive pathway in T-cell leukemia. Cancer Discov. 2016, 6, 973–985. [Google Scholar] [CrossRef]
- Frankel, A.; Zuckero, S.; Mankin, A.; Grable, M.; Mitchell, K.; Lee, Y.; Neville, D.; Woo, J. Anti-CD3 Recombinant Diphtheria Immunotoxin Therapy of Cutaneous T Cell Lymphoma. Curr. Drug Targets 2009, 10, 104–109. [Google Scholar] [CrossRef]
- Chen, K.H.; Wada, M.; Firor, A.E.; Pinz, K.G.; Jares, A.; Liu, H.; Salman, H.; Golightly, M.; Lan, F.; Jiang, X.; et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016, 7, 56219–56232. [Google Scholar] [CrossRef] [PubMed]
- Rasaiyaah, J.; Georgiadis, C.; Preece, R.; Mock, U.; Qasim, W. TCRαβ/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Juillerat, A.; Tkach, D.; Yang, M.; Boyne, A.; Valton, J.; Poirot, L.; Duchateau, P. Straightforward Generation of Ultrapure Off-the-Shelf Allogeneic CAR-T Cells. Front. Bioeng. Biotechnol. 2020, 8, 678. [Google Scholar] [CrossRef] [PubMed]
- Pinz, K.; Liu, H.; Golightly, M.; Jares, A.; Lan, F.; Zieve, G.W.; Hagag, N.; Schuster, M.; Firor, A.E.; Jiang, X.; et al. Preclinical targeting of human T-cell malignancies using CD4-specific chimeric antigen receptor (CAR)-engineered T cells. Leukemia 2016, 30, 701–707. [Google Scholar] [CrossRef]
- Zhou, L.; Chong, M.M.W.; Littman, D.R. Plasticity of CD4+ T Cell Lineage Differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.P.; Deng, T.Z.; Witherden, D.A.; Goldrath, A.W. Origins of CD4+ circulating and tissue-resident memory T-cells. Immunology 2019, 157, 3–12. [Google Scholar] [CrossRef]
- Stewart, S.J.; Fujimoto, J.; Levy, R. Human T lymphocytes and monocytes bear the same Leu-3(T4) antigen. J. Immunol. 1986, 136, 3773–3778. [Google Scholar] [PubMed]
- Wood, G.S.; Warner, N.L.; Warnke, R.A. Anti-Leu-3/T4 antibodies react with cells of monocyte/macrophage and Langerhans lineage. J. Immunol. 1983, 131, 212–216. [Google Scholar]
- Lucey, D.R.; Dorsky, D.I.; Nicholson-Weller, A.; Weller, P.F. Human eosinophils express CD4 protein and bind human immunodeficiency virus 1 gp120. J. Exp. Med. 1989, 169, 327–332. [Google Scholar] [CrossRef]
- Glatzová, D.; Cebecauer, M. Dual role of CD4 in peripheral T lymphocytes. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Knox, S.; Hoppe, R.T.; Maloney, D.; Gibbs, I.; Fowler, S.; Marquez, C.; Cornbleet, P.J.A.; Levy, R. Treatment of cutaneous T-cell lymphoma with chimeric anti-CD4 monoclonal antibody. Blood 1996, 87, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Duvic, M.; Obitz, E.; Gniadecki, R.; Iversen, L.; Österborg, A.; Whittaker, S.; Illidge, T.M.; Schwarz, T.; Kaufmann, R.; et al. Clinical efficacy of zanolimumab (HuMax-CD4): Two phase 2 studies in refractory cutaneous T-cell lymphoma. Blood 2007, 109, 4655–4662. [Google Scholar] [CrossRef]
- Hagberg, H.; Pettersson, M.; Bjerner, T.; Enblad, G. Treatment of a patient with a nodal peripheral T-cell lymphoma (angioimmunoblastic T-cell lymphoma) with a human monoclonal antibody against the CD4 antigen (HuMax-CD4). Med. Oncol. 2005, 22, 191–194. [Google Scholar] [CrossRef]
- D’Amore, F.; Radford, J.; Relander, T.; Jerkeman, M.; Tilly, H.; Österborg, A.; Morschhauser, F.; Gramatzki, M.; Dreyling, M.; Bang, B.; et al. Phase II trial of zanolimumab (HuMax-CD4) in relapsed or refractory non-cutaneous peripheral T cell lymphoma. Br. J. Haematol. 2010, 150, 565–573. [Google Scholar] [CrossRef]
- Pinz, K.G.; Yakaboski, E.; Jares, A.; Liu, H.; Firor, A.E.; Chen, K.H.; Wada, M.; Salman, H.; Tse, W.; Hagag, N.; et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget 2017, 8, 112783–112796. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Shen, J.; Pinz, K.; Wada, M.; Park, J.; Kim, S.; Togano, T.; Tse, W. Targeting T Cell Malignancies Using CD4CAR T-Cells and Implementing a Natural Safety Switch. Stem Cell Rev. Rep. 2019, 15, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Chancellor, A.; Gadola, S.D.; Mansour, S. The versatility of the CD1 lipid antigen presentation pathway. Immunology 2018, 154, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Brigl, M.; Brenner, M.B. CD1: Antigen presentation and T cell function. Annu. Rev. Immunol. 2004, 22, 817–890. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.; Hansen-Hagge, T.E.; Drexler, H.G.; Gramatzki, M. Heterogeneity of T-acute lymphoblastic leukemia (T-ALL) cell lines: Suggestion for classification by immunophenotype and T-cell receptor studies. Leuk. Res. 1999, 23, 19–27. [Google Scholar] [CrossRef]
- Niehues, T.; Kapaun, P.; Harms, D.O.; Burdach, S.; Kramm, C.; Körholz, D.; Janka-Schaub, G.; Göbel, U. A classification based on T cell selection-related phenotypes identifies a subgroup of childhood T-ALL with favorable outcome in the COALL studies. Leukemia 1999, 13, 614–617. [Google Scholar] [CrossRef]
- Sánchez-Martínez, D.; Baroni, M.L.; Gutierrez-Agüera, F.; Roca-Ho, H.; Blanch-Lombarte, O.; González-García, S.; Torrebadell, M.; Junca, J.; Ramírez-Orellana, M.; Velasco-Hernández, T.; et al. Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood 2019, 133, 2291–2304. [Google Scholar] [CrossRef]
- Salamero, J.; Bausinger, H.; Mommaas, A.M.; Lipsker, D.; Proamer, F.; Cazenave, J.P.; Goud, B.; De La Salle, H.; Hanau, D. CD1a molecules traffic through the early recycling endosomal pathway in human Langerhans cells. J. Investig. Dermatol. 2001, 116, 401–408. [Google Scholar] [CrossRef]
- Collin, M.; Mcgovern, N.; Haniffa, M. Human dendritic cell subsets. Immunology 2013, 140, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Cernadas, M.; Lu, J.; Watts, G.; Brenner, M.B. CD1a expression defines an interleukin-12 producing population of human dendritic cells. Clin. Exp. Immunol. 2009, 155, 523–533. [Google Scholar] [CrossRef]
- Kim, J.H.; Hu, Y.; Yongqing, T.; Kim, J.; Hughes, V.A.; Le Nours, J.; Marquez, E.A.; Purcell, A.W.; Wan, Q.; Sugita, M.; et al. CD1a on Langerhans cells controls inflammatory skin disease. Nat. Immunol. 2016, 17, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.C.; Behm, F.G.F.; Singh, B.; Schell, M.J.M.; Williams, D.L.; Rivera, G.K.; Kalwinsky, D.K.; Sandlund, J.T.; Crist, W.M.; Raimondi, S.C. Heterogeneity of presenting features and their relation to treatment outcome in 120 children with T-cell acute lymphoblastic leukemia. Blood 1990, 75, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Went, P.; Agostinelli, C.; Gallamini, A.; Piccaluga, P.P.; Ascani, S.; Sabattini, E.; Bacci, F.; Falini, B.; Motta, T.; Paulli, M.; et al. Marker expression in peripheral T-cell lymphoma: A proposed clinical-pathologic prognostic score. J. Clin. Oncol. 2006, 24, 2472–2479. [Google Scholar] [CrossRef] [PubMed]
- Maciocia, P.M.; Wawrzyniecka, P.A.; Philip, B.; Ricciardelli, I.; Akarca, A.U.; Onuoha, S.C.; Leguţ, M.; Cole, D.K.; Sewell, A.K.; Gritti, G.; et al. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell malignancies. Nat. Med. 2017, 23, 1416–1423. [Google Scholar] [CrossRef]
- Takagi, S.; Fujikawa, K.; Imai, T.; Fukuhara, N.; Fukudome, K.; Minegishi, M.; Tsuchiya, S.; Konno, T.; Hinuma, Y.; Yoshie, O. Identification of a highly specific surface marker of T-cell acute lymphoblastic leukemia and neuroblastoma as a new member of the transmembrane 4 superfamily. Int. J. Cancer 1995, 61, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Orentas, R.J.; Nordlund, J.; He, J.; Sindiri, S.; Mackall, C.; Fry, T.J.; Khan, J. Bioinformatic description of immunotherapy targets for pediatric T-cell leukemia and the impact of normal gene sets used for comparison. Front. Oncol. 2014, 4, 134. [Google Scholar] [CrossRef]
- Peled, A.; Petit, I.; Kollet, O.; Magid, M.; Ponomaryov, T.; Byk, T.; Nagler, A.; Ben-Hur, H.; Many, A.; Shultz, L.; et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 1999, 283, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T.; Hirota, S.; Tachibana, K.; Takakura, N.; Nishikawa, S.I.; Kitamura, Y.; Yoshida, N.; Kikutani, H.; Kishimoto, T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996, 382, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Qing, M.; Jones, D.; Springer, T.A. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity 1999, 10, 463–471. [Google Scholar] [CrossRef]
- Goedhart, M.; Gessel, S.; van der Voort, R.; Slot, E.; Lucas, B.; Gielen, E.; Hoogenboezem, M.; Rademakers, T.; Geerman, S.; van Buul, J.D.; et al. CXCR4, but not CXCR3, drives CD8 + T-cell entry into and migration through the murine bone marrow. Eur. J. Immunol. 2019, 49, 576–589. [Google Scholar] [CrossRef]
- Estes, J.D.; Thacker, T.C.; Hampton, D.L.; Kell, S.A.; Keele, B.F.; Palenske, E.A.; Druey, K.M.; Burton, G.F. Follicular Dendritic Cell Regulation of CXCR4-Mediated Germinal Center CD4 T Cell Migration. J. Immunol. 2004, 173, 6169–6178. [Google Scholar] [CrossRef]
- Jung, S.H.; Won, K.J.; Lee, K.P.; Lee, D.H.; Yu, S.; Lee, D.Y.; Seo, E.H.; Kang, H.; Park, E.S.; Kim, H.J.; et al. DJ-1 protein regulates CD3+ T cell migration via overexpression of CXCR4 receptor. Atherosclerosis 2014, 235, 503–509. [Google Scholar] [CrossRef]
- Bryant, J.; Ahern, D.J.; Brennan, F.M. CXCR4 and vascular cell adhesion molecule 1 are key chemokine/adhesion receptors in the migration of cytokine-activated T cells. Arthritis Rheum. 2012, 64, 2137–2146. [Google Scholar] [CrossRef]
- Liu, K.K.Y.; Dorovini-Zis, K. Regulation of CXCL12 and CXCR4 expression by human brain endothelial cells and their role in CD4+ and CD8+ T cell adhesion and transendothelial migration. J. Neuroimmunol. 2009, 215, 49–64. [Google Scholar] [CrossRef]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Irigoyen, M.; Catherinet, C.; Gachet, S.; Da Costa De Jesus, C.; Lasgi, C.; Tran Quang, C.; Ghysdael, J. CXCR4 Is Required for Leukemia-Initiating Cell Activity in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2015, 27, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Jost, T.R.; Borga, C.; Radaelli, E.; Romagnani, A.; Perruzza, L.; Omodho, L.; Cazzaniga, G.; Biondi, A.; Indraccolo, S.; Thelen, M.; et al. Role of CXCR4-mediated bone marrow colonization in CNS infiltration by T cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2016, 99, 1077–1087. [Google Scholar] [CrossRef]
- Wang, W.; Zimmerman, G.; Huang, X.; Yu, S.; Myers, J.; Wang, Y.; Moreton, S.; Nthale, J.; Awadallah, A.; Beck, R.; et al. Aberrant notch signaling in the bone marrow microenvironment of acute lymphoid leukemia suppresses osteoblast-mediated support of hematopoietic niche function. Cancer Res. 2016, 76, 1641–1652. [Google Scholar] [CrossRef]
- Ferrandino, F.; Bernardini, G.; Tsaouli, G.; Grazioli, P.; Campese, A.F.; Noce, C.; Ciuffetta, A.; Vacca, A.; Besharat, Z.M.; Bellavia, D.; et al. Intrathymic Notch3 and CXCR4 combinatorial interplay facilitates T-cell leukemia propagation. Oncogene 2018, 37, 6285–6298. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef]
- Rettig, M.P.; Ansstas, G.; Dipersio, J.F. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia 2012, 26, 34–53. [Google Scholar] [CrossRef]
- Devine, S.M.; Flomenberg, N.; Vesole, D.H.; Liesveld, J.; Weisdorf, D.; Badel, K.; Calandra, G.; DiPersio, J.F. Rapid mobilization of CD34+ cells following administration of the CXCR4 antagonist AMD3100 to patients with multiple myeloma and non-Hodgkin’s lymphoma. J. Clin. Oncol. 2004, 22, 1095–1102. [Google Scholar] [CrossRef]
- Karpova, D.; Dauber, K.; Spohn, G.; Chudziak, D.; Wiercinska, E.; Schulz, M.; Pettit, A.R.; Levesque, J.P.; Romagnoli, B.; Patel, K.; et al. The novel CXCR4 antagonist POL5551 mobilizes hematopoietic stem and progenitor cells with greater efficiency than Plerixafor. Leukemia 2013, 27, 2322–2331. [Google Scholar] [CrossRef]
- Andreeff, M.; Borthakur, G.; Zeng, Z.; Kelly, M.A.; Wang, R.-Y.; McQueen, T.J.; Qiu, Y.; Mak, D.; Burger, J.A.; Daver, N.G.; et al. Mobilization and elimination of FLT3-ITD + acute myelogenous leukemia (AML) stem/progenitor cells by plerixafor, G-CSF, and sorafenib: Phase I trial results in relapsed/refractory AML patients. J. Clin. Oncol. 2014, 32, 7033. [Google Scholar] [CrossRef]
- Martínez-Cuadrón, D.; Boluda, B.; Martínez, P.; Bergua, J.; Rodríguez-Veiga, R.; Esteve, J.; Vives, S.; Serrano, J.; Vidriales, B.; Salamero, O.; et al. A phase I–II study of plerixafor in combination with fludarabine, idarubicin, cytarabine, and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann. Hematol. 2018, 97, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Andritsos, L.A.; Byrd, J.C.; Cheverton, P.; Wu, J.; Sivina, M.; Kipps, T.J.; Burger, J.A. A multicenter phase 1 study of plerixafor and rituximab in patients with chronic lymphocytic leukemia. Leuk. Lymphoma 2019, 60, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Sison, E.A.R.; Baker, S.D.; Li, L.; Ahmed, A.; Trippett, T.; Gore, L.; Macy, M.E.; Narendran, A.; August, K.; et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: A Pediatric Oncology Experimental Therapeutics Investigators’ Co. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Sison, E.A.R.; Magoon, D.; Li, L.; Annesley, C.E.; Romagnoli, B.; Douglas, G.J.; Tuffin, G.; Zimmermann, J.; Brown, P. POL5551, a novel and potent CXCR4 antagonist, enhances sensitivity to chemotherapy in pediatric ALL. Oncotarget 2015, 6, 30902–30918. [Google Scholar] [CrossRef]
- Zhang, Y.; Saavedra, E.; Tang, R.; Gu, Y.; Lappin, P.; Trajkovic, D.; Liu, S.H.; Smeal, T.; Fantin, V.; De Botton, S.; et al. Targeting primary acute myeloid leukemia with a new CXCR4 antagonist IgG1 antibody (PF-06747143). Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Kashyap, M.K.; Kumar, D.; Jones, H.; Amaya-Chanaga, C.I.; Choi, M.Y.; Melo-Cardenas, J.; Ale-Ali, A.; Kuhne, M.R.; Sabbatini, P.; Cohen, L.J.; et al. Ulocuplumab (BMS-936564/MDX1338): A fully human anti- CXCR4 antibody induces cell death in chronic lymphocytic leukemia mediated through a reactive oxygen speciesdependent pathway. Oncotarget 2016, 7, 2809–2822. [Google Scholar] [CrossRef]
- Kashyap, M.K.; Amaya-Chanaga, C.I.; Kumar, D.; Simmons, B.; Huser, N.; Gu, Y.; Hallin, M.; Lindquist, K.; Yafawi, R.; Choi, M.Y.; et al. Targeting the CXCR4 pathway using a novel anti-CXCR4 IgG1 antibody (PF-06747143) in chronic lymphocytic leukemia. J. Hematol. Oncol. 2017, 10. [Google Scholar] [CrossRef]
- Kuhne, M.R.; Mulvey, T.; Belanger, B.; Chen, S.; Pan, C.; Chong, C.; Cao, F.; Niekro, W.; Kempe, T.; Henning, K.A.; et al. BMS-936564/MDX-1338: A fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin. Cancer Res. 2013, 19, 357–366. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Liu, C.J.; Redd, R.A.; Perez, R.P.; Baz, R.; Zavidij, O.; Sklavenitis-Pistofidis, R.; Richardson, P.G.; Anderson, K.C.; Laubach, J.; et al. A phase Ib/II trial of the first-in-class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma. Clin. Cancer Res. 2020, 26, 344–353. [Google Scholar] [CrossRef]
- Peng, S.B.; Zhang, X.; Paul, D.; Kays, L.M.; Ye, M.; Vaillancourt, P.; Dowless, M.; Stancato, L.F.; Stewart, J.; Uhlik, M.T.; et al. Inhibition of CXCR4 by LY2624587, a fully humanized anti-CXCR4 antibody induces apoptosis of hematologic malignancies. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Park, L.S.; Martin, U.; Garka, K.; Gliniak, B.; Di Santo, J.P.; Muller, W.; Largaespada, D.A.; Copeland, N.G.; Jenkins, N.A.; Farr, A.G.; et al. Cloning of the murine thymic stromal lymphopoietin (TSLP) receptor: Formation of a functional heteromeric complex requires interleukin 7 receptor. J. Exp. Med. 2000, 192, 659–669. [Google Scholar] [CrossRef]
- Namen, A.E.; Lupton, S.; Hjerrild, K.; Wignall, J.; Mochizuki, D.Y.; Schmierer, A.; Mosley, B.; March, C.J.; Urdal, D.; Gillis, S.; et al. Stimulation of B-cell progenitors by cloned murine interleukin-7. Nature 1988, 333, 571–573. [Google Scholar] [CrossRef]
- Morrissey, P.J.; Goodwin, R.G.; Nordan, R.P.; Anderson, D.; Grabstein, K.H.; Cosman, D.; Sims, J.; Lupton, S.; Arces, B.; Reed, S.G.; et al. Recombinant interleukin 7, preB-cell growth factor, has costimulatory activity on purified mature T cells. J. Exp. Med. 1989, 169, 707–716. [Google Scholar] [CrossRef]
- Chazen, G.D.; Pereira, G.M.B.; LeGros, G.; Gillis, S.; Shevach, E.M. Interleukin 7 is a T-cell growth factor. Proc. Natl. Acad. Sci. USA 1989, 86, 5923–5927. [Google Scholar] [CrossRef]
- Peschon, J.J.; Morrissey, P.J.; Grabstein, K.H.; Ramsdell, F.J.; Maraskovsky, E.; Gliniak, B.C.; Park, L.S.; Ziegler, S.F.; Williams, D.E.; Ware, C.B.; et al. Early Lymphocyte Expansion Is Severely Impaired in Interleukin 7 Receptor-deficient Mice. J. Exp. Med. 1994, 180, 1955–1960. [Google Scholar] [CrossRef]
- Plum, J.; De Smedt, M.; Leclercq, G.; Verhasselt, B.; Vandekerckhove, B. Interleukin-7 is a critical growth factor in early human T-cell development. Blood 1996, 88, 4239–4245. [Google Scholar] [CrossRef]
- Maki, K.; Sunaga, S.; Komagata, Y.; Kodaira, Y.; Mabuchi, A.; Karasuyama, H.; Yokomuro, K.; Miyazaki, J.I.; Ikuta, K. Interleukin 7 receptor-deficient mice lack gammadelta T cells. Proc. Natl. Acad. Sci. USA 1996, 93, 7172–7177. [Google Scholar] [CrossRef]
- Roifman, C.M.; Zhang, J.; Chitayat, D.; Sharfe, N. A partial deficiency of interleukin-7Rα is sufficient to abrogate T-cell development and cause severe combined immunodeficiency. Blood 2000, 96, 2803–2807. [Google Scholar] [CrossRef] [PubMed]
- Vella, A.; Teague, T.K.; Ihle, J.; Kappler, J.; Marrack, P. Interleukin 4 (IL-4) or IL-7 prevents the death of resting T cells: Stat6 is probably not required for the effect of IL-4. J. Exp. Med. 1997, 186, 325–330. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Farkash, E.A.; Gao, W.; Thompson, C.B. IL-7 Enhances the Survival and Maintains the Size of Naive T Cells. J. Immunol. 2001, 167, 6869–6876. [Google Scholar] [CrossRef] [PubMed]
- Schluns, K.S.; Kieper, W.C.; Jameson, S.C.; Lefrançois, L. Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat. Immunol. 2000, 1, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Kondrack, R.M.; Harbertson, J.; Tan, J.T.; McBreen, M.E.; Surh, C.D.; Bradley, L.M. Interleukin 7 Regulates the Survival and Generation of Memory CD4 Cells. J. Exp. Med. 2003, 198, 1797–1806. [Google Scholar] [CrossRef]
- Li, J.C.; Huston, G.; Swain, S.L. IL-7 Promotes the Transition of CD4 Effectors to Persistent Memory Cells. J. Exp. Med. 2003, 198, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef]
- Seddon, B.; Tomlinson, P.; Zamoyska, R. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat. Immunol. 2003, 4, 680–686. [Google Scholar] [CrossRef]
- Robinette, M.L.; Bando, J.K.; Song, W.; Ulland, T.K.; Gilfillan, S.; Colonna, M. IL-15 sustains IL-7R-independent ILC2 and ILC3 development. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Yang, J.; Cornelissen, F.; Papazian, N.; Reijmers, R.M.; Llorian, M.; Cupedo, T.; Coles, M.; Seddon, B. IL-7-dependent maintenance of ILC3s is required for normal entry of lymphocytes into lymph nodes. J. Exp. Med. 2018, 215, 1069–1077. [Google Scholar] [CrossRef]
- Touw, I.P.; Pouwels, K.; Van Agthoven, T.; Van Gurp, R.; Budel, L.; Hoogerbrugge, H.; Delwel, R.; Goodwin, R.; Namen, A.; Lowenberg, B. Interleukin-7 is a growth factor of precursor B and T acute lymphoblastic leukemia. Blood 1990, 75, 2097–2101. [Google Scholar] [CrossRef]
- Karawajew, L.; Ruppert, V.; Wuchter, C.; Kösser, A.; Schrappe, M.; Dörken, B.; Ludwig, W.D. Inhibition of in vitro spontaneous apoptosis by IL-7 correlates, with Bcl-2 up-regulation, cortical/mature immunophenotype, and better early cytoreduction of childhood T-cell acute lymphoblastic leukemia. Blood 2000, 96, 297–306. [Google Scholar] [CrossRef]
- Barata, J.T.; Silva, A.; Nadler, L.M.; Cardoso, A.A.; Boussiotis, V.A.; Keenan, T.D. Common gamma chain-signaling cytokines promote proliferation of T-cell acute lymphoblastic leukemia. Haematologica 2004, 89, 1459–1467. [Google Scholar]
- González-García, S.; Mosquera, M.; Fuentes, P.; Palumbo, T.; Escudero, A.; Pérez-Martínez, A.; Ramírez, M.; Corcoran, A.E.; Toribio, M.L. IL-7R is essential for leukemia-initiating cell activity of T-cell acute lymphoblastic leukemia. Blood 2019, 134, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- González-García, S.; García-Peydró, M.; Martín-Gayo, E.; Ballestar, E.; Esteller, M.; Bornstein, R.; De La Pompa, J.L.; Ferrando, A.A.; Toribio, M.L. CSL-MAML-dependent Notch1 signaling controls t lineage-specifc IL-7Rα gene expression in early human thymopoiesis and leukemia. J. Exp. Med. 2009, 206, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zang, C.; Taing, L.; Arnett, K.L.; Wong, Y.J.; Pear, W.S.; Blacklow, S.C.; Liu, X.S.; Aster, J.C. NOTCH1-RBPJ complexes drive target gene expressionthrough dynamic interactions with superenhancers. Proc. Natl. Acad. Sci. USA 2014, 111, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, D.; Melão, A.; Barata, J.T. IL-7R-mediated signaling in T-cell acute lymphoblastic leukemia. Adv. Biol. Regul. 2013, 53, 211–222. [Google Scholar] [CrossRef]
- Barata, J.T.; Silva, A.; Brandao, J.G.; Nadler, L.M.; Cardoso, A.A.; Boussiotis, V.A. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J. Exp. Med. 2004, 200, 659–669. [Google Scholar] [CrossRef]
- Eder, M.; Ottmann, O.G.; Hansen-Hagge, T.E.; Bartram, C.R.; Gillis, S.; Hoelzer, D.; Ganser, A. Effects of recombinant human IL-7 on blast cell proliferation in acute lymphoblastic leukemia. Leukemia 1990, 4, 533–540. [Google Scholar]
- Cramer, S.D.; Aplan, P.D.; Durum, S.K. Therapeutic targeting of IL-7Rα signaling pathways in ALL treatment. Blood 2016, 128, 473–478. [Google Scholar] [CrossRef]
- Rich, B.E.; Campos-Torres, J.; Tepper, R.I.; Moreadith, R.W.; Leder, P. Cutaneous lymphoproliferation and lymphomas in interleukin 7 transgenic mice. J. Exp. Med. 1993, 177, 305–316. [Google Scholar] [CrossRef]
- Laouar, Y.; Crispe, I.N.; Flavell, R.A. Overexpression of IL-7Rα provides a competitive advantage during early T-cell development. Blood 2004, 103, 1985–1994. [Google Scholar] [CrossRef]
- Tremblay, C.S.; Brown, F.C.; Collett, M.; Saw, J.; Chiu, S.K.; Sonderegger, S.E.; Lucas, S.E.; Alserihi, R.; Chau, N.; Toribio, M.L.; et al. Loss-of-function mutations of Dynamin 2 promote T-ALL by enhancing IL-7 signalling. Leukemia 2016, 30, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Laranjeira, A.B.A.; Martins, L.R.; Cardoso, B.A.; Demengeot, J.; Andrés Yunes, J.; Seddon, B.; Barata, J.T. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. Cancer Res. 2011, 71, 4780–4789. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Carroll, W.L.; Meshinchi, S.; Arceci, R.J. Biology, risk stratification, and therapy of pediatric acute leukemias: An update. J. Clin. Oncol. 2011, 29, 551–565. [Google Scholar] [CrossRef]
- Shochat, C.; Tal, N.; Bandapalli, O.R.; Palmi, C.; Ganmore, I.; te Kronnie, G.; Cario, G.; Cazzaniga, G.; Kulozik, A.E.; Stanulla, M.; et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J. Exp. Med. 2011, 208, 901–908. [Google Scholar] [CrossRef]
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 2011, 43, 932–941. [Google Scholar] [CrossRef]
- Mazzucchelli, R.I.; Riva, A.; Durum, S.K. The human IL-7 receptor gene: Deletions, polymorphisms and mutations. Semin. Immunol. 2012, 24, 225–230. [Google Scholar] [CrossRef]
- Barata, J.T.; Durum, S.K.; Seddon, B. Flip the coin: IL-7 and IL-7R in health and disease. Nat. Immunol. 2019, 20, 1584–1593. [Google Scholar] [CrossRef]
- Akkapeddi, P.; Fragoso, R.; Hixon, J.A.; Ramalho, A.S.; Oliveira, M.L.; Carvalho, T.; Gloger, A.; Matasci, M.; Corzana, F.; Durum, S.K.; et al. A fully human anti-IL-7Rα antibody promotes antitumor activity against T-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 2155–2168. [Google Scholar] [CrossRef]
- Hixon, J.A.; Andrews, C.; Kashi, L.; Kohnhorst, C.L.; Senkevitch, E.; Czarra, K.; Barata, J.T.; Li, W.; Schneider, J.P.; Walsh, S.T.R.; et al. New anti-IL-7Rα monoclonal antibodies show efficacy against T cell acute lymphoblastic leukemia in pre-clinical models. Leukemia 2020, 34, 35–49. [Google Scholar] [CrossRef]
- Alsadeq, A.; Lenk, L.; Vadakumchery, A.; Cousins, A.; Vokuhl, C.; Khadour, A.; Vogiatzi, F.; Seyfried, F.; Meyer, L.H.; Cario, G.; et al. IL7R is associated with CNS infiltration and relapse in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 2018, 132, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Duś, D.; Krawczenko, A.; Załȩcki, P.; Paprocka, M.; Wiȩdłocha, A.; Goupille, C.; Kieda, C. IL-7 receptor is present on human microvascular endothelial cells. Immunol. Lett. 2003, 86, 163–168. [Google Scholar] [CrossRef]
- Iolyeva, M.; Aebischer, D.; Proulx, S.T.; Willrodt, A.H.; Ecoiffier, T.; Häner, S.; Bouchaud, G.; Krieg, C.; Onder, L.; Ludewig, B.; et al. Interleukin-7 is produced by afferent lymphatic vessels and supports lymphatic drainage. Blood 2013, 122, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Ziegler, S.F.; Buckley, R.H.; Leonard, W.J. Defective IL7R expression in T-B+NK+ severe combined immunodeficiency. Nat. Genet. 1998, 20, 394–397. [Google Scholar] [CrossRef]
- Yasunaga, M.; Manabe, S.; Matsumura, Y. Immunoregulation by IL-7R-targeting antibody-drug conjugates: Overcoming steroid-resistance in cancer and autoimmune disease. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Ellis, J.; van Maurik, A.; Fortunato, L.; Gisbert, S.; Chen, K.; Schwartz, A.; McHugh, S.; Want, A.; Santos Franco, S.; Oliveira, J.J.; et al. Anti-IL-7 receptor α monoclonal antibody (GSK2618960) in healthy subjects—A randomized, double-blind, placebo-controlled study. Br. J. Clin. Pharmacol. 2019, 85, 304–315. [Google Scholar] [CrossRef]
- Aruffo, A.; Stamenkovic, I.; Melnick, M.; Underhill, C.B.; Seed, B. CD44 is the principal cell surface receptor for hyaluronate. Cell 1990, 61, 1303–1313. [Google Scholar] [CrossRef]
- Liao, H.X.; Lee, D.M.; Levesque, M.C.; Haynes, B.F. N-terminal and central regions of the human CD44 extracellular domain participate in cell surface hyaluronan binding. J. Immunol. 1995, 155, 3938–3945. [Google Scholar]
- Stamenkovic, I.; Amiot, M.; Pesando, J.M.; Seed, B. A lymphocyte molecule implicated in lymph node homing is a member of the cartilage link protein family. Cell 1989, 56, 1057–1062. [Google Scholar] [CrossRef]
- Goldstein, L.A.; Zhou, D.F.H.; Picker, L.J.; Minty, C.N.; Bargatze, R.F.; Ding, J.F.; Butcher, E.C. A human lymphocyte homing receptor, the Hermes antigen, is related to cartilage proteoglycan core and link proteins. Cell 1989, 56, 1063–1072. [Google Scholar] [CrossRef]
- Idzerda, R.L.; Carter, W.G.; Nottenburg, C.; Wayner, E.A.; Gallatin, W.M.; John, T.S. Isolation and DNA sequence of a cDNA clone encoding a lymphocyte adhesion receptor for high endothelium. Proc. Natl. Acad. Sci. USA 1989, 86, 4659–4663. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.; Schwärzler, C.; Günthert, U. CD44 isoforms during differentiation and development. BioEssays 1995, 17, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Horst, E.; Heider, K.H.; Wielenga, V.J.M.; Adolf, G.R.; Herrlich, P.; Pals, S.T. Expression of CD44 splice variants during lymphocyte activation and tumor progression. Cell Commun. Adhes. 1994, 2, 195–200. [Google Scholar] [CrossRef]
- Zöller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef]
- Tjhay, F.; Motohara, T.; Tayama, S.; Narantuya, D.; Fujimoto, K.; Guo, J.; Sakaguchi, I.; Honda, R.; Tashiro, H.; Katabuchi, H. CD44 variant 6 is correlated with peritoneal dissemination and poor prognosis in patients with advanced epithelial ovarian cancer. Cancer Sci. 2015, 106, 1421–1428. [Google Scholar] [CrossRef]
- Fang, M.; Wu, J.; Lai, X.; Ai, H.; Tao, Y.; Zhu, B.; Huang, L. CD44 and CD44v6 are Correlated with Gastric Cancer Progression and Poor Patient Prognosis: Evidence from 42 Studies. Cell. Physiol. Biochem. 2016, 40, 567–578. [Google Scholar] [CrossRef]
- Avigdor, A.; Goichberg, P.; Shivtiel, S.; Dar, A.; Peled, A.; Samira, S.; Kollet, O.; Hershkoviz, R.; Alon, R.; Hardan, I.; et al. CD44 and hyaluronic acid cooperate with SDF-1 in the trafficking of human CD34+ stem/progenitor cells to bone marrow. Blood 2004, 103, 2981–2989. [Google Scholar] [CrossRef]
- Lee-Sayer, S.S.M.; Dougan, M.N.; Cooper, J.; Sanderson, L.; Dosanjh, M.; Maxwell, C.A.; Johnson, P. CD44-mediated hyaluronan binding marks proliferating hematopoietic progenitor cells and promotes bone marrow engraftment. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Krause, D.S.; Lazarides, K.; Von Andrian, U.H.; Van Etten, R.A. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat. Med. 2006, 12, 1175–1180. [Google Scholar] [CrossRef]
- Márquez, C.; Trigueros, C.; FerńIndez, E.; Luisa Toribio, M. The development of T and non-T cell lineages from cd34+ human thymic precursors can be traced by the differential expression of cd44. J. Exp. Med. 1995, 181, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Canté-Barrett, K.; Mendes, R.D.; Li, Y.; Vroegindeweij, E.; Pike-Overzet, K.; Wabeke, T.; Langerak, A.W.; Pieters, R.; Staal, F.J.T.; Meijerink, J.P.P. Loss of CD44dim expression from early progenitor cells marks T-cell lineage commitment in the human thymus. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Márquez, C.; Trigueros, C.; Franco, J.M.; Ramiro, A.R.; Carrasco, Y.R.; López-Botet, M.; Toribio, M.L. Identification of a common developmental pathway for thymic natural killer cells and dendritic cells. Blood 1998, 91, 2760–2771. [Google Scholar] [CrossRef]
- García-Peydró, M.; Fuentes, P.; Mosquera, M.; García-León, M.J.; Alcain, J.; Rodríguez, A.; De Miguel, P.G.; Menéndez, P.; Weijer, K.; Spits, H.; et al. The NOTCH1/CD44 axis drives pathogenesis in a T cell acute lymphoblastic leukemia model. J. Clin. Investig. 2018, 128, 2802–2818. [Google Scholar] [CrossRef]
- Marques, L.V.C.; Noronha, E.P.; Andrade, F.G.; Dos Santos-Bueno, F.V.; Mansur, M.B.; Terra-Granado, E.; Pombo-De-Oliveira, M.S. CD44 expression profile varies according to maturational subtypes and molecular profiles of pediatric T-cell lymphoblastic leukemia. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Giambra, V.; Jenkins, C.R.; Wang, H.; Lam, S.H.; Shevchuk, O.O.; Nemirovsky, O.; Wai, C.; Gusscott, S.; Chiang, M.Y.; Aster, J.C.; et al. NOTCH1 promotes T cell leukemia-initiating activity by RUNX-mediated regulation of PKC-θ and reactive oxygen species. Nat. Med. 2012, 18, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Hoofd, C.; Wang, X.; Lam, S.; Jenkins, C.; Wood, B.; Giambra, V.; Weng, A.P. CD44 promotes chemoresistance in T-ALL by increased drug efflux. Exp. Hematol. 2016, 44, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Vey, N.; Delaunay, J.; Martinelli, G.; Fiedler, W.; Raffoux, E.; Prebet, T.; Gomez-Roca, C.; Papayannidis, C.; Kebenko, M.; Paschka, P.; et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 2016, 7, 32532–32542. [Google Scholar] [CrossRef]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef]
- Charrad, R.S.; Li, Y.; Delpech, B.; Balitrand, N.; Clay, D.; Jasmin, C.; Chomienne, C.; Smadja-Joffe, F. Ligation of the CD44 adhesion molecule reverses blockage of differentiation in human acute myeloid leukemia. Nat. Med. 1999, 5, 669–676. [Google Scholar] [CrossRef]
- Charrad, R.S.; Gadhoum, Z.; Qi, J.; Glachant, A.; Allouche, M.; Jasmin, C.; Chomienne, C.; Smadja-Joffe, F. Effects of anti-CD44 monoclonal antibodies on differentiation and apoptosis of human myeloid leukemia cell lines. Blood 2002, 99, 290–299. [Google Scholar] [CrossRef]
- Gadhoum, Z.; Leibovitch, M.P.; Oi, J.; Dumenil, D.; Durand, L.; Leibovitch, S.; Smadja-Joffe, F. CD44: A new means to inhibit acute myeloid leukemia cell proliferation via p27Kip1. Blood 2004, 103, 1059–1068. [Google Scholar] [CrossRef]
- Tijink, B.M.; Buter, J.; De Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; Van Dongen, G.A.M.S. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 2006, 12, 6064–6072. [Google Scholar] [CrossRef] [PubMed]
- Riechelmann, H.; Sauter, A.; Golze, W.; Hanft, G.; Schroen, C.; Hoermann, K.; Erhardt, T.; Gronau, S. Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral Oncol. 2008, 44, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Rupp, U.; Schoendorf-Holland, E.; Eichbaum, M.; Schuetz, F.; Lauschner, I.; Schmidt, P.; Staab, A.; Hanft, G.; Huober, J.; Sinn, H.P.; et al. Safety and pharmacokinetics of bivatuzumab mertansine in patients with CD44v6-positive metastatic breast cancer: Final results of a phase I study. Anticancer Drugs 2007, 18, 477–485. [Google Scholar] [CrossRef]
- Dürkop, H.; Latza, U.; Hummel, M.; Eitelbach, F.; Seed, B.; Stein, H. Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin’s disease. Cell 1992, 68, 421–427. [Google Scholar] [CrossRef]
- Nikolaenko, L.; Zain, J.; Rosen, S.T.; Querfeld, C. CD30-positive lymphoproliferative disorders. Cancer Treat. Res. 2019, 176, 249–268. [Google Scholar] [CrossRef]
- Falini, B.; Pileri, S.; Pizzolo, G.; Dürkop, H.; Flenghi, L.; Stirpe, F.; Martelli, M.F.; Stein, H. CD30 (Ki-1) molecule: A new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood 1995, 85, 1–14. [Google Scholar] [CrossRef]
- Andreesen, R.; Osterholz, J.; Lohr, G.W.; Bross, K.J. A Hodgkin cell-specific antigen is expressed on a subset of auto- and alloactivated T (helper) lymphoblasts. Blood 1984, 63, 1299–1302. [Google Scholar] [CrossRef]
- Del Prete, G.; De Carli, M.; Almerigogna, F.; Daniel, G.K.; Delios, M.M.; Zancuoghi, G.; Vinante, F.; Pizzolo, G.; Romagnani, S. Preferential expression of CD30 by human CD4 + T cells producing Th2-type cytokines. FASEB J. 1995, 9, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, B.; Reddish, M.; Longenecker, B.M. CD30 expression on human CD8+ T cells isolated from peripheral blood lymphocytes of normal donors. J. Immunol. 1996, 157, 3229–3234. [Google Scholar]
- Sperling, S.; Fiedler, P.; Lechner, M.; Pollithy, A.; Ehrenberg, S.; Schiefer, A.I.; Kenner, L.; Feuchtinger, A.; Kühn, R.; Swinerd, G.; et al. Chronic CD30 signaling in B cells results in lymphomagenesis by driving the expansion of plasmablasts and B1 cells. Blood 2019, 133, 2597–2609. [Google Scholar] [CrossRef]
- Hombach, A.A.; Görgens, A.; Chmielewski, M.; Murke, F.; Kimpel, J.; Giebel, B.; Abken, H. Superior Therapeutic Index in Lymphoma Therapy: CD30+ CD34+ Hematopoietic Stem Cells Resist a Chimeric Antigen Receptor T-cell Attack. Mol. Ther. 2016, 24, 1423–1434. [Google Scholar] [CrossRef]
- Savoldo, B.; Rooney, C.M.; Di Stasi, A.; Abken, H.; Hombach, A.; Foster, A.E.; Zhang, L.; Heslop, H.E.; Brenner, M.K.; Dotti, G. Epstein Barr virus-specific cytotoxic T lymphocytes expressing the anti-CD30ζ artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood 2007, 110, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Ono, K.; Nohgawa, M. Successful treatment with brentuximab vedotin for relapsed and refractory adult T cell leukemia. Anticancer Drugs 2020, 31, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Bartlett, N.L.; Forero-Torres, A.; Younes, A.; Chen, R.; Friedberg, J.W.; Matous, J.V.; Shustov, A.R.; Smith, S.E.; Zain, J.; et al. Brentuximab vedotin in patients aged 60 years or older with relapsed or refractory CD30-positive lymphomas: A retrospective evaluation of safety and efficacy. Leuk. Lymphoma 2014, 55, 2328–2334. [Google Scholar] [CrossRef]
- Wang, C.M.; Wu, Z.Q.; Wang, Y.; Guo, Y.L.; Dai, H.R.; Wang, X.H.; Li, X.; Zhang, Y.J.; Zhang, W.Y.; Chen, M.X.; et al. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory hodgkin lymphoma: An open-label phase i trial. Clin. Cancer Res. 2017, 23, 1156–1166. [Google Scholar] [CrossRef]
- Ramos, C.A.; Ballard, B.; Zhang, H.; Dakhova, O.; Gee, A.P.; Mei, Z.; Bilgi, M.; Wu, M.F.; Liu, H.; Grilley, B.; et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J. Clin. Investig. 2017, 127, 3462–3471. [Google Scholar] [CrossRef]
- Zheng, W.; Medeiros, L.J.; Young, K.H.; Goswami, M.; Powers, L.; Kantarjian, H.H.; Thomas, D.A.; Cortes, J.E.; Wang, S.A. CD30 expression in acute lymphoblastic leukemia as assessed by flow cytometry analysis. Leuk. Lymphoma 2014, 55, 624–627. [Google Scholar] [CrossRef][Green Version]
- Hahn, J.H.; Kim, M.K.; Choi, E.Y.; Kim, S.H.; Sohn, H.W.; Ham, D.I.; Chung, D.H.; Kim, T.J.; Lee, W.J.; Park, C.K.; et al. CD99 (MIC2) regulates the LFA-1/ICAM-1-mediated adhesion of lymphocytes, and its gene encodes both positive and negative regulators of cellular adhesion. J. Immunol. 1997, 159, 2250–2258. [Google Scholar] [PubMed]
- Kwon, I.O.; Byoung, K.K.; Young, L.B.; Eun, Y.C.; Kyeong, C.J.; Lee, I.S.; Seong, H.P. CD99 activates T cells via a costimulatory function that promotes raft association of TCR complex and tyrosine phosphorylation of TCR ζ. Exp. Mol. Med. 2007, 39, 176–184. [Google Scholar] [CrossRef]
- Watson, R.L.; Buck, J.; Levin, L.R.; Winger, R.C.; Wang, J.; Arase, H.; Muller, W.A. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. J. Exp. Med. 2015, 212, 1021–1041. [Google Scholar] [CrossRef]
- Vaikari, V.P.; Du, Y.; Wu, S.; Zhang, T.; Metzeler, K.; Batcha, A.M.N.; Herold, T.; Hiddemann, W.; Akhtari, M.; Alachkar, H. Clinical and preclinical characterization of CD99 isoforms in acute myeloid leukemia. Haematologica 2020, 105, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, R.D.; Bernard, G.; Olafsen, M.K.; Pourtein, M.; Lie, S.O. CD99 Signals Caspase-Independent T Cell Death. J. Immunol. 2001, 166, 4931–4942. [Google Scholar] [CrossRef]
- Husak, Z.; Printz, D.; Schumich, A.; Potschger, U.; Dworzak, M.N. Death induction by CD99 ligation in TEL/AML1-positive acute lymphoblastic leukemia and normal B cell precursors. J. Leukoc. Biol. 2010, 88, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Dworzak, M.; Fröschl, G.; Printz, D.; De Zen, L.; Gaipa, G.; Ratei, R.; Basso, G.; Biondi, A.; Ludwig, W.D.; Gadner, H. CD99 expression in T-lineage ALL: Implications for flow cytometric detection of minimal residual disease. Leukemia 2004, 18, 703–708. [Google Scholar] [CrossRef]
- Cox, C.V.; Diamanti, P.; Moppett, J.P.; Blair, A. Investigating CD99 expression in leukemia propagating cells in childhood t cell acute lymphoblastic leukemia. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Enein, A.A.A.; Rahman, H.A.A.; El Sharkawy, N.; Abd Elhamid, S.M.; Abbas, S.M.A.; Abdelfaatah, R.; Khalil, M.; Fathalla, L.A. Significance of CD99 expression in T-lineage acute lymphoblastic leukemia. Cancer Biomark. 2016, 17, 117–123. [Google Scholar] [CrossRef]
- Husak, Z.; Dworzak, M.N. CD99 ligation upregulates HSP70 on acute lymphoblastic leukemia cells and concomitantly increases NK cytotoxicity. Cell Death Dis. 2012, 3. [Google Scholar] [CrossRef][Green Version]




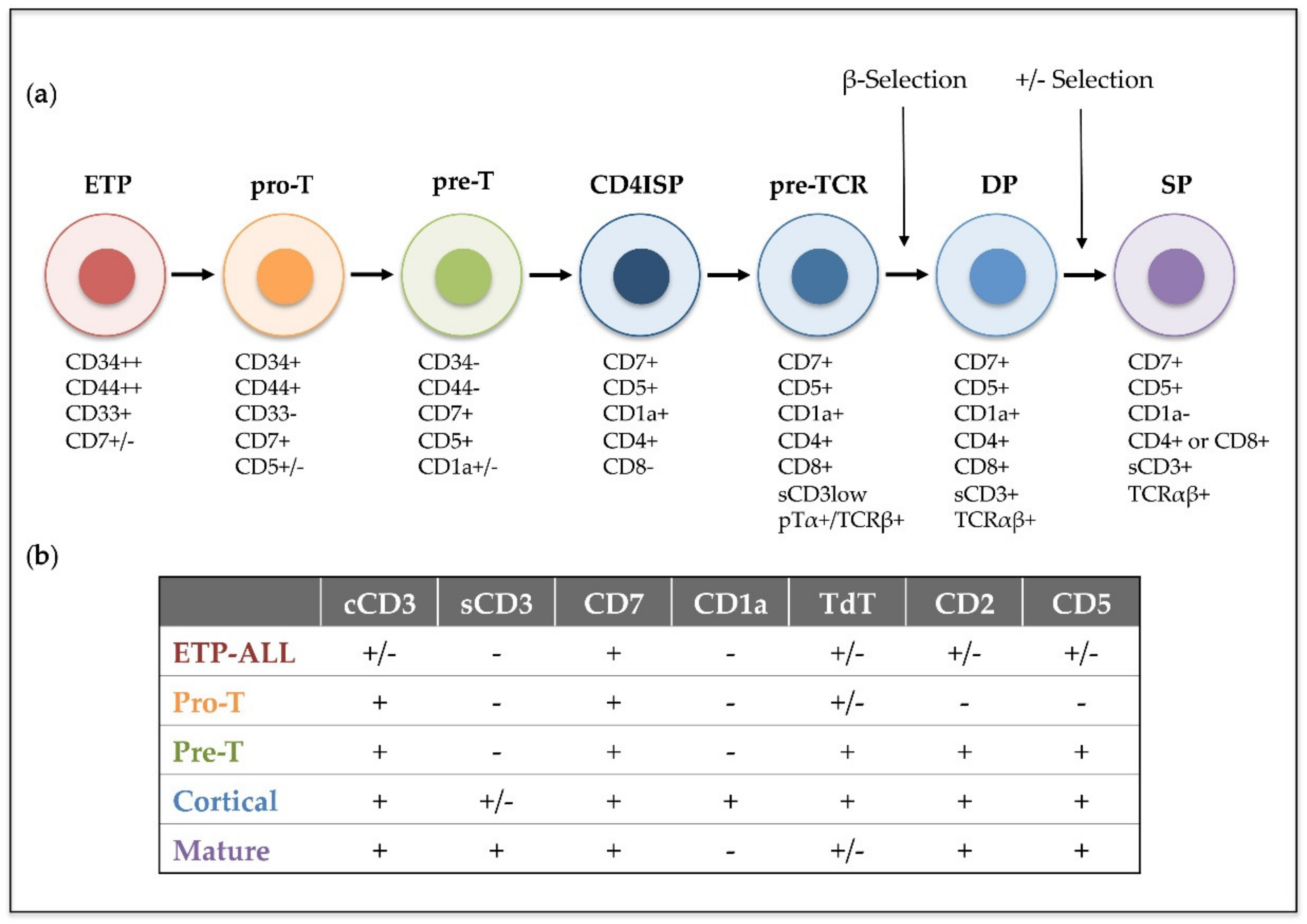{kind=link}
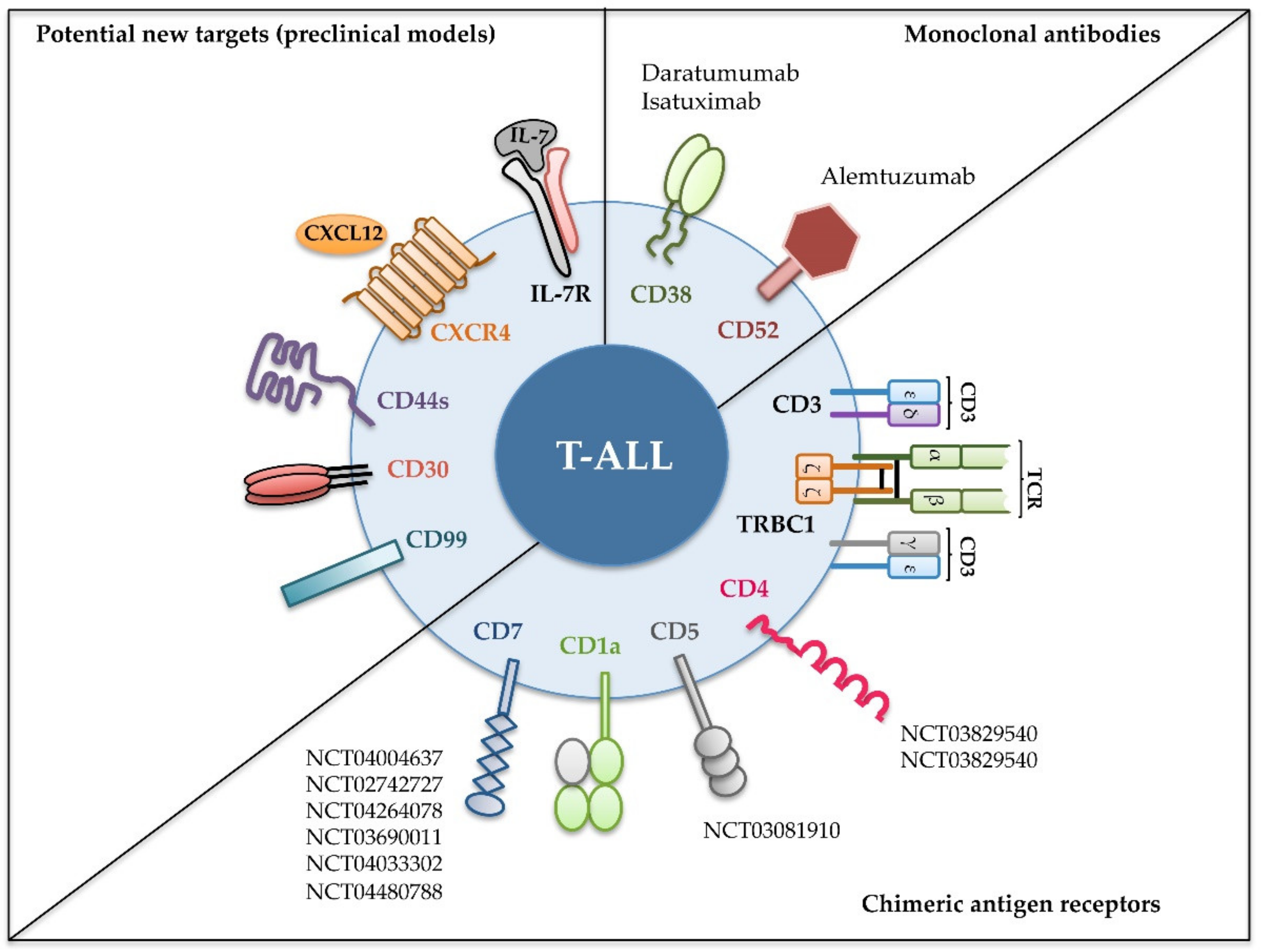{kind=link}
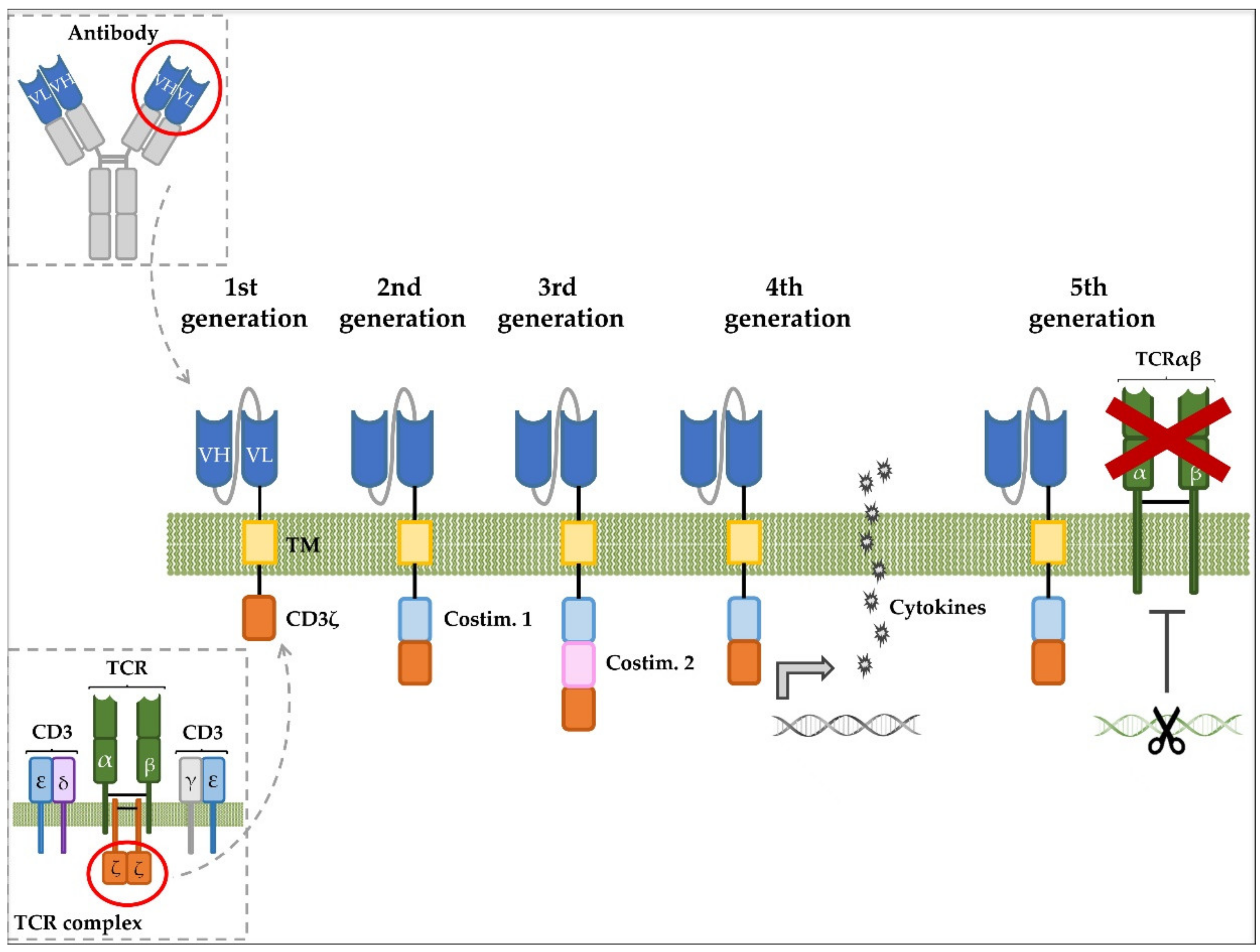{kind=link}
| Monoclonal Antibodies | |||||
| Identifier | Disease | Intervention | Age of Patients | Phase | Status * |
| NCT02999633 | T-ALL/T-LBL | Isatuximab (anti-CD38) | 16 yr and older | Phase II | Terminated |
| NCT00199030 | T-ALL/T-LBL | Alemtuzumab (anti-CD52) | 18 yr and older | Phase II | Completed |
| NCT00061048 | Adult T-cell leukemia | Alemtuzumab (anti-CD52) | 18 yr and older | Phase II | Completed |
| NCT03384654 | T-ALL/B-ALL | Daratumumab (anti-CD38) | 11 to 30 yr | Phase II | Recruiting |
| NCT00061945 | B-/T-precursor lymphoblastic leukemia | Alemtuzumab (anti-CD52) | 15 yr and older | Phase I/II | Completed |
| NCT03860844 | T-ALL/B-ALL/AML | Isatuximab (anti-CD38) | Up to 17 yr | Phase II | Recruiting |
| Chimeric Antigen Receptors | |||||
| Identifier | Disease | Intervention | Age of patients | Phase | Status * |
| NCT03829540 | CD4 + T-cell leukemia | CD4 CAR T cells | 18 yr and older | Phase I | Recruiting |
| NCT04162340 | CD4 + T-cell malignancies | CD4 CAR T cells | 18 yr and older | Phase I | Recruiting |
| NCT03081910 | CD5 + T-cell malignancies | CD5 CAR T cells | Up to 75 yr | Phase I | Recruiting |
| NCT04004637 | CD7 + NK/T lymphoma; T-LBL; T-ALL | CD7 CAR T cells | 7 to 70 yr | Phase I | Recruiting |
| NCT02742727 | CD7 + r/r leukemia or lymphoma | CD7 NK cells | 18 yr and older | Phase I/II | Unknown |
| NCT04264078 | CD7 + NK or T-cell malignancies | Universal CD7 CAR T cells | 2 to 70 yr | Early Phase I | Not yet recruiting |
| NCT03690011 | CD7 + T-cell leukemia/lymphoma | CD7 KO-CD7 CAR T cells | Up to 75 yr | Phase I | Not yet recruiting |
| NCT04033302 | CD7 + hematological malignancies | CD7 CAR T cells | 6 mo to 75 yr | Phase I/II | Recruiting |
| NCT04480788 | CD7 + r/r hematological malignancies | CD7 CAR T cells | 7 to 70 yr | Phase I | Not yet recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayón-Calderón, F.; Toribio, M.L.; González-García, S. Facts and Challenges in Immunotherapy for T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 7685. https://doi.org/10.3390/ijms21207685
Bayón-Calderón F, Toribio ML, González-García S. Facts and Challenges in Immunotherapy for T-Cell Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2020; 21(20):7685. https://doi.org/10.3390/ijms21207685
Chicago/Turabian StyleBayón-Calderón, Fátima, María L. Toribio, and Sara González-García. 2020. "Facts and Challenges in Immunotherapy for T-Cell Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 21, no. 20: 7685. https://doi.org/10.3390/ijms21207685
APA StyleBayón-Calderón, F., Toribio, M. L., & González-García, S. (2020). Facts and Challenges in Immunotherapy for T-Cell Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences, 21(20), 7685. https://doi.org/10.3390/ijms21207685