Sodium-Glucose Co-Transporter 2 Inhibitors Correct Metabolic Maladaptation of Proximal Tubular Epithelial Cells in High-Glucose Conditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
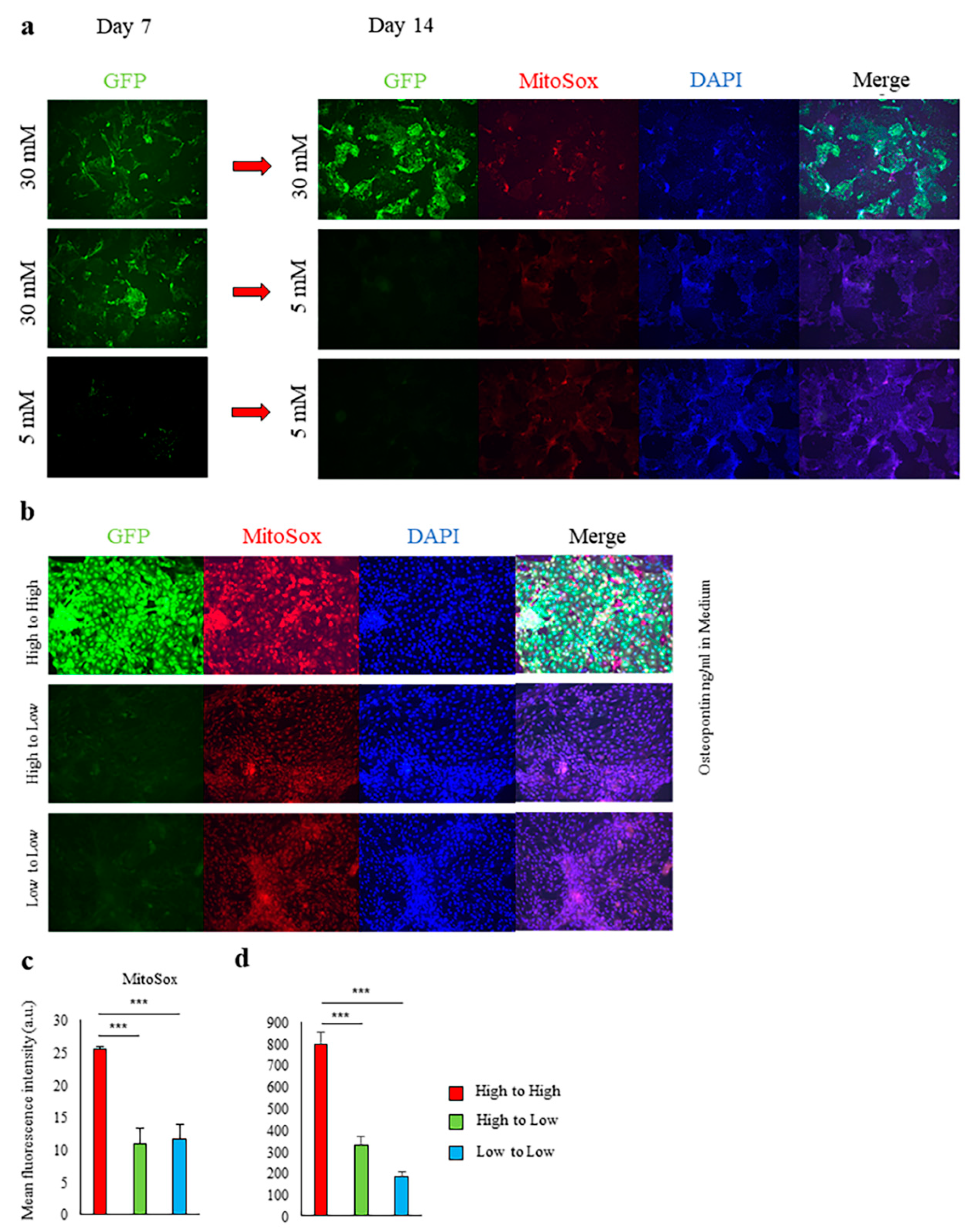
2.1. High-Glucose Conditions Induce Spp1 Transcriptional Activity in PTEC
2.2. Induction of Spp1 Transcriptional Activity in PTEC by High Glucose Was Reversible
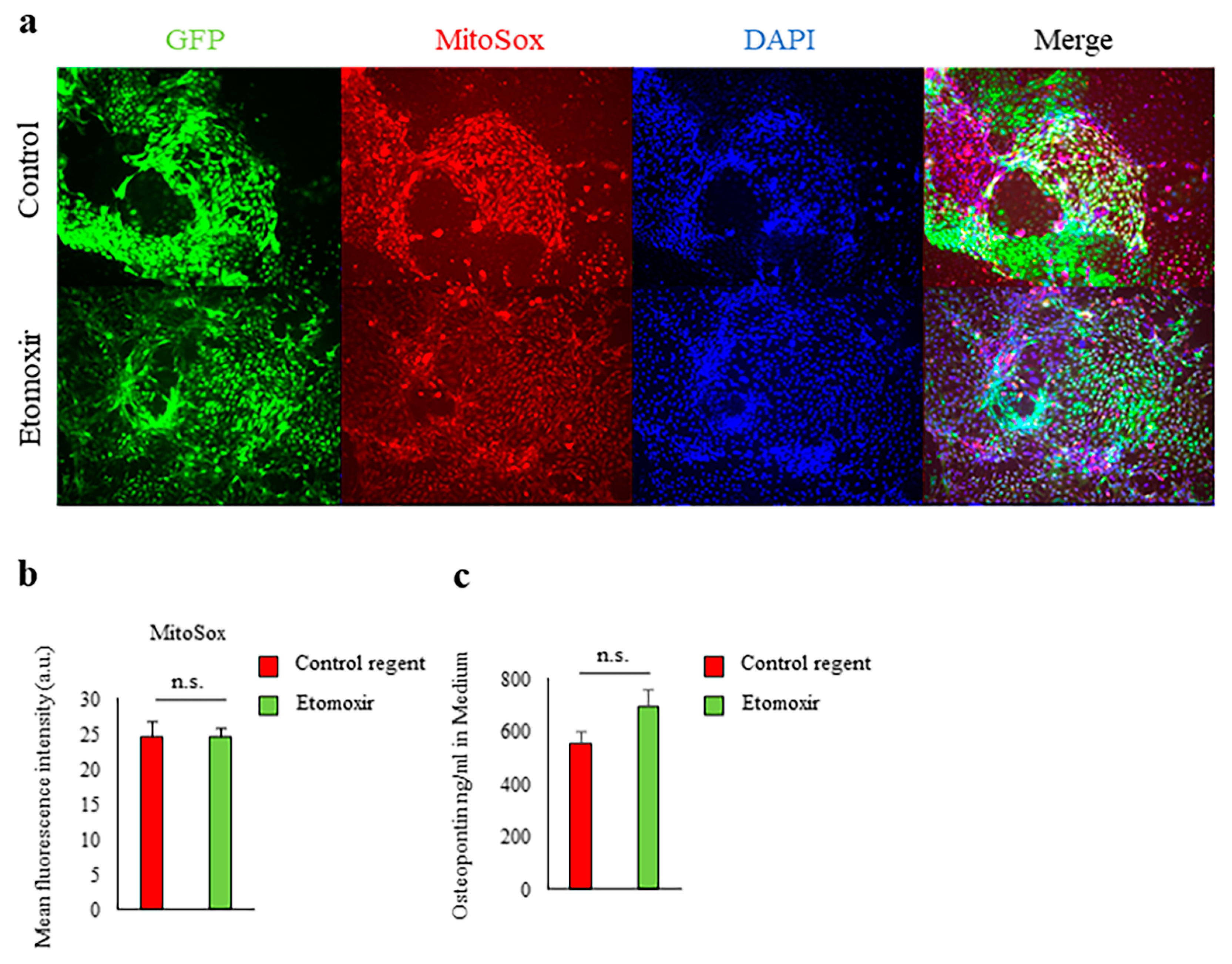
2.3. Fatty Acid Beta-Oxidation Was Not Involved in Spp1 Transcriptional Activation in High-Glucose Conditions
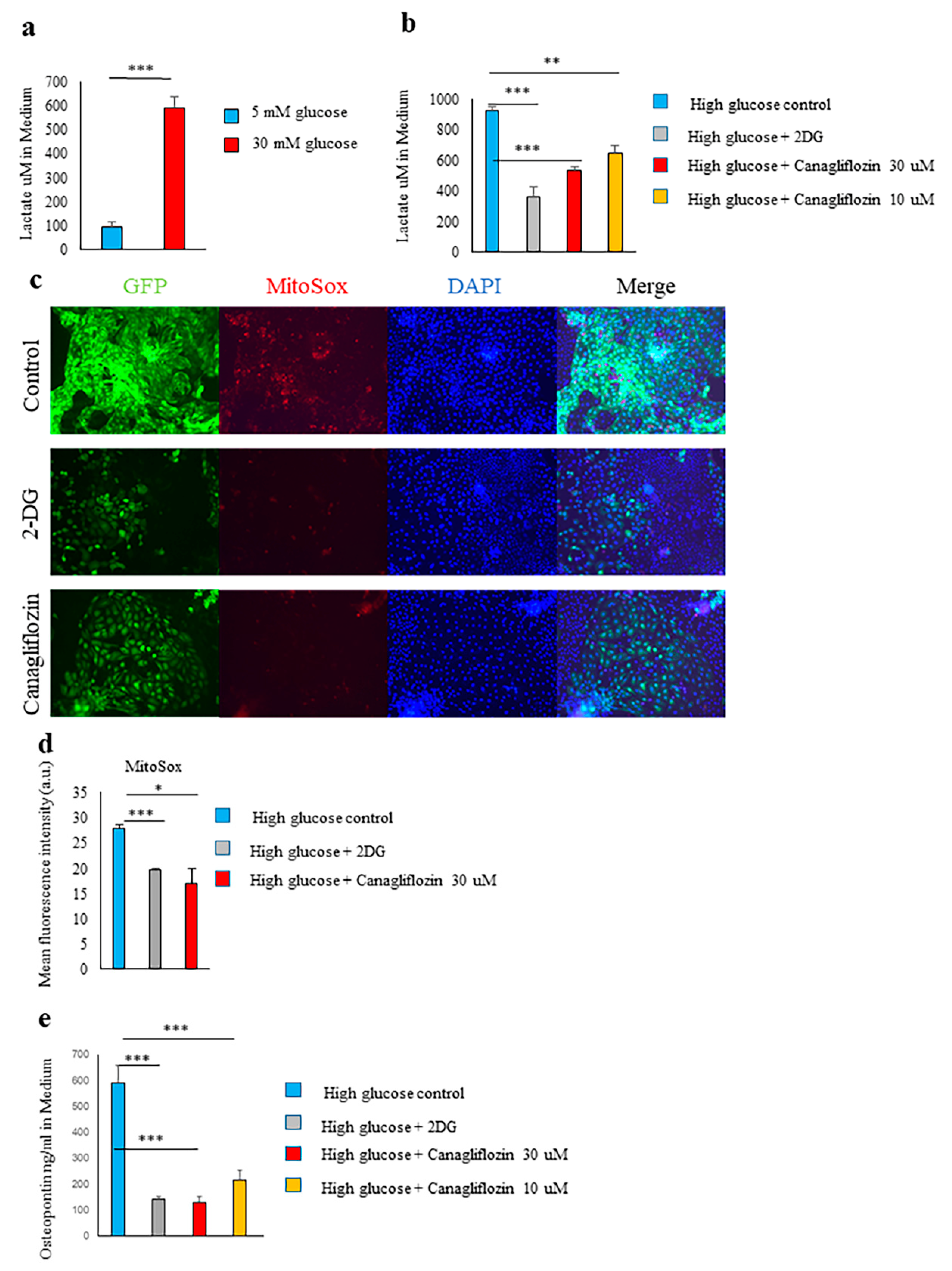
2.4. Glucose Transporter and SGLT2-Mediated Glucose Influx and Activation of the Glycolytic Pathway Were Involved in Spp1 Transcriptional Activation in the High-Glucose Conditions
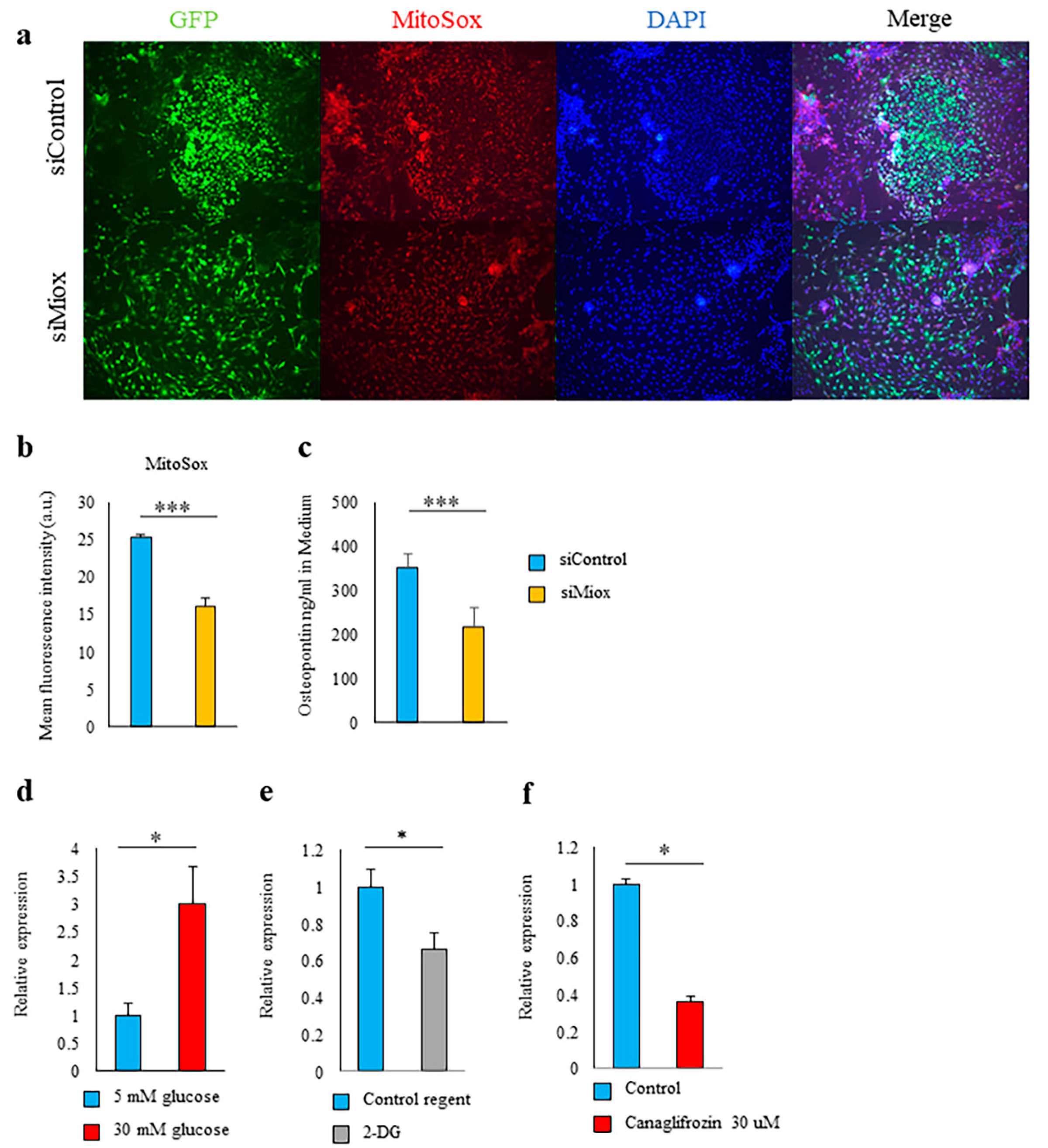
2.5. Canagliflozin and 2-DG Inhibited High Glucose-Induced MIOX Upregulation
3. Discussion
4. Materials and Methods
4.1. Animal Care
4.2. Proximal Tubule Isolation and Primary Culture
4.3. Primary Culture of PTEC with Varying Concentrations of Glucose in the Culture Medium
4.4. Genetic Knock-Down and Expression of PTEC
4.5. Detection of ROS in Primary Culture of PTEC
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Lactate Assay
4.8. Quantitative Real-Time Polymerase Chain Reaction
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| EGFP | Enhanced green fluorescent protein |
| DKD | Diabetic kidney disease |
| GLUT2 | Glucose transporter 2 |
| Miox | Myo-inositol-degrading enzyme myo-inositol oxygenase |
| OPN | Osteopontin |
| PTEC | Proximal tubular epithelial cells |
| SGLT2 | Sodium-glucose cotransporter 2 |
| siRNA | Small interfering RNA |
| ROS | Reactive oxygen species |
References
- Gilbert, R.E. Proximal tubulopathy: Prime mover and key therapeutic target in diabetic kidney disease. Diabetes 2017, 66, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in Type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in Type 2 diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and cardiovascular and renal events in Type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; Fulcher, G.; Erondu, N.; Shaw, W.; Barrett, T.D.; Weidner-Wells, M.; Deng, H.; Matthews, D.R.; et al. Canagliflozin and renal outcomes in type 2 diabetes: Results from the CANVAS Program randomised clinical trials. Lancet Diabetes Endocrinol. 2018, 6, 691–704. [Google Scholar] [CrossRef]
- Mahaffey, K.W.; Jardine, M.J.; Bompoint, S.; Cannon, C.P.; Neal, B.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; et al. Canagliflozin and cardiovascular and renal outcomes in Type 2 diabetes mellitus and chronic kidney disease in primary and secondary cardiovascular prevention groups. Circulation 2019, 140, 739–750. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; DeMets, D.L.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Langkilde, A.M.; Martinez, F.A.; Bengtsson, O.; Ponikowski, P.; Sabatine, M.S.; et al. The dapagliflozin and prevention of adverse-outcomes in heart failure (DAPA-HF) trial: Baseline characteristics. Eur. J. Heart Fail. 2019, 21, 1402–1411. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Neuen, B.L.; Young, T.; Heerspink, H.J.L.; Neal, B.; Perkovic, V.; Billot, L.; Mahaffey, K.W.; Charytan, D.M.; Wheeler, D.C.; Arnott, C.; et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019, 7, 845–854. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and renal outcomes in Type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Heerspink, H.J.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.; McMurray, J.J.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M.; Loo, D.D.F.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Distribution of glucose transporters in renal diseases. J. Biomed. Sci. 2017, 24, 64. [Google Scholar] [CrossRef]
- Hansell, P.; Welch, W.J.; Blantz, R.C.; Palm, F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 2013, 40, 123–137. [Google Scholar] [CrossRef]
- O’Neill, J.; Fasching, A.; Pihl, L.; Patinha, D.; Franzén, S.; Palm, F. Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol. Physiol. 2015, 309, F227–F234. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef]
- Sas, K.M.; Kayampilly, P.; Byun, J.; Nair, V.; Hinder, L.M.; Hur, J.; Zhang, H.; Lin, C.; Qi, N.R.; Michailidis, G.; et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016, 1, e86976. [Google Scholar] [CrossRef]
- Chang, H.-H.G.; Chao, H.-N.; Walker, C.S.; Choong, S.Y.; Phillips, A.R.J.; Loomes, K.M. Renal depletion of myo-inositol is associated with its increased degradation in animal models of metabolic disease. Am. J. Physiol. Physiol. 2015, 309, F755–F763. [Google Scholar] [CrossRef]
- Nayak, B.; Kondeti, V.K.; Xie, P.; Lin, S.; Viswakarma, N.; Raparia, K.; Kanwar, Y.S. Transcriptional and post-translational modulation of myo-inositol oxygenase by high glucose and related pathobiological stresses. J. Biol. Chem. 2011, 286, 27594–27611. [Google Scholar] [CrossRef] [PubMed]
- Nayak, B.; Xie, P.; Akagi, S.; Yang, Q.; Sun, L.; Wada, J.; Thakur, A.; Danesh, F.R.; Chugh, S.S.; Kanwar, Y.S. Modulation of renal-specific oxidoreductase/myo-inositol oxygenase by high-glucose ambience. Proc. Natl. Acad. Sci. USA 2005, 102, 17952–17957. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304–1321. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yoshida, N.; Yamamoto, T.; Isobe, S.; Moriyama, H.; Goto, S.; Kitakata, H.; et al. IL (Interleukin)-10-STAT3-Galectin-3 Axis is essential for osteopontin-producing reparative macrophage polarization after myocardial infarction. Circulation 2018, 138, 2021–2035. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Anzai, A.; Moriyama, H.; Kitakata, H.; Hiraide, T.; Ko, S.; Goto, S.; et al. MerTK Expression and ERK activation are essential for the functional maturation of osteopontin-producing reparative macrophages after myocardial infarction. J. Am. Heart Assoc. 2020, e017071. [Google Scholar] [CrossRef]
- Xie, Y.; Sakatsume, M.; Nishi, S.; Narita, I.; Arakawa, M.; Gejyo, F. Expression, roles, receptors, and regulation of osteopontin in the kidney. Kidney Int. 2001, 60, 1645–1657. [Google Scholar] [CrossRef]
- Liu, J.; Kumar, S.; Dolzhenko, E.; Alvarado, G.F.; Guo, J.; Lu, C.; Chen, Y.; Li, M.; Dessing, M.C.; Parvez, R.K.; et al. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Zhang, Z.-X.; Shek, K.; Wang, S.; Huang, X.; Lau, A.; Yin, Z.; Liu, W.; Garcia, B.; Rittling, S.; Jevnikar, A.M. Osteopontin expressed in tubular epithelial cells regulates NK cell-mediated kidney ischemia reperfusion injury. J. Immunol. 2010, 185, 967–973. [Google Scholar] [CrossRef]
- Susztak, K.; Böttinger, E.; Novetsky, A.P.; Liang, D.; Zhu, Y.; Ciccone, E.; Wu, D.; Dunn, S.; McCue, P.; Sharma, K.; et al. Molecular profiling of diabetic mouse kidney reveals novel genes linked to glomerular disease. Diabetes 2004, 53, 784–794. [Google Scholar] [CrossRef][Green Version]
- Marks, J.; Carvou, N.J.C.; Debnam, E.S.; Srai, S.K.; Unwin, R.J. Diabetes increases facilitative glucose uptake and GLUT2 expression at the rat proximal tubule brush border membrane. J. Physiol. 2003, 553, 137–145. [Google Scholar] [CrossRef]
- Aft, R.L.; Zhang, F.W.; Gius, D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: Mechanism of cell death. Br. J. Cancer. 2002, 87, 805–812. [Google Scholar] [CrossRef]
- Sharma, I.; Deng, F.; Liao, Y.; Kanwar, Y.S. Myo-inositol oxygenase (MIOX) overexpression drives the progression of renal tubulointerstitial injury in diabetes. Diabetes 2020, 69, 1248–1263. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Johnson, E.J.; Tuttle, K.R. SGLT2 Inhibition for the prevention and treatment of diabetic kidney disease: A review. Am. J. Kidney Dis. 2018, 72, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Goto, S. Possible mechanism of hematocrit elevation by sodium glucose cotransporter 2 inhibitors and associated beneficial renal and cardiovascular effects. Circulation 2019, 139, 1985–1987. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-K.; Xin, K.-Y.; Ge, H.-W.; Kong, F.-J.; Zhao, G. Upregulation of Renal GLUT2 and SGLT2 is involved in high-fat diet-induced gestational diabetes in mice. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2095–2105. [Google Scholar] [CrossRef] [PubMed]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: A meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet 2020. [Google Scholar] [CrossRef]
- Lorenzen, J.; Krämer, R.; Kliem, V.; Bode-Boeger, S.M.; Veldink, H.; Haller, H.; Fliser, D.; Kielstein, J.T. Circulating levels of osteopontin are closely related to glomerular filtration rate and cardiovascular risk markers in patients with chronic kidney disease. Eur. J. Clin. Investig. 2010, 40, 294–300. [Google Scholar] [CrossRef]
- Rosenberg, M.; Zugck, C.; Nelles, M.; Juenger, C.; Frank, D.; Remppis, A.; Giannitsis, E.; Katus, H.A.; Frey, N. Osteopontin, a new prognostic biomarker in patients with chronic heart fail. Circ. Heart Fail. 2008, 1, 43–49. [Google Scholar] [CrossRef]
- Francia, P.; Adduci, C.; Semprini, L.; Borro, M.; Ricotta, A.; Sensini, I.; Santini, D.; Caprinozzi, M.; Balla, C.; Simmaco, M.; et al. Osteopontin and galectin-3 predict the risk of ventricular tachycardia and fibrillation in heart failure patients with implantable defibrillators. J. Cardiovasc. Electrophysiol. 2014, 25, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Yousefi, K.; Shehadeh, L.A. Isolation, characterization, and high throughput extracellular flux analysis of mouse primary renal tubular epithelial cells. J. Vis. Exp. 2018, 136. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirakawa, K.; Sano, M. Sodium-Glucose Co-Transporter 2 Inhibitors Correct Metabolic Maladaptation of Proximal Tubular Epithelial Cells in High-Glucose Conditions. Int. J. Mol. Sci. 2020, 21, 7676. https://doi.org/10.3390/ijms21207676
Shirakawa K, Sano M. Sodium-Glucose Co-Transporter 2 Inhibitors Correct Metabolic Maladaptation of Proximal Tubular Epithelial Cells in High-Glucose Conditions. International Journal of Molecular Sciences. 2020; 21(20):7676. https://doi.org/10.3390/ijms21207676
Chicago/Turabian StyleShirakawa, Kohsuke, and Motoaki Sano. 2020. "Sodium-Glucose Co-Transporter 2 Inhibitors Correct Metabolic Maladaptation of Proximal Tubular Epithelial Cells in High-Glucose Conditions" International Journal of Molecular Sciences 21, no. 20: 7676. https://doi.org/10.3390/ijms21207676
APA StyleShirakawa, K., & Sano, M. (2020). Sodium-Glucose Co-Transporter 2 Inhibitors Correct Metabolic Maladaptation of Proximal Tubular Epithelial Cells in High-Glucose Conditions. International Journal of Molecular Sciences, 21(20), 7676. https://doi.org/10.3390/ijms21207676