
Pleiotropic Effects of Sodium-Glucose Cotransporter-2 Inhibitors: Renoprotective Mechanisms beyond Glycemic Control



Abstract
1. Diabetic Kidney Disease and the Treatment Strategy
2. Clinical Evidence on SGLT2 Inhibitors
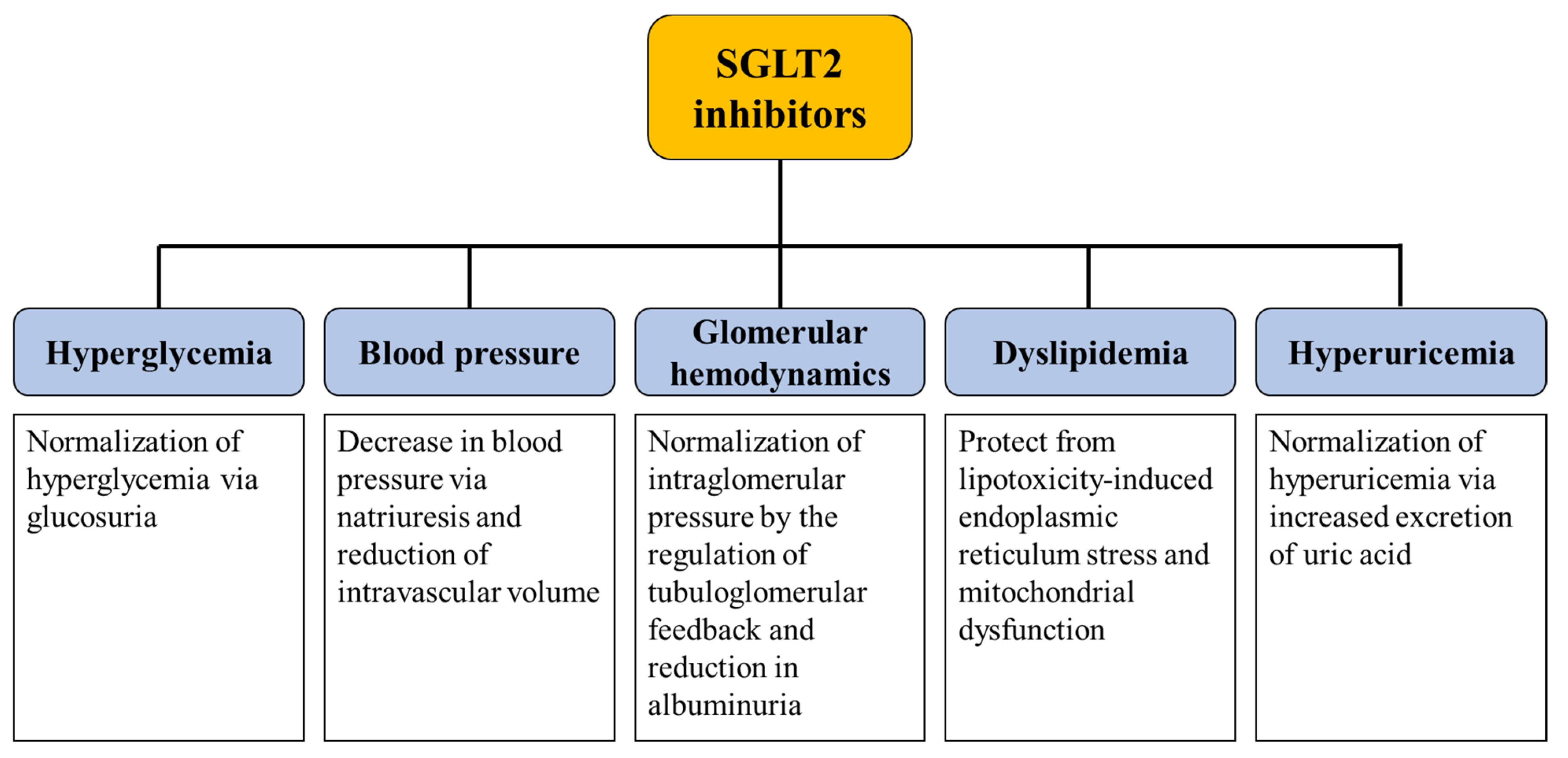
3. Mechanisms Underlying the Renoprotective Effect of SGLT2 Inhibitors
3.1. Glycemic Control
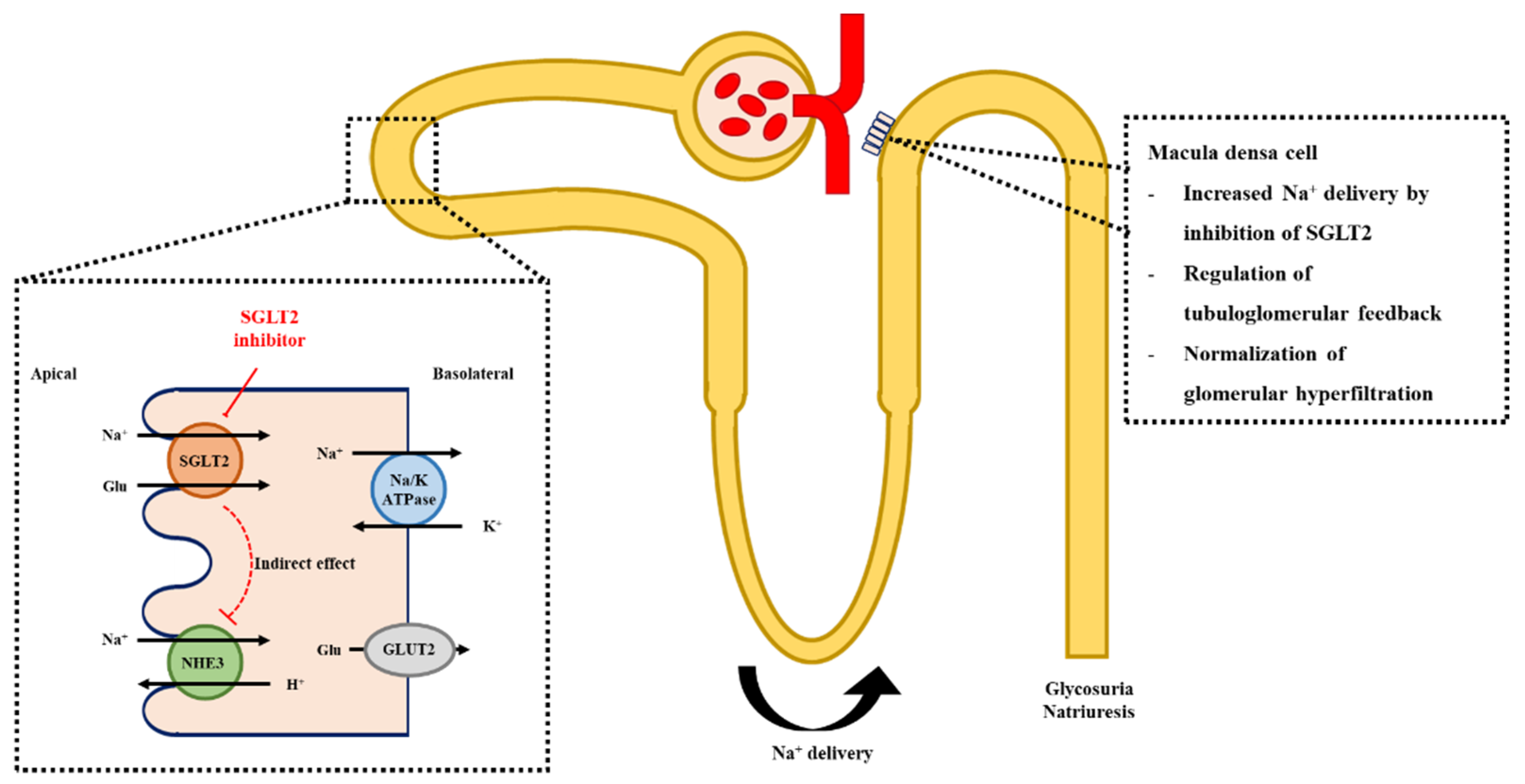
3.2. Glomerular Hemodynamics, Natriuresis, and Tubuloglomerular Feedback
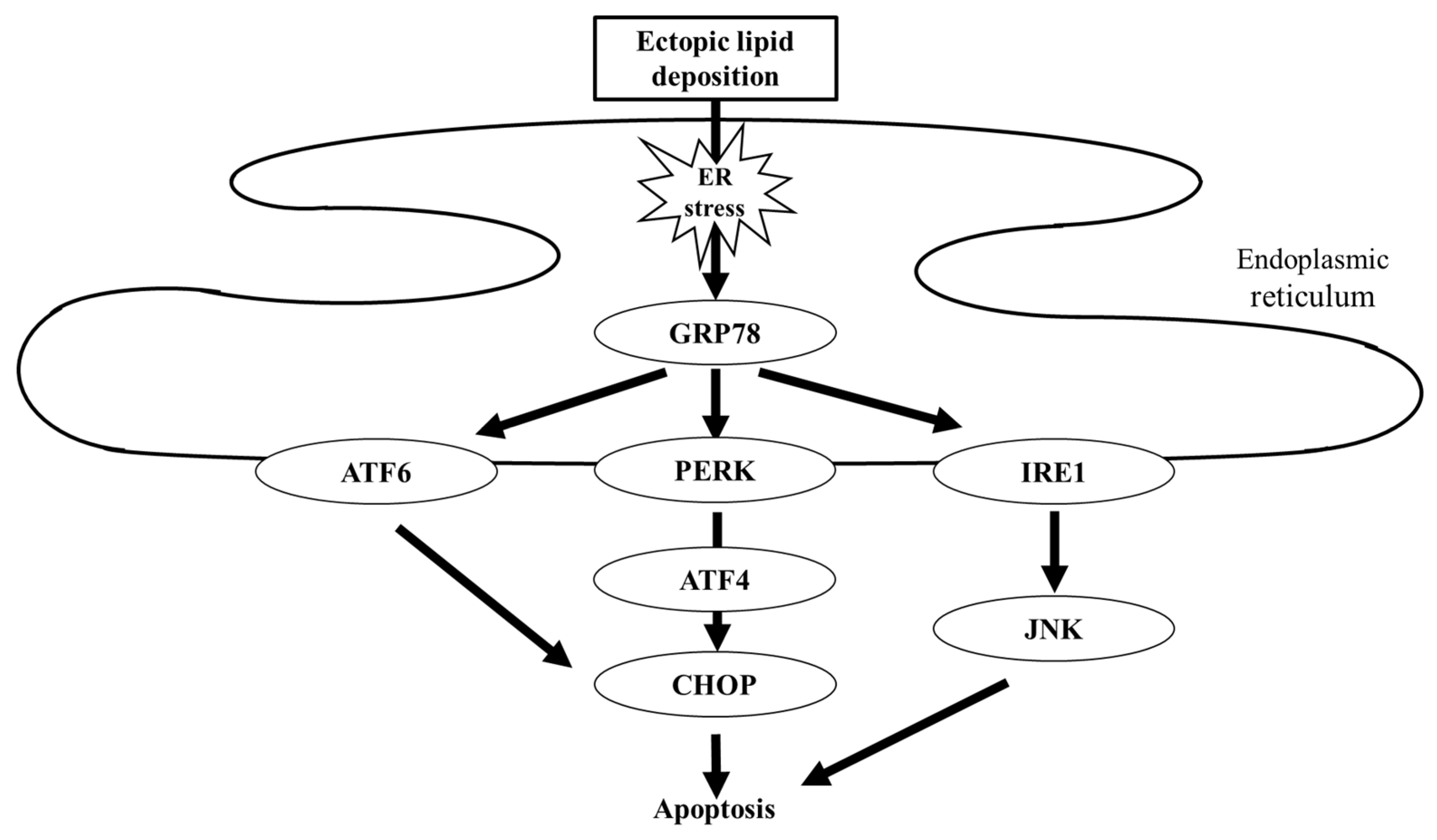
3.3. Protection from Lipotoxicity
3.4. Uric Acid Control
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xie, Y.; Bowe, B.; Mokdad, A.H.; Xian, H.; Yan, Y.; Li, T.; Maddukuri, G.; Tsai, C.Y.; Floyd, T.; Al-Aly, Z. Analysis of the global Burden of disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney Int. 2018, 94, 567–581. [Google Scholar] [CrossRef]
- Wu, B.; Bell, K.; Stanford, A.; Kern, D.M.; Tunceli, O.; Vupputuri, S.; Kalsekar, I.; Willey, V. Understanding C.K.D. among patients with T2DM: Prevalence, temporal trends, and treatment patterns-N.H.A.N.E.S. 2007–2012. BMJ Open Diabetes Res. Care 2016, 4, e000154. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Chronic Kidney Disease in the United States, 2019; US Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019.
- Nitta, K.; Goto, S.; Masakane, I.; Hanafusa, N.; Taniguchi, M.; Hasegawa, T.; Nakai, S.; Wada, A.; Hamano, T.; Hoshino, J.; et al. Annual dialysis data report for 2018, JSDT Renal Data Registry: Survey methods, facility data, incidence, prevalence, and mortality. Ren. Replace. Ther. 2020, 6, 1–8. [Google Scholar] [CrossRef]
- Yokoyama, H.; Sone, H.; Oishi, M.; Kawai, K.; Fukumoto, Y.; Kobayashi, M.; Japan Diabetes Clinical Data Management Study Group. Prevalence Prevalence of albuminuria and renal insufficiency and associated clinical factors in type 2 diabetes: The Japan Diabetes Clinical Data Management study (JDDM15). Nephrol. Dial. Transplant. 2009, 24, 1212–1219. [Google Scholar] [CrossRef]
- Martínez-Castelao, A.; Navarro-González, J.F.; Górriz, J.L.; de Alvaro, F. The concept and the epidemiology of diabetic nephropathy have changed in recent years. J. Clin. Med. 2015, 4, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Mimura, I.; Tanaka, T.; Nangaku, M. Treatment of diabetic kidney disease: Current and future. Diabetes Metab. J. 2021, 45, 11–26. [Google Scholar] [CrossRef]
- Opazo-Ríos, L.; Plaza, A.; Sánchez Matus, Y.; Bernal, S.; Lopez-Sanz, L.; Jimenez-Castilla, L.; Carpio, D.; Droguett, A.; Mezzano, S.; Egido, J.; et al. Targeting NF-κB by the Cell-Permeable N.E.M.O.-Binding Domain Peptide Improves Albuminuria and Renal Lesions in an Experimental Model of Type 2 Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 4225. [Google Scholar] [CrossRef] [PubMed]
- Donate-Correa, J.; Luis-Rodríguez, D.; Martín-Núñez, E.; Tagua, V.G.; Hernández-Carballo, C.; Ferri, C.; Rodríguez-Rodríguez, A.E.; Mora-Fernández, C.; Navarro-González, J.F. Inflammatory Targets in Diabetic Nephropathy. J. Clin. Med. 2020, 9, 458. [Google Scholar] [CrossRef]
- Górriz, J.L.; Nieto, J.; Navarro-González, J.F.; Molina, P.; Martínez-Castelao, A.; Pallardó, L.M. Nephroprotection by hypoglycemic agents: Do we have supporting data? J. Clin. Med. 2015, 4, 1866–1889. [Google Scholar] [CrossRef]
- Heo, C.U.; Choi, C.I. Current progress in pharmacogenetics of second-line antidiabetic medications: Towards precision medicine for Type 2 diabetes. J. Clin. Med. 2019, 8, 393. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group K.D.I.G.O. 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2020, 98, S1–S115. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef]
- Haller, H.; Ito, S.; Izzo, J.L.; Januszewicz, A.; Katayama, S.; Menne, J.; Mimran, A.; Rabelink, T.J.; Ritz, E.; Ruilope, L.M.; et al. Olmesartan for the delay or prevention of microalbuminuria in type 2 diabetes. N. Engl. J. Med. 2011, 364, 907–917. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in Type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and cardiovascular and renal events in Type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in Type 2 diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and renal outcomes in Type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and progression of kidney disease in Type 2 diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Wright, E.M.; Loo, D.D.F.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef]
- Bertrand, L.; Auquier, J.; Renguet, E.; Angé, M.; Cumps, J.; Horman, S.; Beauloye, C. Glucose transporters in cardiovascular system in health and disease. Pflugers Arch. 2020, 472, 1385–1399. [Google Scholar]
- Barfuss, D.W.; Schafer, J.A. Differences in active and passive glucose transport along the proximal nephron. Am. J. Physiol. 1981, 241, F322–F332. [Google Scholar] [CrossRef]
- Rieg, T.; Masuda, T.; Gerasimova, M.; Mayoux, E.; Platt, K.; Powell, D.R.; Thomson, S.C.; Koepsell, H.; Vallon, V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol. Renal Physiol. 2014, 306, F188–F193. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, G.; Hach, T.; Crowe, S.; Sanghvi, A.; Hall, K.D.; Ferrannini, E. Energy balance after sodium-glucose cotransporter 2 inhibition. Diabetes Care 2015, 38, 1730–1735. [Google Scholar] [CrossRef]
- Dominguez Rieg, J.A.; Rieg, T. What does sodium-glucose co-transporter 1 inhibition add: Prospects for dual inhibition. Diabetes Obes. Metab. 2019, 21 (Suppl. 2), 43–52. [Google Scholar] [CrossRef]
- Gilbert, R.E. Sodium-glucose linked transporter-2 inhibitors: Potential for renoprotection beyond blood glucose lowering? Kidney Int. 2014, 86, 693–700. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Kosiborod, M.; Inzucchi, S.E.; Cherney, D.Z.I. Renoprotective effects of sodium-glucose cotransporter-2 inhibitors. Kidney Int. 2018, 94, 26–39. [Google Scholar] [CrossRef]
- Giunti, S.; Barit, D.; Cooper, M.E. Mechanisms of diabetic nephropathy: Role of hypertension. Hypertension 2006, 48, 519–526. [Google Scholar] [CrossRef]
- Scholtes, R.A.; van Baar, M.J.B.; Kok, M.D.; Bjornstad, P.; Cherney, D.Z.I.; Joles, J.A.; van Raalte, D.H. Renal haemodynamic and protective effects of renoactive drugs in type 2 diabetes: Interaction with SGLT2 inhibitors. Nephrology (Carlton) 2020. [Google Scholar] [CrossRef]
- Jacobsen, P.; Andersen, S.; Rossing, K.; Jensen, B.R.; Parving, H.H. Dual blockade of the renin-angiotensin system versus maximal recommended dose of A.C.E. inhibition in diabetic nephropathy. Kidney Int. 2003, 63, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Schnermann, J.; Briggs, J.P. Tubuloglomerular feedback: Mechanistic insights from gene-manipulated mice. Kidney Int. 2008, 74, 418–426. [Google Scholar] [CrossRef]
- Vallon, V.; Rose, M.; Gerasimova, M.; Satriano, J.; Platt, K.A.; Koepsell, H.; Cunard, R.; Sharma, K.; Thomson, S.C.; Rieg, T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Renal Physiol. 2013, 304, F156–F167. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, T.D.; Campos, L.C.G.; Carraro-Lacroix, L.; Girardi, A.C.C.; Malnic, G. Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J. Am. Soc. Nephrol. 2014, 25, 2028–2039. [Google Scholar] [CrossRef]
- Onishi, A.; Fu, Y.; Patel, R.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Freeman, B.; Kim, Y.C.; Soleimani, M.; et al. A role for tubular Na+/H+ exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol. Renal Physiol. 2020, 319, F712–F728. [Google Scholar] [CrossRef] [PubMed]
- Komoroski, B.; Vachharajani, N.; Feng, Y.; Li, L.; Kornhauser, D.; Pfister, M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin. Pharmacol. Ther. 2009, 85, 513–519. [Google Scholar] [CrossRef]
- Tanaka, H.; Takano, K.; Iijima, H.; Kubo, H.; Maruyama, N.; Hashimoto, T.; Arakawa, K.; Togo, M.; Inagaki, N.; Kaku, K. Factors affecting canagliflozin-induced transient urine volume increase in patients with type 2 diabetes mellitus. Adv. Ther. 2017, 34, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.C.; Rieg, T.; Miracle, C.; Mansoury, H.; Whaley, J.; Vallon, V.; Singh, P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R75–R83. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, K.; Cherney, D.Z.I.; Bozovic, A.; Nagasu, H.; Satoh, M.; Kanda, E.; Sasaki, T.; Kashihara, N. Evaluation of glomerular hemodynamic function by empagliflozin in diabetic mice using in vivo imaging. Circulation 2019, 140, 303–315. [Google Scholar] [CrossRef]
- Cassis, P.; Locatelli, M.; Cerullo, D.; Corna, D.; Buelli, S.; Zanchi, C.; Villa, S.; Morigi, M.; Remuzzi, G.; Benigni, A.; et al. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight 2018, 3, e98720. [Google Scholar] [CrossRef]
- Takiyama, Y.; Sera, T.; Nakamura, M.; Ishizeki, K.; Saijo, Y.; Yanagimachi, T.; Maeda, M.; Bessho, R.; Takiyama, T.; Kitsunai, H.; et al. Impacts of Diabetes and an SGLT2 Inhibitor on the Glomerular Number and Volume in db/db Mice, as Estimated by Synchrotron Radiation Micro-CT at SPring-8. EBiomedicine 2018, 36, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, S.; Jinnouchi, H.; Kurinami, N.; Hieshima, K.; Yoshida, A.; Jinnouchi, K.; Tanaka, M.; Nishimura, H.; Suzuki, T.; Miyamoto, F.; et al. Impact of dapagliflozin therapy on renal protection and kidney morphology in patients with uncontrolled type 2 diabetes mellitus. J. Clin. Med. Res. 2018, 10, 466–477. [Google Scholar] [CrossRef]
- Takata, T.; Koda, M.; Sugihara, T.; Sugihara, S.; Okamoto, T.; Miyoshi, K.; Hodotsuka, M.; Fujise, Y.; Matono, T.; Okano, J.; et al. Left renal cortical thickness measured by ultrasound can predict early progression of chronic kidney disease. Nephron 2016, 132, 25–32. [Google Scholar] [CrossRef]
- Hoi, S.; Takata, T.; Sugihara, T.; Ida, A.; Ogawa, M.; Mae, Y.; Fukuda, S.; Munemura, C.; Isomoto, H. Predictive value of cortical thickness measured by ultrasonography for renal impairment: A longitudinal study in chronic kidney disease. J. Clin. Med. 2018, 7, 527. [Google Scholar] [CrossRef]
- Opazo-Ríos, L.; Mas, S.; Marín-Royo, G.; Mezzano, S.; Gómez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and diabetic nephropathy: Novel mechanistic insights and therapeutic opportunities. Int. J. Mol. Sci. 2020, 21, 2632. [Google Scholar] [CrossRef]
- Praga, M.; Morales, E. The fatty kidney: Obesity and renal disease. Nephron 2017, 136, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Lahuerta, A.; Martínez-García, C.; Medina-Gómez, G. Lipotoxicity as a trigger factor of renal disease. J. Nephrol. 2016, 29, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Jao, T.M.; Nangaku, M.; Wu, C.H.; Sugahara, M.; Saito, H.; Maekawa, H.; Ishimoto, Y.; Aoe, M.; Inoue, T.; Tanaka, T.; et al. ATF6α downregulation of PPARα promotes lipotoxicity-induced tubulointerstitial fibrosis. Kidney Int. 2019, 95, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.C.; Chen, C.R.; Chen, C.Y.; Lin, Y.C.; Chen, K.C.; Peng, R.Y. Nifedipine upregulates ATF6-α, caspases -12, -3, and -7 Implicating Lipotoxicity-Associated Renal E.R. Stress. Int. J. Mol. Sci. 2020, 21, 3147. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauléon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.K.; Mathur, A.; Kakkar, P. Emerging role of Unfolded Protein Response (U.P.R.) mediated proteotoxic apoptosis in diabetes. Life Sci. 2019, 216, 246–258. [Google Scholar] [CrossRef]
- Hosokawa, K.; Takata, T.; Sugihara, T.; Matono, T.; Koda, M.; Kanda, T.; Taniguchi, S.; Ida, A.; Mae, Y.; Yamamoto, M.; et al. Ipragliflozin ameliorates endoplasmic reticulum stress and apoptosis through preventing ectopic lipid deposition in renal tubules. Int. J. Mol. Sci. 2019, 21, 190. [Google Scholar] [CrossRef]
- Shibusawa, R.; Yamada, E.; Okada, S.; Nakajima, Y.; Bastie, C.C.; Maeshima, A.; Kaira, K.; Yamada, M. Dapagliflozin rescues endoplasmic reticulum stress-mediated cell death. Sci. Rep. 2019, 9, 9887. [Google Scholar] [CrossRef]
- Wang, D.; Luo, Y.; Wang, X.; Orlicky, D.J.; Myakala, K.; Yang, P.; Levi, M. The sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents renal and liver disease in western diet induced obesity mice. Int. J. Mol. Sci. 2018, 19, 137. [Google Scholar] [CrossRef]
- Lin, P.H.; Duann, P. Dyslipidemia in kidney disorders: Perspectives on mitochondria homeostasis and therapeutic opportunities. Front. Physiol. 2020, 11, 1050. [Google Scholar] [CrossRef]
- Tang, C.; Cai, J.; Dong, Z. Mitochondrial dysfunction in obesity-related kidney disease: A novel therapeutic target. Kidney Int. 2016, 90, 930–933. [Google Scholar] [CrossRef]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective mitochondrial fatty acid oxidation and lipotoxicity in kidney diseases. Front. Med. (Lausanne) 2020, 7, 65. [Google Scholar] [CrossRef]
- Ge, M.; Fontanesi, F.; Merscher, S.; Fornoni, A. The vicious cycle of renal lipotoxicity and mitochondrial dysfunction. Front. Physiol. 2020, 11, 732. [Google Scholar] [CrossRef]
- Wei, D.; Liao, L.; Wang, H.; Zhang, W.; Wang, T.; Xu, Z. Canagliflozin ameliorates obesity by improving mitochondrial function and fatty acid oxidation via PPARα in vivo and in vitro. Life Sci. 2020, 247, 117414. [Google Scholar] [CrossRef]
- Takagi, S.; Li, J.; Takagaki, Y.; Kitada, M.; Nitta, K.; Takasu, T.; Kanasaki, K.; Koya, D. Ipragliflozin improves mitochondrial abnormalities in renal tubules induced by a high-fat diet. J. Diabetes Investig. 2018, 9, 1025–1032. [Google Scholar] [CrossRef]
- Mauer, M.; Doria, A. Uric acid and risk of diabetic kidney disease. J. Nephrol. 2020, 33, 995–999. [Google Scholar] [CrossRef]
- Romi, M.M.; Arfian, N.; Tranggono, U.; Setyaningsih, W.A.W.; Sari, D.C.R. Uric acid causes kidney injury through inducing fibroblast expansion, endothelin-1 expression, and inflammation. BMC Nephrol. 2017, 18, 326. [Google Scholar] [CrossRef]
- Bose, B.; Badve, S.V.; Hiremath, S.S.; Boudville, N.; Brown, F.G.; Cass, A.; de Zoysa, J.R.; Fassett, R.G.; Faull, R.; Harris, D.C.; et al. Effects of uric acid-lowering therapy on renal outcomes: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2014, 29, 406–413. [Google Scholar] [CrossRef]
- Kanji, T.; Gandhi, M.; Clase, C.M.; Yang, R. Urate lowering therapy to improve renal outcomes in patients with chronic kidney disease: Systematic review and meta-analysis. BMC Nephrol. 2015, 16, 58. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2018, 20, 458–462. [Google Scholar] [CrossRef]
- Fralick, M.; Chen, S.K.; Patorno, E.; Kim, S.C. Assessing the risk for gout with sodium-glucose Cotransporter-2 inhibitors in patients with Type 2 diabetes: A population-based cohort study. Ann. Intern. Med. 2020, 172, 186–194. [Google Scholar] [CrossRef]
- Auberson, M.; Stadelmann, S.; Stoudmann, C.; Seuwen, K.; Koesters, R.; Thorens, B.; Bonny, O. SLC2A9 (GLUT9) mediates urate reabsorption in the mouse kidney. Pflugers Arch. 2018, 470, 1739–1751. [Google Scholar] [CrossRef]
- Bailey, C.J. Uric acid and the cardio-renal effects of SGLT2 inhibitors. Diabetes Obes. Metab. 2019, 21, 1291–1298. [Google Scholar] [CrossRef]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clinical Trial | Year | Drug (Dose) | N | Age (Years) | Median Follow-Up Period (Years) | CVD | DM | Mean eGFR (mL/min/1.73 m2) |
|---|---|---|---|---|---|---|---|---|
| EMPA-REG | 2015 | Empagliflozin (10 mg, 25 mg) | 7020 | 63.1 | 3.1 | 100% | 100% | 74.1 |
| CANVAS | 2017 | Canagliflozin (100 mg, 300 mg) | 10142 | 63.3 | 2.4 | 65.6% | 100% | 76.5 |
| DECLARE-TIMI 58 | 2019 | Dapagliflozin (10 mg) | 17160 | 63.9 | 4.2 | 40.6% | 100% | 85.2 |
| CREDENCE | 2019 | Canagliflozin (100 mg) | 4401 | 63.0 | 2.6 | 50.40% | 100% | 56.2 |
| DAPA-HF | 2019 | Dapagliflozin (10 mg) | 4401 | 66.3 | 1.5 | 100% | 41.8% | 65.8 |
| EMPEROR-Reduced | 2020 | Empagliflozin (10 mg) | 4401 | 66.8 | 1.3 | 100% | 49.8% | 62.0 |
| DAPA-CKD | 2020 | Dapagliflozin (10 mg) | 4401 | 61.9 | 2.4 | 37.4% | 67.5% | 43.1 |
| Clinical Trial | Definition of Renal Outcome | HR (95% CI) |
|---|---|---|
| EMPA-REG | Progression to macroalbuminuria, doubling of sCr, initiation of RRT, or death from renal disease | 0.61 (0.53–0.70) |
| CANVAS | 40% reduction in eGFR, requirement for RRT, or death from renal causes | 0.60 (0.47–0.77) |
| DECLARE-TIMI 58 | 40% decrease in eGFR, ESRD, death from renal or cardiovascular causes | 0.76 (0.67–0.87) |
| CREDENCE | ESRD, doubling of the sCr, or death from renal or cardiovascular causes | 0.70 (0.59–0.82) |
| DAPA-HF | 50% decline in the eGFR, ESRD, or renal death | 0.71 (0.44–1.16) |
| EMPEROR-Reduced | Hemodialysis, renal transplantation, or profound sustained reduction in eGFR | 0.50 (0.32–0.77) |
| DAPA-CKD | Sustained decline in the eGFR, ESRD, or death from renal or cardiovascular causes | 0.61 (0.51–0.72) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takata, T.; Isomoto, H. Pleiotropic Effects of Sodium-Glucose Cotransporter-2 Inhibitors: Renoprotective Mechanisms beyond Glycemic Control. Int. J. Mol. Sci. 2021, 22, 4374. https://doi.org/10.3390/ijms22094374
Takata T, Isomoto H. Pleiotropic Effects of Sodium-Glucose Cotransporter-2 Inhibitors: Renoprotective Mechanisms beyond Glycemic Control. International Journal of Molecular Sciences. 2021; 22(9):4374. https://doi.org/10.3390/ijms22094374
Chicago/Turabian StyleTakata, Tomoaki, and Hajime Isomoto. 2021. "Pleiotropic Effects of Sodium-Glucose Cotransporter-2 Inhibitors: Renoprotective Mechanisms beyond Glycemic Control" International Journal of Molecular Sciences 22, no. 9: 4374. https://doi.org/10.3390/ijms22094374
APA StyleTakata, T., & Isomoto, H. (2021). Pleiotropic Effects of Sodium-Glucose Cotransporter-2 Inhibitors: Renoprotective Mechanisms beyond Glycemic Control. International Journal of Molecular Sciences, 22(9), 4374. https://doi.org/10.3390/ijms22094374