Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression


Abstract
1. Introduction
2. Results
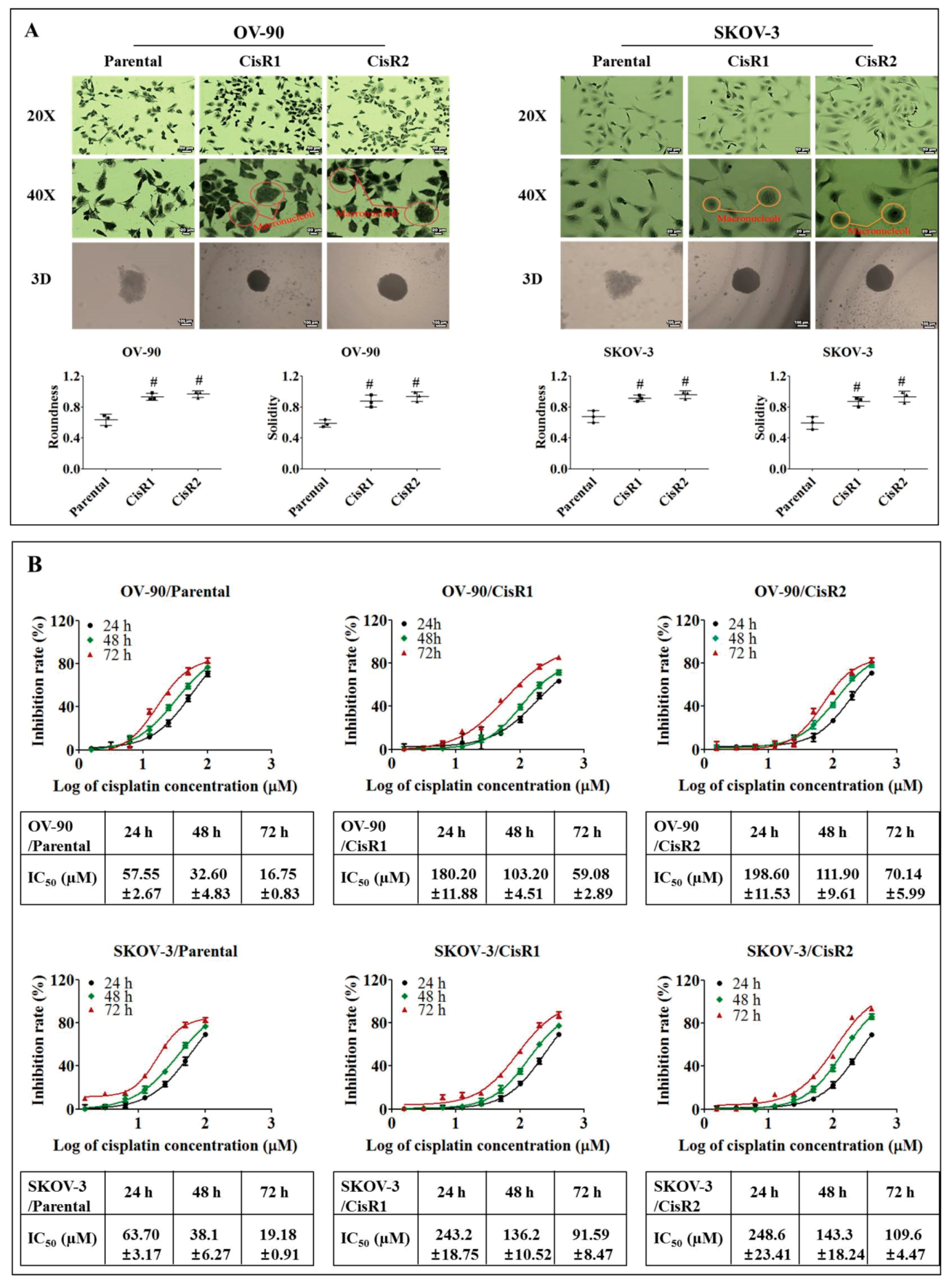
2.1. Generation and Characterization of Cisplatin Resistance in Ovarian Cancer Cell Lines
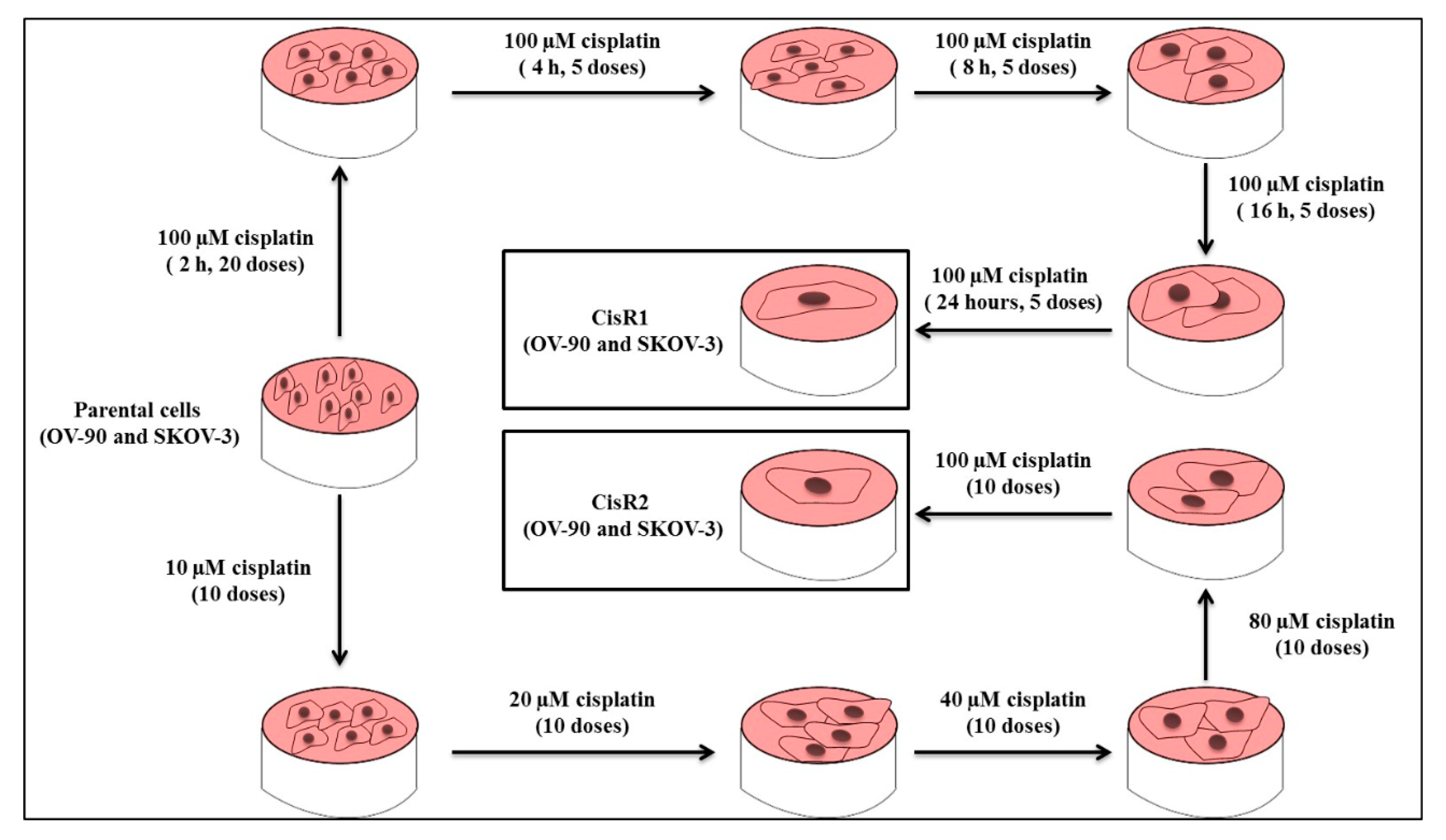
2.1.1. Generation of Cisplatin Resistance in (CisR) Ovarian Cancer (OC) Cell Lines
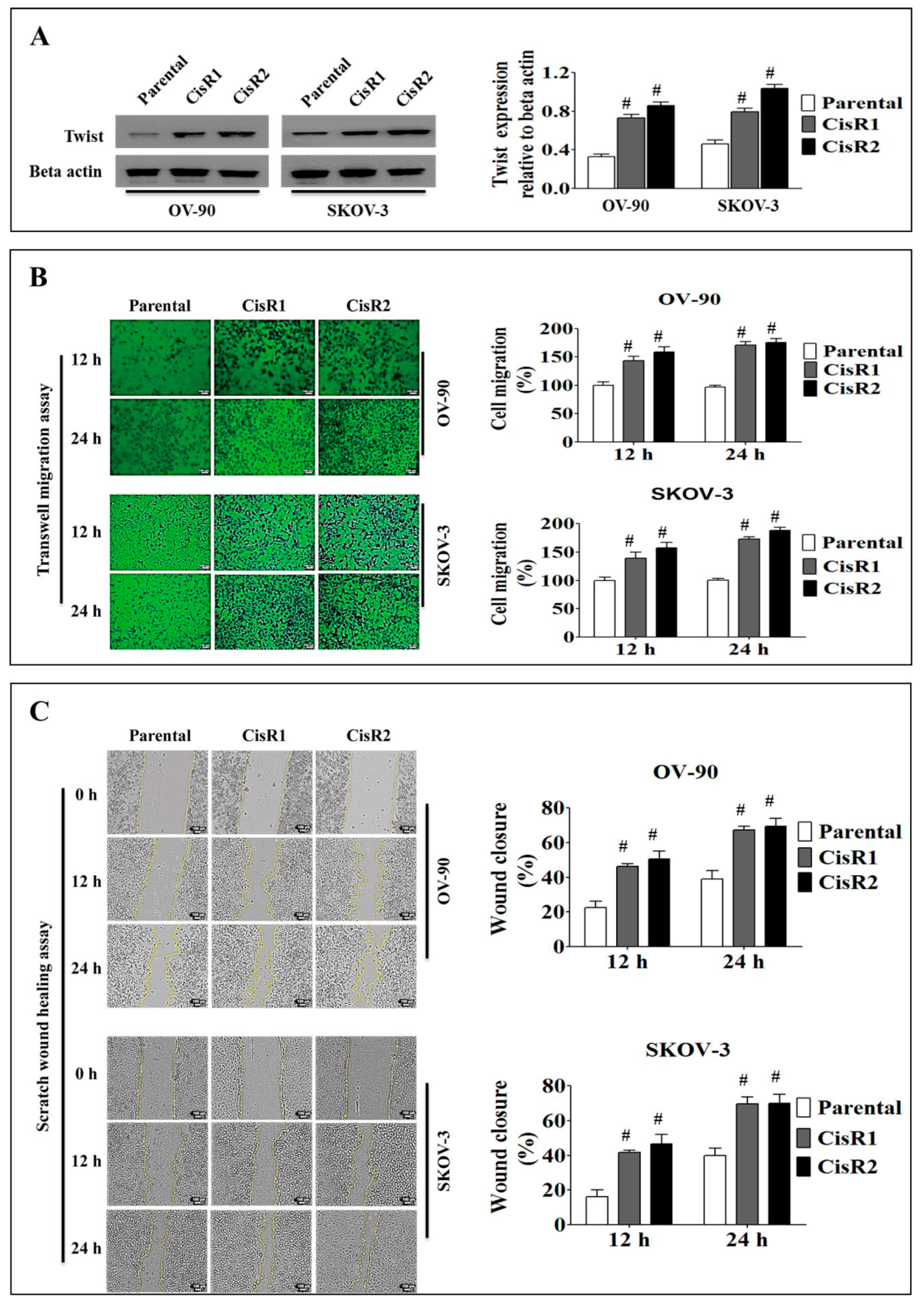
2.1.2. The CisR OC Cells Display Higher Twist Expression with Increased Metastasis Abilities than Their Parental OC Cells
2.1.3. The CisR OC Cells Exhibit Higher Metastasis Ability with an EMT Phenotype
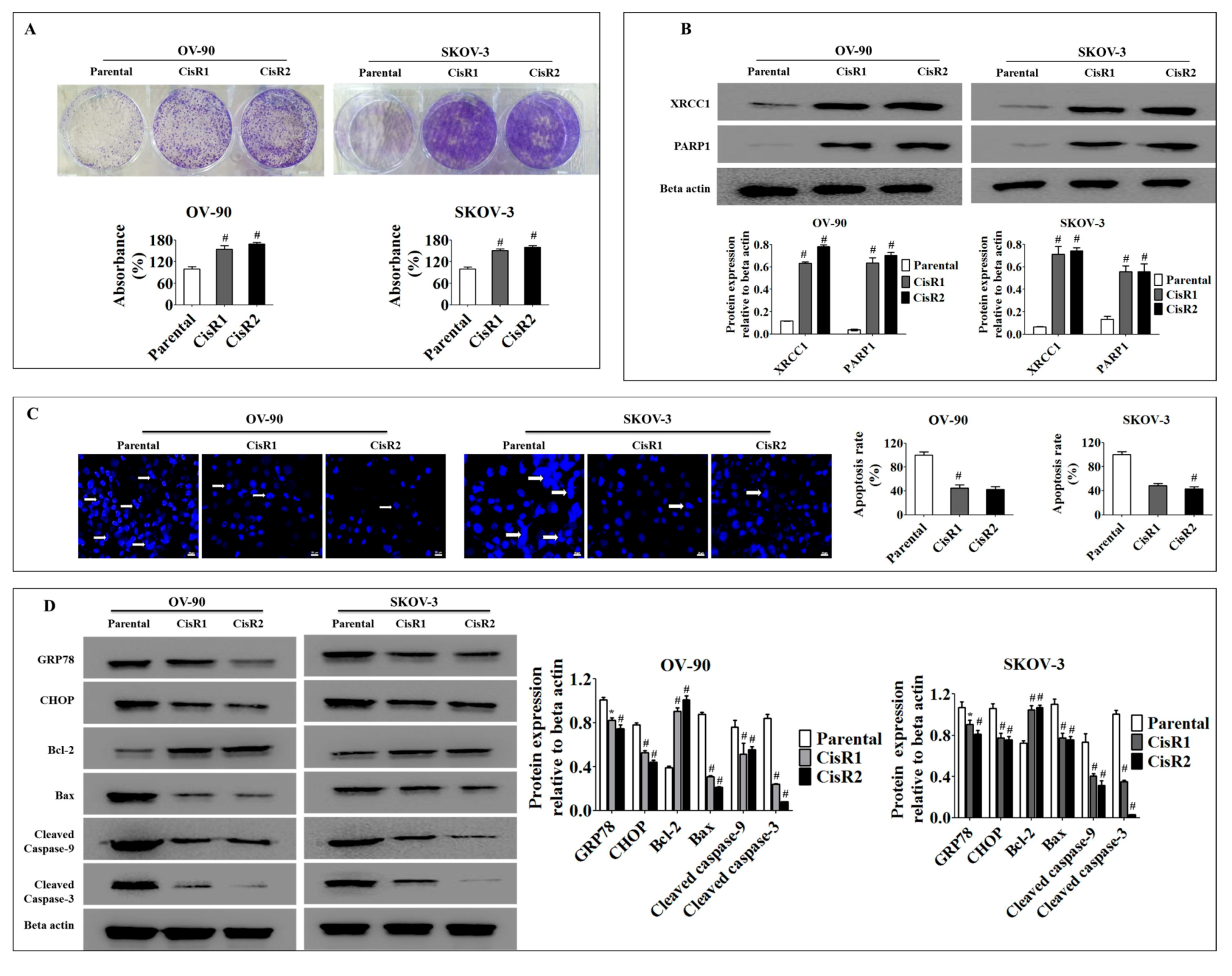
2.1.4. Acquisition of Cisplatin Resistance in OC Results in Higher Cell Colonization and Poor Apoptotic Signatures Associated with Activation of the DNA Excision Repair Pathway and Suppression of ER Stress Mediated Apoptosis
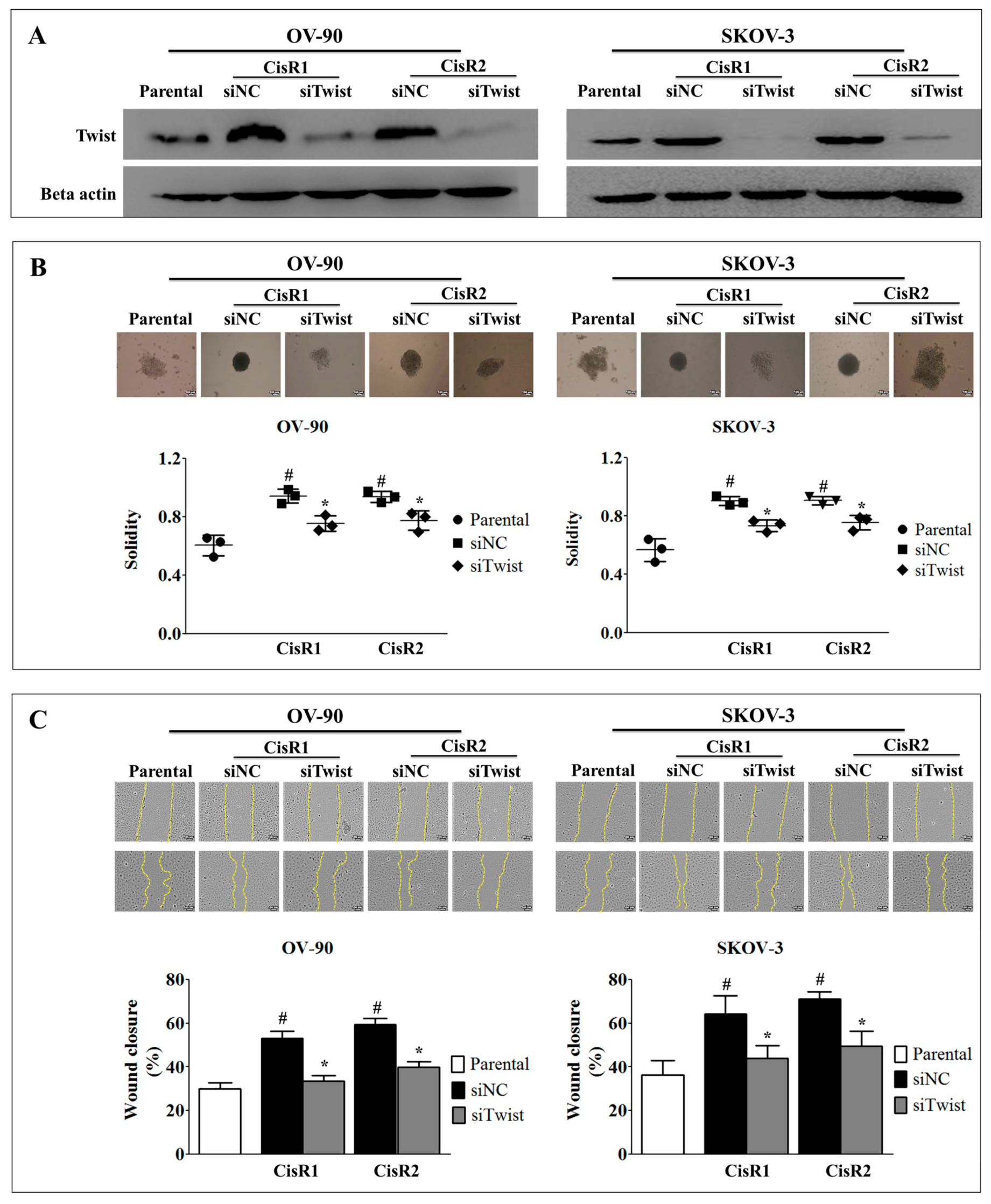
2.2. Twist Knockdown Can Affect the Metastasis Potential of Acquired CisR OC Cells
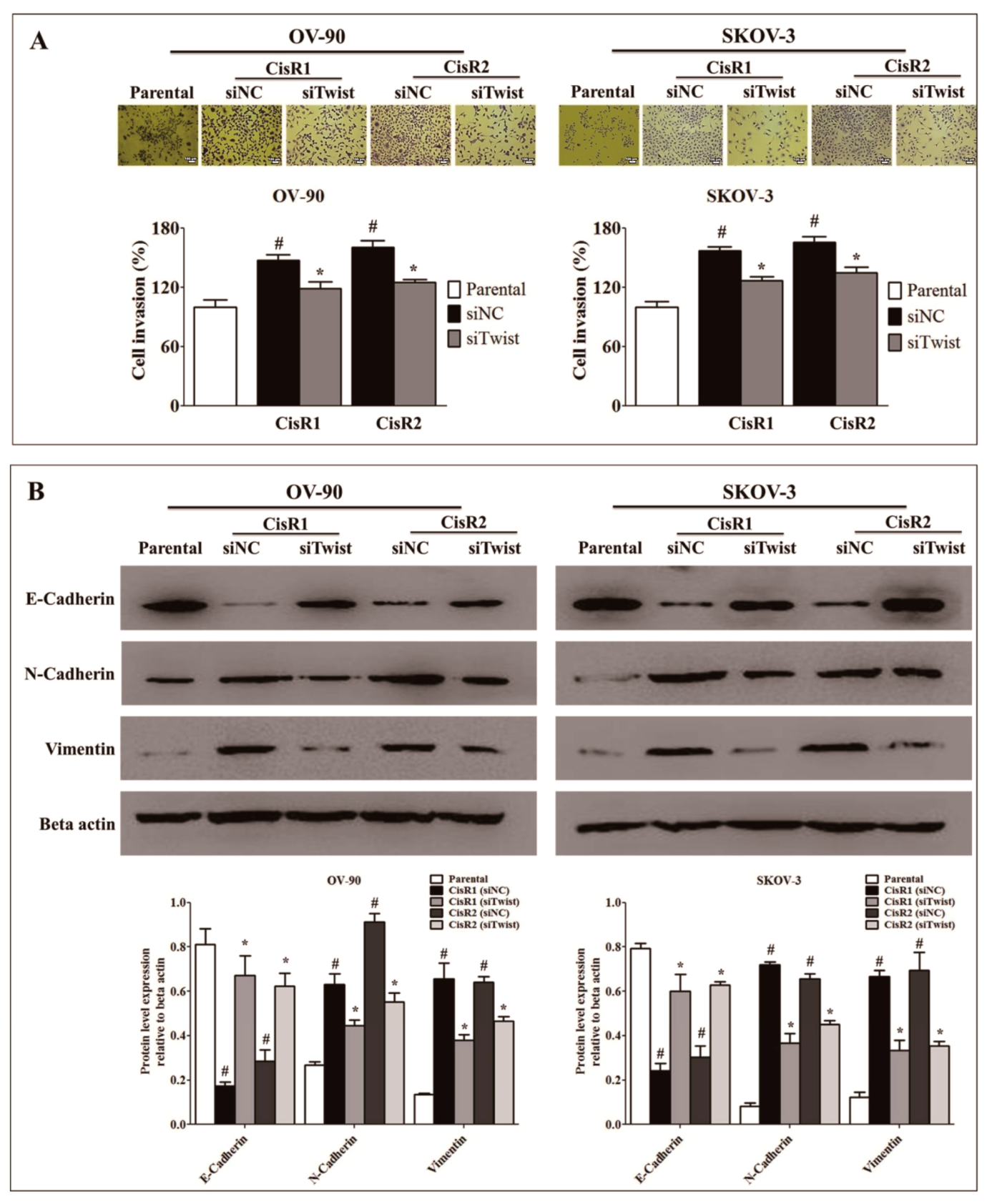
2.3. Twist Knockdown Attenuated Cell Metastasis Properties via Suppression of Cell Invasion and EMT Phenotype in Acquired CisR OC Cells
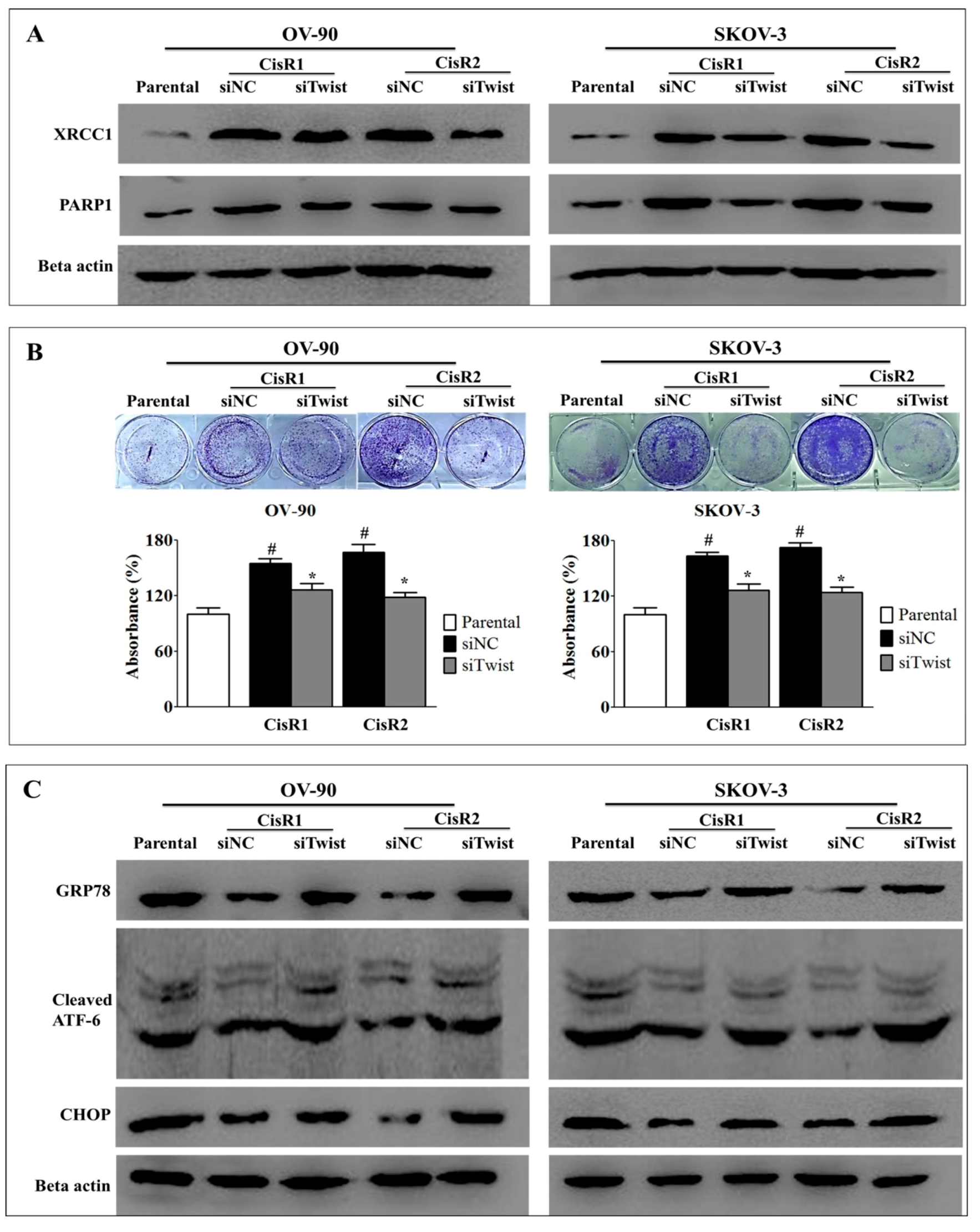
2.4. Twist Knockdown Reduces Cell Survival Potential via Downregulation of DNA Repair Pathway and Activation of ER-Stress-Mediated Cell Death in CisR OC Cells
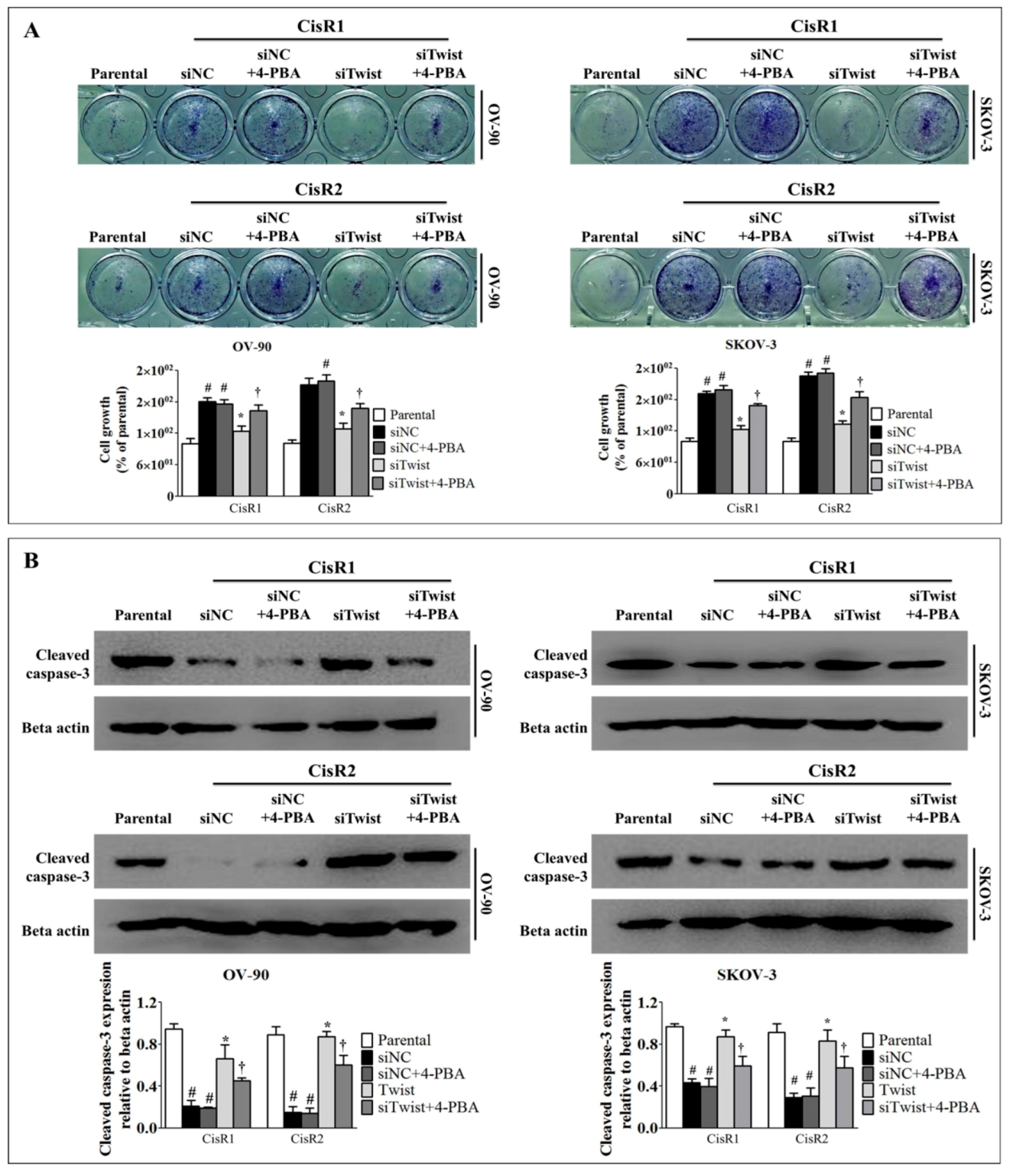
2.5. ER Stress Inhibition Reversed the Twist Knockdown-Induced Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Generation of Acquired CisR Ovarian Cancer (OC) Cell Lines
4.3. CisR Subline 1 (OV-90/CisR1 and SKOV-3/CisR1)
4.4. CisR Subline 2 (OV-90/CisR2 and SKOV-3/CisR2)
4.5. siRNA Transfection
4.6. Determination of 50% Inhibitory Concentration (IC50)
4.7. Morphological Evaluation and 3-Dimensional (3d) Spheres Generation
4.8. Colony Formation Assay
4.9. Cell Migration Assays
4.10. Cell Invasion Assays
4.11. Wound Healing Assay
4.12. Adhesion Assay
4.13. Apoptosis Assay
4.14. Western Blotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Womens Health 2019, 11, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Budiana, I.N.G.; Angelina, M.; Pemayun, T.G.A. Ovarian cancer: Pathogenesis and current recommendations for prophylactic surgery. J. Turk. Ger. Gynecol. Assoc. 2019, 20, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy resistance in advanced ovarian cancer patients. Biomark Cancer 2019, 11, 1179299X19860815. [Google Scholar] [CrossRef]
- Zhang, D.F.; Dou, P.H.; Zhao, D.X.; Li, J.; Hu, Y.H. Weekly cisplatin for the treatment of patients with ovarian cancer: A protocol for a systematic review of randomized controlled trial. Medicine 2019, 98, e15001. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Maliepaard, M.; Kolker, H.J.; Verweij, J.; Schellens, J.H. Abrogated energy-dependent uptake of cisplatin in a cisplatin-resistant subline of the human ovarian cancer cell line IGROV-1. Cancer Chemother. Pharmacol. 1998, 41, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Pectasides, D.; Pectasides, M.; Farmakis, D.; Gaglia, A.; Koumarianou, A.; Nikolaou, M.; Koumpou, M.; Kountourakis, P.; Papaxoinis, G.; Mitrou, P.; et al. Oxaliplatin plus high-dose leucovorin and 5-fluorouracil (FOLFOX 4) in platinum-resistant and taxane-pretreated ovarian cancer: A phase II study. Gynecol. Oncol. 2004, 95, 165–172. [Google Scholar] [CrossRef]
- Grabosch, S.; Bulatovic, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Loh, S.Y.; Mistry, P.; Kelland, L.R.; Abel, G.; Harrap, K.R. Reduced drug accumulation as a major mechanism of acquired resistance to cisplatin in a human ovarian carcinoma cell line: Circumvention studies using novel platinum (II) and (IV) ammine/amine complexes. Br. J. Cancer 1992, 66, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Righetti, S.C.; Perego, P.; Carenini, N.; Corna, E.; Bo, L.D.; Cedrola, S.; La Porta, C.A.; Zunino, F. Molecular alterations of cells resistant to platinum drugs: Role of PKCalpha. Biochim. Biophys. Acta 2006, 1763, 93–100. [Google Scholar] [CrossRef]
- Gore, M.E.; Fryatt, I.; Wiltshaw, E.; Dawson, T.; Robinson, B.A.; Calvert, A.H. Cisplatin/carboplatin cross-resistance in ovarian cancer. Br. J. Cancer 1989, 60, 767–769. [Google Scholar] [CrossRef]
- Jones, M.; Siracky, J.; Kelland, L.R.; Harrap, K.R. Acquisition of platinum drug resistance and platinum cross resistance patterns in a panel of human ovarian carcinoma xenografts. Br. J. Cancer 1993, 67, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, F.L.; Kerr, I.D. ABC transporter research: Going strong 40 years on. Biochem. Soc. Trans. 2015, 43, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.J.; Eastman, A.; Bostick-Bruton, F.; Reed, E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatin-DNA lesions and reduced drug accumulation. J. Clin. Investig. 1991, 87, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef]
- Robey, R.W.; Honjo, Y.; van de Laar, A.; Miyake, K.; Regis, J.T.; Litman, T.; Bates, S.E. A functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2). Biochim. Biophys. Acta 2001, 1512, 171–182. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Yoon, H. Chemotherapy resistance explained through endoplasmic reticulum stress-dependent signaling. Cancers 2019, 11, 338. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to overcome drug resistance in cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef]
- Jiang, Z.S.; Sun, Y.Z.; Wang, S.M.; Ruan, J.S. Epithelial-mesenchymal transition: Potential regulator of ABC transporters in tumor progression. J. Cancer 2017, 8, 2319–2327. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, Y.K.; Yun, P.Y. Upregulation of MDR- and EMT-related molecules in cisplatin-resistant human oral squamous cell carcinoma cell lines. Int. J. Mol. Sci. 2019, 20, 3034. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Calcagno, A.M.; Varma, S.; Marino, M.; Green, L.J.; Vora, M.I.; Patel, C.; Orina, J.N.; Eliseeva, T.A.; Singal, V.; et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 18708–18713. [Google Scholar] [CrossRef] [PubMed]
- Coley, H.M. Development of drug-resistant models. Methods Mol. Med. 2004, 88, 267–273. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro development of chemotherapy and targeted therapy drug-resistant cancer cell lines: A practical guide with case studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhang, G.; Zhang, Y. A novel method to establish glucocorticoid resistant acute lymphoblastic leukemia cell lines. J. Exp. Clin. Cancer Res. 2019, 38, 269. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.B.; Lind, M.J.; Cawkwell, L. Establishment of in-vitro models of chemotherapy resistance. Anticancer Drugs 2007, 18, 749–754. [Google Scholar] [CrossRef]
- Ru, G.Q.; Wang, H.J.; Xu, W.J.; Zhao, Z.S. Upregulation of Twist in gastric carcinoma associated with tumor invasion and poor prognosis. Pathol. Oncol. Res. 2011, 17, 341–347. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Ohuchida, K.; Mizumoto, K.; Ohhashi, S.; Yamaguchi, H.; Konomi, H.; Nagai, E.; Yamaguchi, K.; Tsuneyoshi, M.; Tanaka, M. Twist, a novel oncogene, is upregulated in pancreatic cancer: Clinical implication of Twist expression in pancreatic juice. Int. J. Cancer 2007, 120, 1634–1640. [Google Scholar] [CrossRef]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef]
- Yu, J.; Xie, F.; Bao, X.; Chen, W.; Xu, Q. miR-300 inhibits epithelial to mesenchymal transition and metastasis by targeting Twist in human epithelial cancer. Mol. Cancer 2014, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.H.; Park, H.J.; Choi, Y.; Won, Y.J.; Lee, S.J.; Park, D.Y. TWIST mediates resistance to paclitaxel by regulating Akt and Bcl-2 expression in gastric cancer cells. Tumour Biol. 2017, 39, 1010428317722070. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Lu, H.W.; Li, Y.M.; Lu, L.; Wang, J.L.; Zhang, Y.F.; Shang, H. Twist promotes invasion and cisplatin resistance in pancreatic cancer cells through growth differentiation factor 15. Mol. Med. Rep. 2015, 12, 3841–3848. [Google Scholar] [CrossRef]
- Wang, X.; Ling, M.T.; Guan, X.Y.; Tsao, S.W.; Cheung, H.W.; Lee, D.T.; Wong, Y.C. Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene 2004, 23, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Tran, M.A.; Pitruzzello, M.C.; Wen, W.; Loeza, J.; Dellinger, T.H.; Mor, G.; Glackin, C.A. TWIST1 drives cisplatin resistance and cell survival in an ovarian cancer model, via upregulation of GAS6, L1CAM, and Akt signalling. Sci. Rep. 2016, 6, 37652. [Google Scholar] [CrossRef]
- Wu, J.; Liao, Q.; He, H.; Zhong, D.; Yin, K. TWIST interacts with beta-catenin signaling on osteosarcoma cell survival against cisplatin. Mol. Carcinog. 2014, 53, 440–446. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef]
- Song, Y.H.; Shiota, M.; Yokomizo, A.; Uchiumi, T.; Kiyoshima, K.; Kuroiwa, K.; Oda, Y.; Naito, S. Twist1 and Y-box-binding protein-1 are potential prognostic factors in bladder cancer. Urol. Oncol. 2014, 32, 31.e1-7. [Google Scholar] [CrossRef]
- Shiota, M.; Song, Y.; Yokomizo, A.; Kiyoshima, K.; Tada, Y.; Uchino, H.; Uchiumi, T.; Inokuchi, J.; Oda, Y.; Kuroiwa, K.; et al. Foxo3a suppression of urothelial cancer invasiveness through Twist1, Y-box-binding protein 1, and E-cadherin regulation. Clin. Cancer Res. 2010, 16, 5654–5663. [Google Scholar] [CrossRef]
- Hung, J.J.; Yang, M.H.; Hsu, H.S.; Hsu, W.H.; Liu, J.S.; Wu, K.J. Prognostic significance of hypoxia-inducible factor-1alpha, TWIST1 and Snail expression in resectable non-small cell lung cancer. Thorax 2009, 64, 1082–1089. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; Van Ijcken, W.F.J.; Heine, A.A.J.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Shim, H.J.; Yun, J.Y.; Hwang, J.E.; Bae, W.K.; Cho, S.H.; Lee, J.H.; Kim, H.N.; Shin, M.H.; Kweon, S.S.; Lee, J.H.; et al. BRCA1 and XRCC1 polymorphisms associated with survival in advanced gastric cancer treated with taxane and cisplatin. Cancer Sci. 2010, 101, 1247–1254. [Google Scholar] [CrossRef]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef] [PubMed]
- Yadunandam, A.K.; Yoon, J.S.; Seong, Y.A.; Oh, C.W.; Kim, G.D. Prospective impact of 5-FU in the induction of endoplasmic reticulum stress, modulation of GRP78 expression and autophagy in Sk-Hep1 cells. Int. J. Oncol. 2012, 41, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef]
- Kamata, K. Pharmacological studies on the interrelation between the dopaminergic, GABAergic and opioid peptidergic systems in the central nervous system of the rat. Jpn. J. Pharmacol. 1987, 45, 439–447. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef]
- Cho, H.J.; Baek, K.E.; Park, S.M.; Kim, I.K.; Nam, I.K.; Choi, Y.L.; Park, S.H.; Im, M.J.; Choi, J.; Ryu, J.; et al. RhoGDI2 confers gastric cancer cells resistance against cisplatin-induced apoptosis by upregulation of Bcl-2 expression. Cancer Lett. 2011, 311, 48–56. [Google Scholar] [CrossRef]
- Sun, X.; Wei, Q.; Cheng, J.; Bian, Y.; Tian, C.; Hu, Y.; Li, H. Enhanced Stim1 expression is associated with acquired chemo-resistance of cisplatin in osteosarcoma cells. Hum. Cell 2017, 30, 216–225. [Google Scholar] [CrossRef]
- Shen, L.; Wen, N.; Xia, M.; Zhang, Y.U.; Liu, W.; Xu, Y.E.; Sun, L. Calcium efflux from the endoplasmic reticulum regulates cisplatin-induced apoptosis in human cervical cancer HeLa cells. Oncol. Lett. 2016, 11, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Dragonetti, E.; Vincenzi, B.; Onori, N.; Citro, G.; Baldi, A. Pulse-mediated chemotherapy enhances local control and survival in a spontaneous canine model of primary mucosal melanoma. Melanoma Res. 2006, 16, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Berry, S.R.; Cosby, R.; Asmis, T.; Chan, K.; Hammad, N.; Krzyzanowska, M.K. Cancer care Ontario’s gastrointestinal disease site, G. Continuous versus intermittent chemotherapy strategies in metastatic colorectal cancer: A systematic review and meta-analysis. Ann. Oncol. 2015, 26, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.J.; Shen, D.W.; Garfield, S.; Gottesman, M.M. Mislocalization of membrane proteins associated with multidrug resistance in cisplatin-resistant cancer cell lines. Cancer Res. 2003, 63, 5909–5916. [Google Scholar] [PubMed]
- Shen, D.W.; Akiyama, S.; Schoenlein, P.; Pastan, I.; Gottesman, M.M. Characterisation of high-level cisplatin-resistant cell lines established from a human hepatoma cell line and human KB adenocarcinoma cells: Cross-resistance and protein changes. Br. J. Cancer 1995, 71, 676–683. [Google Scholar] [CrossRef]
- Clynes, M.; Redmond, A.; Moran, E.; Gilvarry, U. Multiple drug-resistance in variant of a human non-small cell lung carcinoma cell line, DLKP-A. Cytotechnology 1992, 10, 75–89. [Google Scholar] [CrossRef]
- Yan, X.D.; Li, M.; Yuan, Y.; Mao, N.; Pan, L.Y. Biological comparison of ovarian cancer resistant cell lines to cisplatin and Taxol by two different administrations. Oncol. Rep. 2007, 17, 1163–1169. [Google Scholar] [CrossRef]
- Zhang, H.; Qian, G.; Zong, D.; Fan, S.; Owonikoko, T.K.; Ramalingam, S.S.; Sun, S.Y. Overcoming acquired resistance of epidermal growth factor receptor-mutant non-small-cell lung cancer cells to osimertinib by combining osimertinib with the histone deacetylase inhibitor panobinostat (LBH589). Cancer 2020, 126, 2024–2033. [Google Scholar] [CrossRef]
- Letourneau, I.J.; Quinn, M.C.; Wang, L.L.; Portelance, L.; Caceres, K.Y.; Cyr, L.; Delvoye, N.; Meunier, L.; de Ladurantaye, M.; Shen, Z.; et al. Derivation and characterization of matched cell lines from primary and recurrent serous ovarian cancer. BMC Cancer 2012, 12, 379. [Google Scholar] [CrossRef]
- Brigulova, K.; Cervinka, M.; Tosner, J.; Sedlakova, I. Chemoresistance testing of human ovarian cancer cells and its in vitro model. Toxicol. Vitr. 2010, 24, 2108–2115. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.; Michor, F. Evolution of resistance to targeted anti-cancer therapies during continuous and pulsed administration strategies. PLoS Comput. Biol. 2009, 5, e1000557. [Google Scholar] [CrossRef]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Bellat, V.; Law, B. Chemotherapy induces adaptive drug resistance and metastatic potentials via phenotypic CXCR4-expressing cell state transition in ovarian cancer. PLoS ONE 2017, 12, e0171044. [Google Scholar] [CrossRef] [PubMed]
- Tahtamouni, L.; Ahram, M.; Koblinski, J.; Rolfo, C. Molecular regulation of cancer cell migration, invasion, and metastasis. Anal. Cell. Pathol. 2019, 2019, 1356508. [Google Scholar] [CrossRef]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Entschladen, F.; Drell, T.L., 4th; Lang, K.; Joseph, J.; Zaenker, K.S. Tumour-cell migration, invasion, and metastasis: Navigation by neurotransmitters. Lancet Oncol. 2004, 5, 254–258. [Google Scholar] [CrossRef]
- Alluri, P.G.; Speers, C.; Chinnaiyan, A.M. Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res. 2014, 16, 494. [Google Scholar] [CrossRef]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28 (Suppl. 1), S24–S31. [Google Scholar] [CrossRef]
- Liang, Y.; McDonnell, S.; Clynes, M. Examining the relationship between cancer invasion/metastasis and drug resistance. Curr. Cancer Drug Targets 2002, 2, 257–277. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, Y.; Oue, N.; Fujiwara-Tani, R.; Sasaki, T.; Ohmori, H.; Kishi, S.; Mori, S.; Mori, T.; Ikeda, N.; Matsumoto, S.; et al. Role of metastasis-related genes in cisplatin chemoresistance in gastric cancer. Int. J. Mol. Sci. 2019, 21, 254. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.Y.; Farrand, L.; Kim, J.Y.; Byun, S.; Suh, J.Y.; Lee, H.J.; Tsang, B.K. Molecular determinants of ovarian cancer chemoresistance: New insights into an old conundrum. Ann. N. Y. Acad. Sci. 2012, 1271, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Nakajima, S.; Hsieh, C.L.; Kanno, S.; Masutani, M.; Levine, A.S.; Yasui, A.; Lan, L. Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose). J. Cell Sci. 2013, 126, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Brem, R.; Hall, J. XRCC1 is required for DNA single-strand break repair in human cells. Nucleic Acids Res. 2005, 33, 2512–2520. [Google Scholar] [CrossRef]
- Kothandapani, A.; Sawant, A.; Dangeti, V.S.; Sobol, R.W.; Patrick, S.M. Epistatic role of base excision repair and mismatch repair pathways in mediating cisplatin cytotoxicity. Nucleic Acids Res. 2013, 41, 7332–7343. [Google Scholar] [CrossRef]
- Lin, Z.P.; Lee, Y.; Lin, F.; Belcourt, M.F.; Li, P.; Cory, J.G.; Glazer, P.M.; Sartorelli, A.C. Reduced level of ribonucleotide reductase R2 subunits increases dependence on homologous recombination repair of cisplatin-induced DNA damage. Mol. Pharmacol. 2011, 80, 1000–1012. [Google Scholar] [CrossRef]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.; Fousteri, M.I. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol. Cell 2007, 27, 311–323. [Google Scholar] [CrossRef]
- Keil, C.; Grobe, T.; Oei, S.L. MNNG-induced cell death is controlled by interactions between PARP-1, poly(ADP-ribose) glycohydrolase, and XRCC1. J. Biol. Chem. 2006, 281, 34394–34405. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, C.; Zhang, L.; Chi, X.; Ji, J.; Gao, X.; Wang, Y.; Zhao, Z.; Liu, L.; Cao, X.; et al. Triptolide interferes with XRCC1/PARP1-mediated DNA repair and confers sensitization of triple-negative breast cancer cells to cisplatin. Biomed. Pharmacother. 2019, 109, 1541–1546. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xiong, J.; Qiu, D.; Zhao, X.; Yan, D.; Xu, W.; Wang, Z.; Chen, Q.; Panday, S.; Li, A.; et al. Inhibition of PARP1 activity enhances chemotherapeutic efficiency in cisplatin-resistant gastric cancer cells. Int. J. Biochem. Cell Biol. 2017, 92, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, B.; Lin, D.; Tan, W.; Leaw, S.; Hong, X.; Hu, X. XRCC1 polymorphisms and severe toxicity in lung cancer patients treated with cisplatin-based chemotherapy in Chinese population. Lung Cancer 2008, 62, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef] [PubMed]
- Slyskova, J.; Sabatella, M.; Ribeiro-Silva, C.; Stok, C.; Theil, A.F.; Vermeulen, W.; Lans, H. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018, 46, 9537–9549. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.K.; Watson, M.; Stefanick, D.F.; Shaughnessy, D.T.; Taylor, J.A.; Wilson, S.H. XRCC1 and DNA polymerase beta in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008, 18, 48–63. [Google Scholar] [CrossRef]
- Annunziata, C.M.; O’Shaughnessy, J. Poly (ADP-ribose) polymerase as a novel therapeutic target in cancer. Clin. Cancer Res 2010, 16, 4517–4526. [Google Scholar] [CrossRef]
- Bixel, K.; Hays, J.L. Olaparib in the management of ovarian cancer. Pharmgenomics Pers. Med. 2015, 8, 127–135. [Google Scholar] [CrossRef]
- Yang, X.; Zheng, F.; Xing, H.; Gao, Q.; Wei, W.; Lu, Y.; Wang, S.; Zhou, J.; Hu, W.; Ma, D. Resistance to chemotherapy-induced apoptosis via decreased caspase-3 activity and overexpression of antiapoptotic proteins in ovarian cancer. J. Cancer Res. Clin. Oncol. 2004, 130, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.; Sultana, R.; Abbotts, R.; Hawkes, C.; Seedhouse, C.; Chan, S.; Madhusudan, S. Clinicopathological and functional significance of XRCC1 expression in ovarian cancer. Int. J. Cancer 2013, 132, 2778–2786. [Google Scholar] [CrossRef]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef]
- Pyrko, P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Xiang, C.; Cazacu, S.; Finniss, S.; Kazimirsky, G.; Lemke, N.; Lehman, N.L.; Rempel, S.A.; Mikkelsen, T.; Brodie, C. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro Oncol. 2008, 10, 236–243. [Google Scholar] [CrossRef]
- Wang, J.; Kho, D.H.; Zhou, J.Y.; Davis, R.J.; Wu, G.S. MKP-1 suppresses PARP-1 degradation to mediate cisplatin resistance. Oncogene 2017, 36, 5939–5947. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.T.; Yang, M.H. Revisiting epithelial-mesenchymal transition in cancer metastasis: The connection between epithelial plasticity and stemness. Mol. Oncol. 2017, 11, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Mani, S.A.; Levine, H. Hybrid epithelial/mesenchymal phenotype(s): The ‘fittest’ for metastasis? Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 151–157. [Google Scholar] [CrossRef]
- Norouzi, S.; Valokala, M.G.; Mosaffa, F.; Zirak, M.R.; Zamani, P.; Behravan, J. Crosstalk in cancer resistance and metastasis. Crit. Rev. Oncol. Hematol. 2018, 132, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef]
- Iseri, O.D.; Kars, M.D.; Arpaci, F.; Atalay, C.; Pak, I.; Gunduz, U. Drug resistant MCF-7 cells exhibit epithelial-mesenchymal transition gene expression pattern. Biomed. Pharmacother. 2011, 65, 40–45. [Google Scholar] [CrossRef]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi-Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef]
- Jolly, M.K.; Tripathi, S.C.; Somarelli, J.A.; Hanash, S.M.; Levine, H. Epithelial/mesenchymal plasticity: How have quantitative mathematical models helped improve our understanding? Mol. Oncol. 2017, 11, 739–754. [Google Scholar] [CrossRef]
- Bitting, R.L.; Schaeffer, D.; Somarelli, J.A.; Garcia-Blanco, M.A.; Armstrong, A.J. The role of epithelial plasticity in prostate cancer dissemination and treatment resistance. Cancer Metastasis Rev. 2014, 33, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Sinha, S.K.; Levine, H.; Jolly, M.K.; Dutta, P.S. Anticipating critical transitions in epithelial-hybrid-mesenchymal cell-fate determination. Proc. Natl. Acad. Sci. USA 2019, 116, 26343–26352. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Jolly, M.K.; Kulkarni, P.; Levine, H. Phenotypic plasticity and cell fate decisions in cancer: Insights from dynamical systems theory. Cancers 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.; Jolly, M.K.; Ghosh, A. Stability and mean residence times for hybrid epithelial/mesenchymal phenotype. Phys. Biol. 2019, 16, 025003. [Google Scholar] [CrossRef] [PubMed]
- Garg, M. Epithelial, mesenchymal and hybrid epithelial/mesenchymal phenotypes and their clinical relevance in cancer metastasis. Expert Rev. Mol. Med. 2017, 19, e3. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Du, P.; Ge, Z.; Jin, Y.; Ding, D.; Liu, X.; Zou, Q. TWIST1 and BMI1 in cancer metastasis and chemoresistance. J. Cancer 2016, 7, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Yang, A.D.; Fan, F.; Camp, E.R.; van Buren, G.; Liu, W.; Somcio, R.; Gray, M.J.; Cheng, H.; Hoff, P.M.; Ellis, L.M. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin. Cancer Res. 2006, 12, 4147–4153. [Google Scholar] [CrossRef]
- Deng, J.J.; Zhang, W.; Xu, X.M.; Zhang, F.; Tao, W.P.; Ye, J.J.; Ge, W. Twist mediates an aggressive phenotype in human colorectal cancer cells. Int. J. Oncol. 2016, 48, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Kwok, W.K.; Ling, M.T.; Lee, T.W.; Lau, T.C.; Zhou, C.; Zhang, X.; Chua, C.W.; Chan, K.W.; Chan, F.L.; Glackin, C.; et al. Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res. 2005, 65, 5153–5162. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Ying, X.; Lin, P.C.; Zhou, B.P. Twist-mediated epithelial-mesenchymal transition promotes breast tumor cell invasion via inhibition of hippo pathway. Sci. Rep. 2016, 6, 24606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Wei, X.L.; Liang, Y.K.; Chen, W.L.; Zhang, F.; Bai, J.W.; Qiu, S.Q.; Du, C.W.; Huang, W.H.; Zhang, G.J. Over-expressed twist associates with markers of epithelial mesenchymal transition and predicts poor prognosis in breast cancers via ERK and Akt activation. PLoS ONE 2015, 10, e0135851. [Google Scholar] [CrossRef]
- Liu, Y.-R.; Liang, L.; Zhao, J.M.; Zhang, Y.; Zhang, M.; Zhong, W.-L.; Zhang, Q.; Wei, J.-J.; Li, M.; Yuan, J.; et al. Twist1 confers multidrug resistance in colon cancer through upregulation of ATP-binding cassette transporters. Oncotarget 2017, 8, 52901–52912. [Google Scholar] [CrossRef]
- Wu, Y.H.; Huang, Y.F.; Chang, T.H.; Chou, C.Y. Activation of TWIST1 by COL11A1 promotes chemoresistance and inhibits apoptosis in ovarian cancer cells by modulating NF-kappaB-mediated IKKbeta expression. Int. J. Cancer 2017, 141, 2305–2317. [Google Scholar] [CrossRef]
- Sun, K.X.; Jiao, J.W.; Chen, S.; Liu, B.L.; Zhao, Y. MicroRNA-186 induces sensitivity of ovarian cancer cells to paclitaxel and cisplatin by targeting ABCB1. J. Ovarian Res. 2015, 8, 80. [Google Scholar] [CrossRef]
- Xiang, Y.; Chen, Y.J.; Yan, Y.B.; Liu, Y.; Qiu, J.; Tan, R.Q.; Tian, Q.; Guan, L.; Niu, S.S.; Xin, H.W. MiR-186 bidirectionally regulates cisplatin sensitivity of ovarian cancer cells via suppressing targets PIK3R3 and PTEN and upregulating APAF1 expression. J. Cancer 2020, 11, 3446–3453. [Google Scholar] [CrossRef]
- Li, C.; Duan, P.; Wang, J.; Lu, X.; Cheng, J. miR-320 inhibited ovarian cancer oncogenicity via targeting TWIST1 expression. Am. J. Transl. Res. 2017, 9, 3705–3713. [Google Scholar]
- Zhu, X.; Shen, H.; Yin, X.; Long, L.; Xie, C.; Liu, Y.; Hui, L.; Lin, X.; Fang, Y.; Cao, Y.; et al. miR-186 regulation of Twist1 and ovarian cancer sensitivity to cisplatin. Oncogene 2016, 35, 323–332. [Google Scholar] [CrossRef]
- Roberts, C.M.; Shahin, S.A.; Wen, W.; Finlay, J.B.; Dong, J.; Wang, R.; Dellinger, T.H.; Zink, J.I.; Tamanoi, F.; Glackin, C.A. Nanoparticle delivery of siRNA against TWIST to reduce drug resistance and tumor growth in ovarian cancer models. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wu, C.; Liang, H.; Zhao, Y.; Lin, C.; Zhang, X.; Ye, C. Knockdown of TWIST enhances the cytotoxicity of chemotherapeutic drugs in doxorubicin-resistant HepG2 cells by suppressing MDR1 and EMT. Int. J. Oncol. 2018, 53, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.Y.; Trujillo, J.M. Biological characterization of multidrug-resistant human colon carcinoma sublines induced/selected by two methods. Cancer Res. 1990, 50, 3218–3225. [Google Scholar] [PubMed]
- Phung, Y.T.; Barbone, D.; Broaddus, V.C.; Ho, M. Rapid generation of in vitro multicellular spheroids for the study of monoclonal antibody therapy. J. Cancer 2011, 2, 507–514. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Calculation |
|---|---|
| Cell migration (%) | ACisR/Ap × 100 |
| Cell migration speed (%/h) | M/∆T |
| Parameter | Calculation |
|---|---|
| Cell invasion (%) | ACisR/Ap × 100 |
| Cell invasion speed (%/h) | I/∆T |
| Parameter | Calculation |
|---|---|
| Would closure (%) | W0 − Wt/W0 × 100 |
| Healing speed (µm2/h) | W0 − Wt/∆T |
| Relative wound area | W0/Wt |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahar, E.; Kim, J.-Y.; Kim, H.-S.; Yoon, H. Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression. Int. J. Mol. Sci. 2020, 21, 7613. https://doi.org/10.3390/ijms21207613
Bahar E, Kim J-Y, Kim H-S, Yoon H. Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression. International Journal of Molecular Sciences. 2020; 21(20):7613. https://doi.org/10.3390/ijms21207613
Chicago/Turabian StyleBahar, Entaz, Ji-Ye Kim, Hyun-Soo Kim, and Hyonok Yoon. 2020. "Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression" International Journal of Molecular Sciences 21, no. 20: 7613. https://doi.org/10.3390/ijms21207613
APA StyleBahar, E., Kim, J.-Y., Kim, H.-S., & Yoon, H. (2020). Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression. International Journal of Molecular Sciences, 21(20), 7613. https://doi.org/10.3390/ijms21207613