The Role of the RANKL/RANK Axis in the Prevention and Treatment of Breast Cancer with Immune Checkpoint Inhibitors and Anti-RANKL


 ,
, 
 and
and 
Abstract
1. RANKL/RANK Signaling Pathway
2. The Role of the RANKL/RANK Signaling Pathway in Normal Mammary Gland Development
3. The Role of the RANKL/RANK Signaling Pathway in Breast Cancer
4. Role of RANKL in the Survival, Resistance Development and Metastatic Capacity of Breast Cancer Cells
5. RANKL/RANK Pathway and Members of the ERBB Family in Breast Carcinogenesis
6. Clinical Trials with Denosumab
7. RANKL/RANK Signaling Pathway and Tumor Immunomodulation
8. Coinhibition of RANKL and Immune Checkpoint Inhibitors (ICIs) as a Putative Therapeutic Strategy
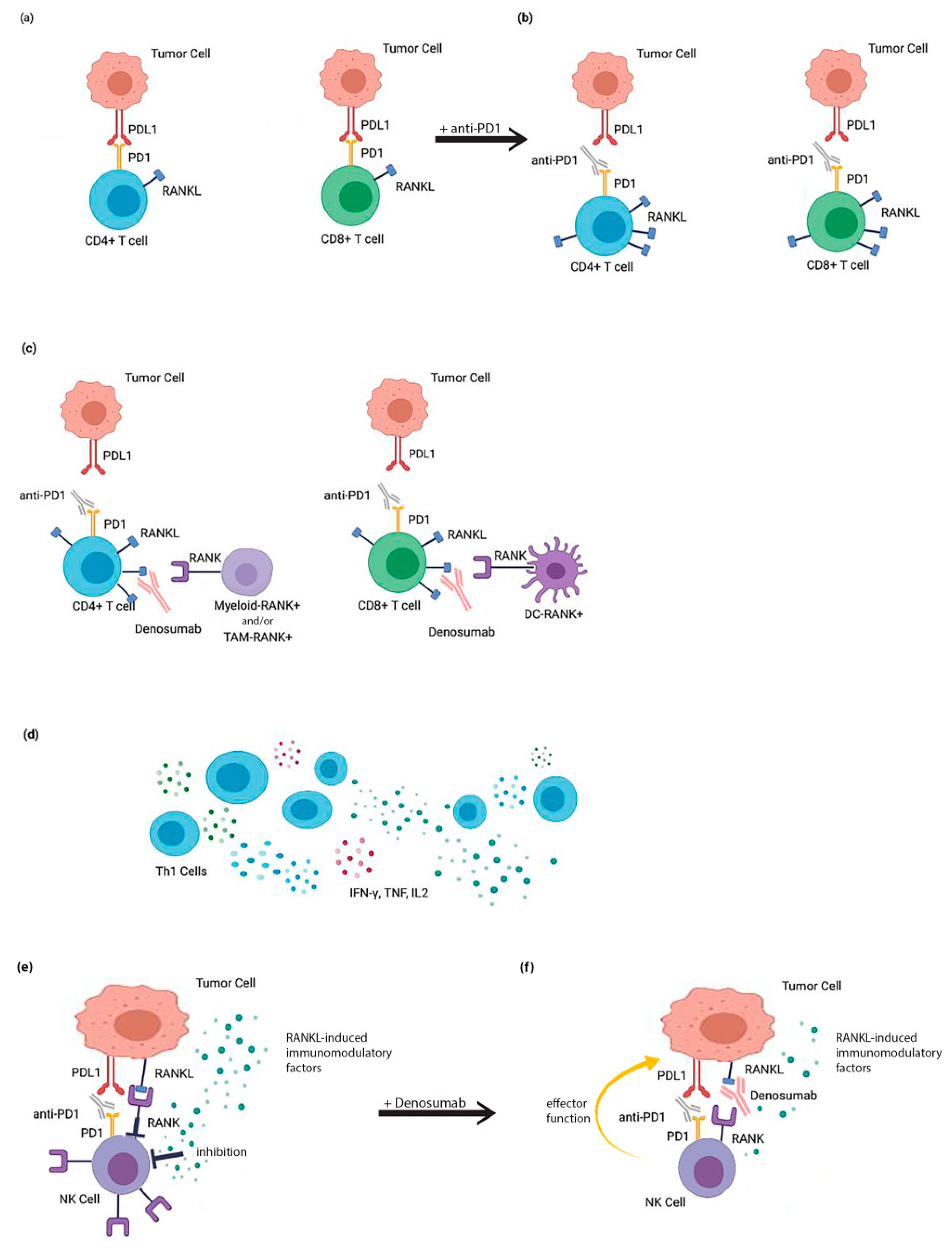
9. Mechanisms Involved in the Treatment with RANKL and ICIs
10. Conclusions
- (1)
- The evaluation of change in chemokine production (TNF-alpha, interleukins, IFN gamma) both in serum and in the TME under the conditions of RANKL/RANK inhibition.
- (2)
- A change in intratumoral T-cell (CD4, CD8 and Treg) numbers and function to determine the intratumor ratio of effectors to regulators.
- (3)
- A change in myeloid cell (M1/M2 macrophage, myeloid-derived suppressor cell (MDSC), DC) numbers and function.
- (4)
- Functional markers of myeloid cells expressing RANK. The identification of the main subpopulations of myeloid-derived suppressor cells (MDSCs) expressing RANK may serve as biomarkers for identifying patients who would benefit more from this therapeutic approach.
Author Contributions
Funding
Conflicts of Interest
References
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Luan, X.; Lu, Q.; Jiang, Y.; Zhang, S.; Wang, Q.; Yuan, H.; Zhao, W.; Wang, J.; Wang, X. Crystal Structure of Human RANKL Complexed with Its Decoy Receptor Osteoprotegerin. J. Immunol. 2012, 189, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Cawley, K.; Piemontese, M.; Fujiwara, Y.; Zhao, H.; Goellner, J.J.; O’Brien, C.A. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- González-Suárez, E.; Sanz-Moreno, A. RANK as a therapeutic target in cancer. FEBS J. 2016, 283, 2018–2033. [Google Scholar] [CrossRef]
- Yen, C.H.; Hsu, C.M.; Hsiao, S.Y.; Hsiao, H.H. Pathogenic mechanisms of myeloma bone disease and possible roles for nrf2. Int. J. Mol. Sci. 2020, 21, 6723. [Google Scholar] [CrossRef]
- Nagaoka, M.; Maeda, T.; Moriwaki, S.; Nomura, A.; Kato, Y.; Niida, S.; Kruger, M.C.; Suzuki, K. Petunidin, a b-ring 50-o-methylated derivative of delphinidin, stimulates osteoblastogenesis and reduces srankl-induced bone loss. Int. J. Mol. Sci. 2019, 20, 2795. [Google Scholar] [CrossRef]
- Renema, N.; Navet, B.; Heymann, M.F.; Lezot, F.; Heymann, D. RANK-RANKL signalling in cancer. Biosci. Rep. 2016, 36, e00366. [Google Scholar] [CrossRef]
- Ithimakin, S.; Day, K.C.; Malik, F.; Zen, Q.; Dawsey, S.J.; Bersano-Begey, T.F.; Quraishi, A.A.; Ignatoski, K.W.; Daignault, S.; Davis, A.; et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Res. 2013, 73, 1635–1646. [Google Scholar] [CrossRef]
- Lacey, D.L.; Boyle, W.J.; Simonet, W.S.; Kostenuik, P.J.; Dougall, W.C.; Sullivan, J.K.; Martin, J.S.; Dansey, R. Bench to bedside: Elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat. Rev. Drug Discov. 2012, 11, 401–419. [Google Scholar] [CrossRef]
- Castellano, D.; Sepulveda, J.M.; García-Escobar, I.; Rodriguez-Antolín, A.; Sundlöv, A.; Cortes-Funes, H. The Role of RANK-Ligand Inhibition in Cancer: The Story of Denosumab. Oncologist 2011, 16, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Body, J.J.; Lipton, A.; Gralow, J.; Steger, G.G.; Gao, G.; Yeh, H.; Fizazi, K. Effects of denosumab in patients with bone metastases with and without previous bisphosphonate exposure. J. Bone Miner. Res. 2010, 25, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Lipton, A.; Mariette, X.; Body, J.J.; Rahim, Y.; Gralow, J.R.; Gao, G.; Wu, L.; Sohn, W.; Jun, S. Randomized phase II trial of denosumab in patients with bone metastases from prostate cancer, breast cancer, or other neoplasms after intravenous bisphosphonates. J. Clin. Oncol. 2009, 27, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Stopeck, A.T.; Lipton, A.; Body, J.J.; Steger, G.G.; Tonkin, K.; De Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: A randomized, double-blind study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef]
- Van Poznak, C.; Somerfield, M.R.; Moy, B. Role of bone-modifying agents in metastatic breast cancer: An American society of clinical oncology-cancer care Ontario focused guideline update summary. J. Oncol. Pract. 2017, 13, 822–824. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, C.; Fu, X.; Liu, M.; Li, Y.; Pan, J.; Liu, H.; Wang, S.; Xiang, L.; Xiao, G.G.; et al. High-dose diosgenin reduces bone loss in ovariectomized rats via attenuation of the RANKL/OPG ratio. Int. J. Mol. Sci. 2014, 15, 17130–17147. [Google Scholar] [CrossRef]
- Fata, J.E.; Kong, Y.Y.; Li, J.; Sasaki, T.; Irie-Sasaki, J.; Moorehead, R.A.; Elliott, R.; Scully, S.; Voura, E.B.; Lacey, D.L.; et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell 2000, 103, 41–50. [Google Scholar] [CrossRef]
- Gonzalez-Suarez, E.; Branstetter, D.; Armstrong, A.; Dinh, H.; Blumberg, H.; Dougall, W.C. RANK Overexpression in Transgenic Mice with Mouse Mammary Tumor Virus Promoter-Controlled RANK Increases Proliferation and Impairs Alveolar Differentiation in the Mammary Epithelia and Disrupts Lumen Formation in Cultured Epithelial Acini. Mol. Cell. Biol. 2007, 27, 1442–1454. [Google Scholar] [CrossRef]
- Srivastava, S.; Matsuda, M.; Hou, Z.; Bailey, J.P.; Kitazawa, R.; Herbst, M.P.; Horseman, N.D. Receptor Activator of NF-κB Ligand Induction via Jak2 and Stat5a in Mammary Epithelial Cells. J. Biol. Chem. 2003, 278, 46171–46178. [Google Scholar] [CrossRef]
- Mulac-Jericevic, B.; Lydon, J.P.; DeMayo, F.J.; Conneely, O.M. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc. Natl. Acad. Sci. USA 2003, 100, 9744–9749. [Google Scholar] [CrossRef]
- Mukherjee, A.; Soyal, S.M.; Li, J.; Ying, Y.; He, B.; Demayo, F.J.; Lydon, J.P. Targeting RANKL to a specific subset of murine mammary epithelial cells induces ordered branching morphogenesis and alveologenesis in the absence of progesterone receptor expression. FASEB J. 2010, 24, 4408–4419. [Google Scholar] [CrossRef] [PubMed]
- Beleut, M.; Rajaram, R.D.; Caikovski, M.; Ayyanan, A.; Germano, D.; Choi, Y.; Schneider, P.; Brisken, C. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc. Natl. Acad. Sci. USA 2010, 107, 2989–2994. [Google Scholar] [CrossRef]
- Cao, Y.; Luo, J.L.; Karin, M. IκB kinase α kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15852–15857. [Google Scholar] [CrossRef] [PubMed]
- Tanos, T.; Sflomos, G.; Echeverria, P.C.; Ayyanan, A.; Gutierrez, M.; Delaloye, J.F.; Raffoul, W.; Fiche, M.; Dougall, W.; Schneider, P.; et al. Progesterone/RANKL is a major regulatory axis in the human breast. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Joshi, P.A.; Jackson, H.W.; Beristain, A.G.; Di Grappa, M.A.; Mote, P.A.; Clarke, C.L.; Stingl, J.; Waterhouse, P.D.; Khokha, R. Progesterone induces adult mammary stem cell expansion. Nature 2010, 465, 803–807. [Google Scholar] [CrossRef]
- Asselin-Labat, M.L.; Vaillant, F.; Sheridan, J.M.; Pal, B.; Wu, D.; Simpson, E.R.; Yasuda, H.; Smyth, G.K.; Martin, T.J.; Lindeman, G.J.; et al. Control of mammary stem cell function by steroid hormone signalling. Nature 2010, 465, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Hanada, R.; Hanada, T.; Sigl, V.; Schramek, D.; Penninger, J.M. RANKL/RANK-beyond bones. J. Mol. Med. 2011, 89, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Suarez, E.; Jacob, A.P.; Jones, J.; Miller, R.; Roudier-Meyer, M.P.; Erwert, R.; Pinkas, J.; Branstetter, D.; Dougall, W.C. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010, 468, 103–107. [Google Scholar] [CrossRef]
- Schramek, D.; Leibbrandt, A.; Sigl, V.; Kenner, L.; Pospisilik, J.A.; Lee, H.J.; Hanada, R.; Joshi, P.A.; Aliprantis, A.; Glimcher, L.; et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010, 468, 98–102. [Google Scholar] [CrossRef]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef]
- Tsubaki, M.; Komai, M.; Fujimoto, S.I.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Takeda, T.; Ogawa, N.; Mashimo, K.; et al. Activation of NF-κB by the RANKL/RANK system up-regulates snail and twist expressions and induces epithelial-to-mesenchymal transition in mammary tumor cell lines. J. Exp. Clin. Cancer Res. 2013, 32. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancermetastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Sigl, V.; Owusu-Boaitey, K.; Joshi, P.A.; Kavirayani, A.; Wirnsberger, G.; Novatchkova, M.; Kozieradzki, I.; Schramek, D.; Edokobi, N.; Hersl, J.; et al. RANKL/RANK control Brca1 mutation-driven mammary tumors. Cell Res. 2016, 26, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Nolan, E.; Vaillant, F.; Branstetter, D.; Pal, B.; Giner, G.; Whitehead, L.; Lok, S.W.; Mann, G.B.; Rohrbach, K.; Huang, L.Y.; et al. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat. Med. 2016, 22, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; de Almeida, B.P.; Dâmaso, S.; Mansinho, A.; Correia, I.; Henriques, S.; Cruz-Duarte, R.; Vilhais, G.; Félix, P.; Alves, P.; et al. Expression of receptor activator of NFkB (RANK) drives stemness and resistance to therapy in ER+HER2-breast cancer. Oncotarget 2020, 11, 1714–1728. [Google Scholar] [CrossRef]
- Yoldi, G.; Pellegrini, P.; Trinidad, E.M.; Cordero, A.; Gomez-Miragaya, J.; Serra-Musach, J.; Dougall, W.C.; Muñoz, P.; Pujana, M.A.; Planelles, L.; et al. RANK signaling blockade reduces breast cancer recurrence by inducing tumor cell differentiation. Cancer Res. 2016, 76, 5857–5869. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Hyun, K.A.; Goo, K.B.; Han, H.; Sohn, J.; Choi, W.; Kim, S.-I.; Jung, H.-I.; Kim, Y.S. Epithelial-to-mesenchymal transition leads to loss of EpCAM and different physical properties in circulating tumor cells from metastatic breast cancer. Oncotarget 2016, 7, 24677–24687. [Google Scholar] [CrossRef]
- Perez, S.A.; Karamouzis, M.V.; Skarlos, D.V.; Ardavanis, A.; Sotiriadou, N.N.; Iliopoulou, E.G.; Salagianni, M.L.; Orphanos, G.; Baxevanis, C.N.; Rigatos, G.; et al. CD4+CD25+ regulatory T-cell frequency in HER-2/neu (HER)-positive and HER-negative advanced-stage breast cancer patients. Clin. Cancer Res. 2007, 13, 2714–2721. [Google Scholar] [CrossRef]
- Mercatali, L.; La Manna, F.; Miserocchi, G.; Liverani, C.; De Vita, A.; Spadazzi, C.; Bongiovanni, A.; Recine, F.; Amadori, D.; Ghetti, M.; et al. Tumor-stroma crosstalk in bone tissue: The osteoclastogenic potential of a breast cancer cell line in a co-culture system and the role of EGFR inhibition. Int. J. Mol. Sci. 2017, 18, 1655. [Google Scholar] [CrossRef]
- Casimiro, S.; Mohammad, K.S.; Pires, R.; Tato-Costa, J.; Alho, I.; Teixeira, R.; Carvalho, A.; Ribeiro, S.; Lipton, A.; Guise, T.A.; et al. RANKL/RANK/MMP-1 Molecular Triad Contributes to the Metastatic Phenotype of Breast and Prostate Cancer Cells In Vitro. PLoS ONE 2013, 8, e63153. [Google Scholar] [CrossRef]
- Wu, X.; Li, F.; Dang, L.; Liang, C.; Lu, A.; Zhang, G. RANKL/RANK System-Based Mechanism for Breast Cancer Bone Metastasis and Related Therapeutic Strategies. Front. Cell Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef]
- Futakuchi, M.; Fukamachi, K.; Suzui, M. Heterogeneity of tumor cells in the bone microenvironment: Mechanisms and therapeutic targets for bone metastasis of prostate or breast cancer. Adv. Drug Deliv. Rev. 2016, 99, 206–211. [Google Scholar] [CrossRef]
- Mascarau, R.; Bertrand, F.; Labrousse, A.; Gennero, I.; Poincloux, R.; Maridonneau-Parini, I.; Raynaud-Messina, B.; Vérollet, C. Hiv-1-infected human macrophages, by secreting rank-l, contribute to enhanced osteoclast recruitment. Int. J. Mol. Sci. 2020, 21, 3154. [Google Scholar] [CrossRef]
- Maurizi, A.; Rucci, N. The osteoclast in bone metastasis: Player and target. Cancers (Basel) 2018, 10, 218. [Google Scholar] [CrossRef]
- Azim, H.A.; Kamal, N.S.; Azim, H.A. Bone metastasis in breast cancer: The story of RANK-Ligand. J. Egypt. Natl. Canc. Inst. 2012, 24, 107–114. [Google Scholar] [CrossRef]
- Azim, H.; Azim, H.A. Targeting RANKL in breast cancer: Bone metastasis and beyond. Expert Rev. Anticancer Ther. 2013, 13, 195–201. [Google Scholar] [CrossRef]
- Shupp, A.B.; Kolb, A.D.; Mukhopadhyay, D.; Bussard, K.M. Cancer metastases to bone: Concepts, mechanisms, and interactions with bone osteoblasts. Cancers (Basel) 2018, 10, 182. [Google Scholar] [CrossRef]
- Merkhofer, E.C.; Cogswell, P.; Baldwin, A.S. Her2 activates NF-κB and induces invasion through the canonical pathway involving IKKα. Oncogene 2010, 29, 1238–1248. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Normanno, N.; Gullick, W.J. Epidermal growth factor receptor tyrosine kinase inhibitors and bone metastases: Different mechanisms of action for a novel therapeutic application? Endocr. Relat. Cancer 2006, 13, 3–6. [Google Scholar] [CrossRef]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. NF-κB activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef]
- Bailey, S.T.; Miron, P.L.; Choi, Y.J.; Kochupurakkal, B.; Maulik, G.; Rodig, S.J.; Tian, R.; Foley, K.M.; Bowman, T.; Miron, A.; et al. NF-κB activation-induced anti-apoptosis renders HER2-positive cells drug resistant and accelerates tumor growth. Mol. Cancer Res. 2014, 12, 408–420. [Google Scholar] [CrossRef]
- Chen, Y.J.; Yeh, M.H.; Yu, M.C.; Wei, Y.L.; Chen, W.S.; Chen, J.Y.; Shih, C.Y.; Tu, C.Y.; Chen, C.H.; Hsia, T.C.; et al. Lapatinib-induced NF-kappaB activation sensitizes triple-negative breast cancer cells to proteasome inhibitors. Breast Cancer Res. 2013, 15. [Google Scholar] [CrossRef]
- Zoi, I.; Karamouzis, M.V.; Adamopoulos, C.; Papavassiliou, A.G. RANKL Signaling and ErbB Receptors in Breast Carcinogenesis. Trends Mol. Med. 2016, 22, 839–850. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, J.C.; Chung, P.E.D.; Egan, S.E.; Zacksenhaus, E. Targeting HER2+ breast cancer: The TBK1/IKKε axis. Oncoscience 2014, 1, 180–182. [Google Scholar] [CrossRef]
- Zoi, I.; Karamouzis, M.V.; Xingi, E.; Sarantis, P.; Thomaidou, D.; Lembessis, P.; Theocharis, S.; Papavassiliou, A.G. Combining RANK/RANKL and ERBB-2 targeting as a novel strategy in ERBB-2-positive breast carcinomas. Breast Cancer Res. 2019, 21. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current understanding of RANK signaling in osteoclast differentiation and maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar]
- Zhou, B.P.; Hu, M.C.T.; Miller, S.A.; Yu, Z.; Xia, W.; Lin, S.Y.; Hung, M.C. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF- κB pathway. J. Biol. Chem. 2000, 275, 8027–8031. [Google Scholar] [CrossRef]
- Guo, G.; Wang, T.; Gao, Q.; Tamae, D.; Wong, P.; Chen, T.; Chen, W.C.; Shively, J.E.; Wong, J.Y.C.; Li, J.J. Expression of ErbB2 enhances radiation-induced NF-κB activation. Oncogene 2004, 23, 535–545. [Google Scholar] [CrossRef]
- Liu, M.; Ju, X.; Willmarth, N.E.; Casimiro, M.C.; Ojeifo, J.; Sakamaki, T.; Katiyar, S.; Jiao, X.; Popov, V.M.; Yu, Z.; et al. Nuclear factor-κB enhances ErbB2-induced mammary tumorigenesis and neoangiogenesis in vivo. Am. J. Pathol. 2009, 174, 1910–1920. [Google Scholar] [CrossRef]
- Ma, C.; Zuo, W.; Wang, X.; Wei, L.; Guo, Q.; Song, X. Lapatinib inhibits the activation of NF-κB through reducing phosphorylation of IκB-α in breast cancer cells. Oncol. Rep. 2013, 29, 812–818. [Google Scholar] [CrossRef]
- Coleman, R.; Finkelstein, D.M.; Barrios, C.; Martin, M.; Iwata, H.; Hegg, R.; Glaspy, J.; Periañez, A.M.; Tonkin, K.; Deleu, I.; et al. Adjuvant denosumab in early breast cancer (D-CARE): An international, multicentre, randomised, controlled, phase 3 trial. Lancet Oncol. 2020, 21, 60–72. [Google Scholar] [CrossRef]
- Stefanovic, S.; Diel, I.; Sinn, P.; Englert, S.; Hennigs, A.; Mayer, C.; Schott, S.; Wallwiener, M.; Blumenstein, M.; Golatta, M.; et al. Disseminated Tumor Cells in the Bone Marrow of Patients with Operable Primary Breast Cancer: Prognostic Impact in Immunophenotypic Subgroups and Clinical Implication for Bisphosphonate Treatment. Ann. Surg. Oncol. 2016, 23, 757–766. [Google Scholar] [CrossRef]
- Ellis, G.K.; Bone, H.G.; Chlebowski, R.; Paul, D.; Spadafora, S.; Smith, J.; Fan, M.; Jun, S. Randomized trial of denosumab in patients receiving adjuvant aromatase inhibitors for nonmetastatic breast cancer. J. Clin. Oncol. 2008, 26, 4875–4882. [Google Scholar] [CrossRef]
- Fizazi, K.; Bosserman, L.; Gao, G.; Skacel, T.; Markus, R. Denosumab Treatment of Prostate Cancer With Bone Metastases and Increased Urine N-Telopeptide Levels After Therapy With Intravenous Bisphosphonates: Results of a Randomized Phase II Trial. J. Urol. 2009, 182, 509–516. [Google Scholar] [CrossRef]
- Cheng, M.L.; Fong, L. Effects of RANKL-targeted therapy in immunity and cancer. Front. Oncol. 2014, 3, 329. [Google Scholar] [CrossRef]
- Ferrari-Lacraz, S.; Ferrari, S. Do RANKL inhibitors (denosumab) affect inflammation and immunity? Osteoporos. Int. 2011, 22, 435–446. [Google Scholar] [CrossRef]
- Khan, I.S.; Mouchess, M.L.; Zhu, M.L.; Conley, B.; Fasano, K.J.; Hou, Y.; Fong, L.; Su, M.A.; Anderson, M.S. Enhancement of an anti-tumor immune response by transient blockade of central T cell tolerance. J. Exp. Med. 2014, 211, 761–768. [Google Scholar] [CrossRef]
- Guo, S.; Deng, C.X. Effect of stromal cells in tumor microenvironment on metastasis initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef]
- Yang, D.H.; Yang, M.Y. The role of macrophage in the pathogenesis of osteoporosis. Int. J. Mol. Sci. 2019, 20, 2093. [Google Scholar] [CrossRef]
- Green, E.A.; Choi, Y.; Flavell, R.A. Pancreatic lymph node-derived CD4+CD25+ treg cells: Highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity 2002, 16, 183–191. [Google Scholar] [CrossRef]
- Faget, J.; Contat, C.; Zangger, N.; Peters, S.; Meylan, E. RANKL Signaling Sustains Primary Tumor Growth in Genetically Engineered Mouse Models of Lung Adenocarcinoma. J. Thorac. Oncol. 2018, 13, 387–398. [Google Scholar] [CrossRef]
- Schmiedel, B.J.; Grosse-Hovest, L.; Salih, H.R. A “vicious cycle” of NK-cell immune evasion in acute myeloid leukemia mediated by RANKL? Oncoimmunology 2013, 2. [Google Scholar] [CrossRef]
- Schmiedel, B.J.; Nuebling, T.; Steinbacher, J.; Malinovska, A.; Wende, C.M.; Azuma, M.; Schneider, P.; Grosse-Hovest, L.; Salih, H.R. Receptor Activator for NF-κB Ligand in Acute Myeloid Leukemia: Expression, Function, and Modulation of NK Cell Immunosurveillance. J. Immunol. 2013, 190, 821–831. [Google Scholar] [CrossRef]
- Clar, K.L.; Hinterleitner, C.; Schneider, P.; Salih, H.R.; Maurer, S. Inhibition of NK reactivity against solid tumors by platelet-derived RANKL. Cancers (Basel) 2019, 11, 277. [Google Scholar] [CrossRef]
- Li, H.; Hong, S.; Qian, J.; Zheng, Y.; Yang, J.; Yi, Q. Cross talk between the bone and immune systems: Osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood 2010, 116, 210–217. [Google Scholar] [CrossRef]
- Bekker, P.J.; Holloway, D.L.; Rasmussen, A.S.; Murphy, R.; Martin, S.W.; Leese, P.T.; Holmes, G.B.; Dunstan, C.R.; DePaoli, A.M. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J. Bone Miner. Res. 2004, 19, 1059–1066. [Google Scholar] [CrossRef]
- BOOG Study Center—Studie. Available online: https://www.boogstudycenter.nl/studie/287/perideno.html (accessed on 9 August 2020).
- Van Dam, P.A.; Verhoeven, Y.; Jacobs, J.; Wouters, A.; Tjalma, W.; Lardon, F.; Van Den Wyngaert, T.; Dewulf, J.; Smits, E.; Colpaert, C.; et al. RANK-RANKL signaling in cancer of the uterine cervix: A review. Int. J. Mol. Sci. 2019, 20, 2183. [Google Scholar] [CrossRef]
- de Groot, A.F.; Appelman-Dijkstra, N.M.; van der Burg, S.H.; Kroep, J.R. The anti-tumor effect of RANKL inhibition in malignant solid tumors—A systematic review. Cancer Treat. Rev. 2018, 62, 18–28. [Google Scholar] [CrossRef]
- Thallinger, C.; Füreder, T.; Preusser, M.; Heller, G.; Müllauer, L.; Höller, C.; Prosch, H.; Frank, N.; Swierzewski, R.; Berger, W.; et al. Review of cancer treatment with immune checkpoint inhibitors: Current concepts, expectations, limitations and pitfalls. Wien. Klin. Wochenschr. 2018, 130, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Sarantis, P.; Papavassiliou, A.G.; Karamouzis, M.V. The resistance mechanisms of checkpoint inhibitors in solid tumors. Biomolecules 2020, 10, 666. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.L.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Yagita, H.; Mcarthur, G.A. Combination anti-CTLA-4 and Anti-RANKL in metastatic melanoma. J. Clin. Oncol. 2014, 32. [Google Scholar] [CrossRef]
- Ahern, E.; Harjunpää, H.; Barkauskas, D.; Allen, S.; Takeda, K.; Yagita, H.; Wyld, D.; Dougall, W.C.; Teng, M.W.L.; Smyth, M.J. Co-administration of RANKL and CTLA4 antibodies enhances lymphocyte-mediated antitumor immunity in mice. Clin. Cancer Res. 2017, 23, 5789–5801. [Google Scholar] [CrossRef]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.Z.; Shirai, K. Immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy alone versus immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy in combination with anti-RANKL denosumuab in malignant melanoma: A retrospective analysis at a tertiary care center. Melanoma Res. 2018, 28, 341–347. [Google Scholar] [CrossRef]
- Evaluation of Denosumab in Combination with Immune Checkpoint Inhibitors in Patients with Unresectable or Metastatic Melanoma-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03161756 (accessed on 9 August 2020).
- Denosumab and Pembrolizumab in Clear Cell Renal Carcinoma-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03280667 (accessed on 9 August 2020).
- Ahern, E.; Smyth, M.J.; Dougall, W.C.; Teng, M.W.L. Roles of the RANKL–RANK axis in antitumour immunity—Implications for therapy. Nat. Rev. Clin. Oncol. 2018, 15, 676–693. [Google Scholar] [CrossRef]
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603. [Google Scholar] [CrossRef]
- Ahern, E.; Harjunpää, H.; O’Donnell, J.S.; Allen, S.; Dougall, W.C.; Teng, M.W.L.; Smyth, M.J. RANKL blockade improves efficacy of PD1-PD-L1 blockade or dual PD1-PD-L1 and CTLA4 blockade in mouse models of cancer. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef]
- Dougall, W.C.; Aguilera, A.R.; Smyth, M.J. Dual targeting of RANKL and PD-1 with a bispecific antibody improves anti-tumor immunity. Clin. Transl. Immunol. 2019, 8. [Google Scholar] [CrossRef]


{kind=link}
| Study Number | Intervention | Phase | Cancer Type | Status |
|---|---|---|---|---|
| NCT03324932 | Denosumab | III | Breast cancer | Recruiting |
| NCT02900469 | Denosumab and surgery | I | Breast cancer | Recruiting |
| NCT01077154 | Denosumab, placebo | III | Breast cancer | Terminated, has results [63] |
| NCT02366130 | Ra-223 dichloride, denosumab, hormone therapy | II | Breast cancer | Active, not recruiting |
| NCT01545648 | Denosumab | II | Breast cancer | Terminated, has results [64] |
| NCT00556374 | Placebo, denosumab, nonsteroidal aromatase inhibitor therapy, zoledronic Acid | III | Breast cancer | Active, not recruiting, has results |
| NCT00091832 | Denosumab, IV bisphosphonates | II | Breast cancer | Completed |
| NCT03691311 | Denosumab | I | Breast cancer | Recruiting |
| NCT00089661 | Placebo, AMG 162/denosumab | III | Breast cancer | Completed, has Results [65] |
| NCT03629717 | Denosumab, calcium, vitamin D | I | Breast cancer prevention | Completed |
| NCT02613416 | Denosumab | II | Breast cancer | Recruiting |
| NCT01419717 | Denosumab | III | Bone metastasis | Completed |
| NCT02051218 | Denosumab (reduced/standard dose) | III | Metastatic disease | Recruiting |
| NCT02721433 | Pamidronate, denosumab, zoledronic acid | IV | Metastatic disease | Active, not recruiting |
| NCT00950911 | AMG 162 (denosumab) | III | Bone metastasis | Completed |
| NCT00104650 | AMG 162 | II | Bone metastasis | Completed, has results [13,66] |
| Study Number | Intervention | Phase | Cancer Type |
|---|---|---|---|
| NCT03161756 | Denosumab, nivolumab, ipilimumab | I/II | Melanoma |
| NCT03280667 | Pembrolizumab plus denosumab | II | Renal cell carcinoma, clear cell metastatic kidney cancer |
| NCT03620019 | Denosumab, pembrolizumab, nivolumab | II | Melanoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simatou, A.; Sarantis, P.; Koustas, E.; Papavassiliou, A.G.; Karamouzis, M.V. The Role of the RANKL/RANK Axis in the Prevention and Treatment of Breast Cancer with Immune Checkpoint Inhibitors and Anti-RANKL. Int. J. Mol. Sci. 2020, 21, 7570. https://doi.org/10.3390/ijms21207570
Simatou A, Sarantis P, Koustas E, Papavassiliou AG, Karamouzis MV. The Role of the RANKL/RANK Axis in the Prevention and Treatment of Breast Cancer with Immune Checkpoint Inhibitors and Anti-RANKL. International Journal of Molecular Sciences. 2020; 21(20):7570. https://doi.org/10.3390/ijms21207570
Chicago/Turabian StyleSimatou, Aristofania, Panagiotis Sarantis, Evangelos Koustas, Athanasios G. Papavassiliou, and Michalis V. Karamouzis. 2020. "The Role of the RANKL/RANK Axis in the Prevention and Treatment of Breast Cancer with Immune Checkpoint Inhibitors and Anti-RANKL" International Journal of Molecular Sciences 21, no. 20: 7570. https://doi.org/10.3390/ijms21207570
APA StyleSimatou, A., Sarantis, P., Koustas, E., Papavassiliou, A. G., & Karamouzis, M. V. (2020). The Role of the RANKL/RANK Axis in the Prevention and Treatment of Breast Cancer with Immune Checkpoint Inhibitors and Anti-RANKL. International Journal of Molecular Sciences, 21(20), 7570. https://doi.org/10.3390/ijms21207570