HGF/MET Signaling in Malignant Brain Tumors

Abstract
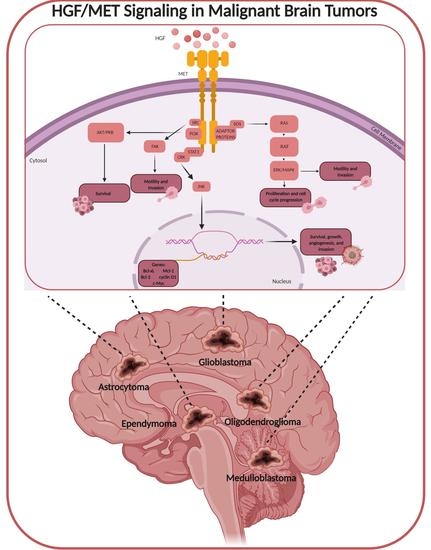
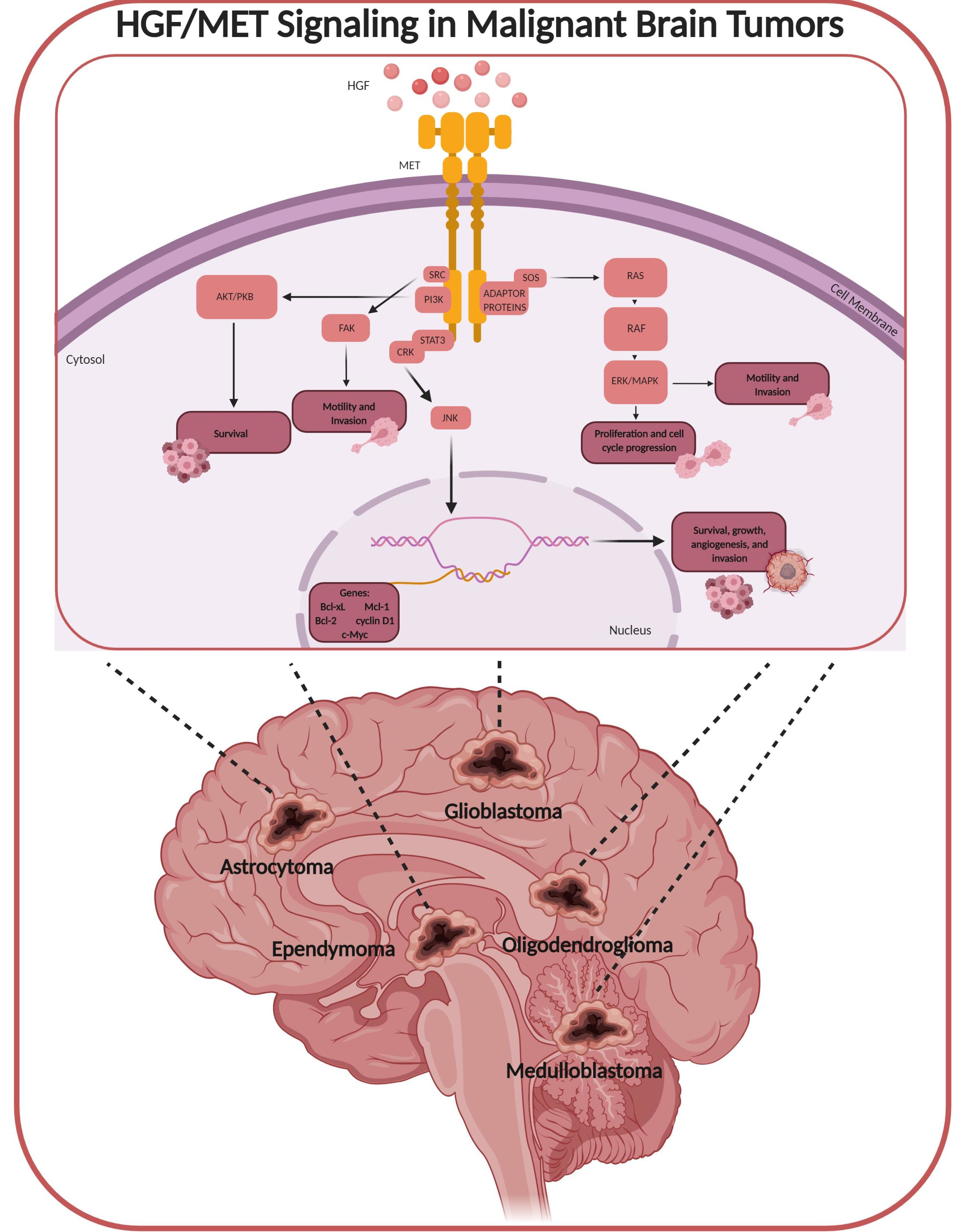{kind=link}
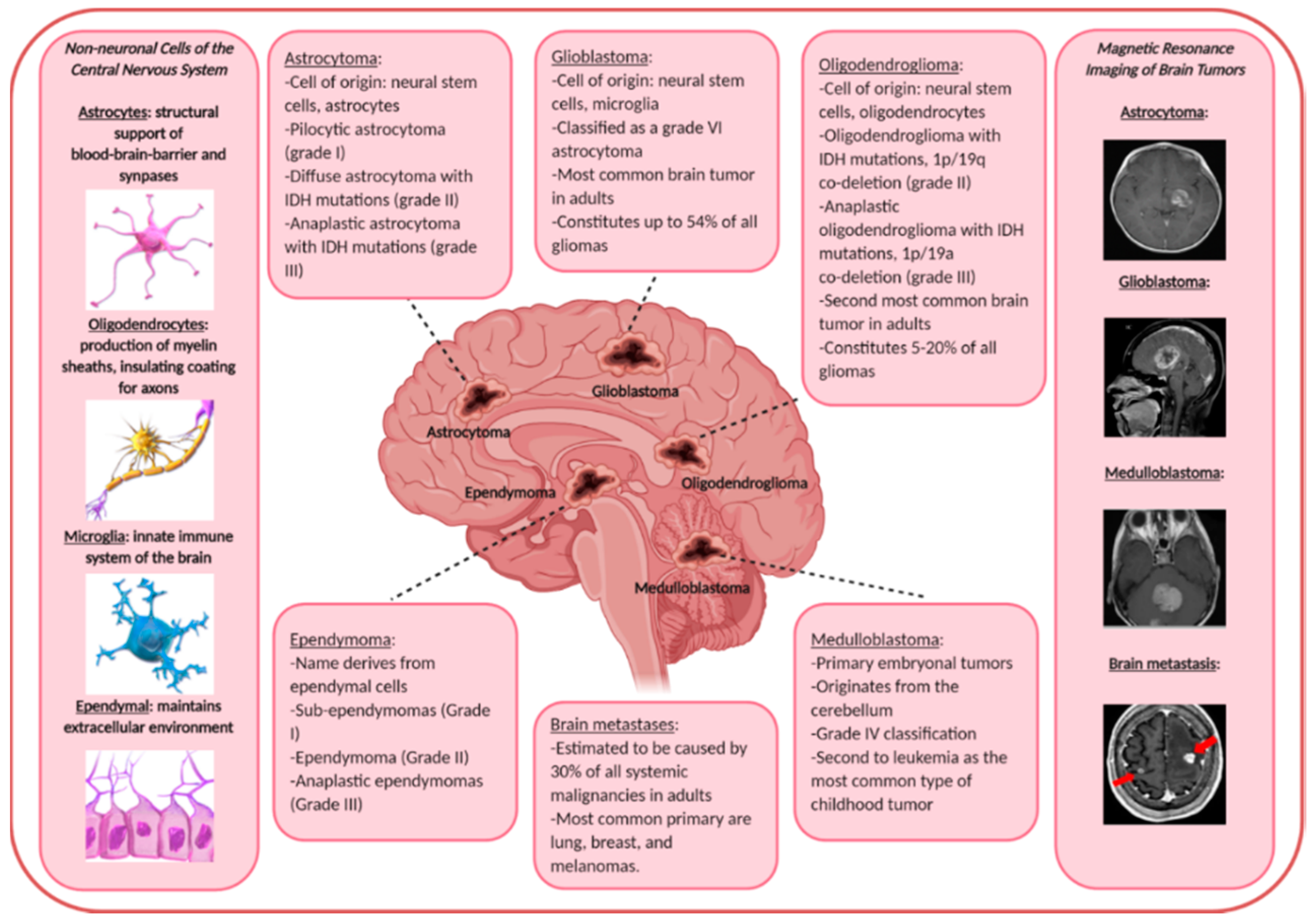{kind=link}
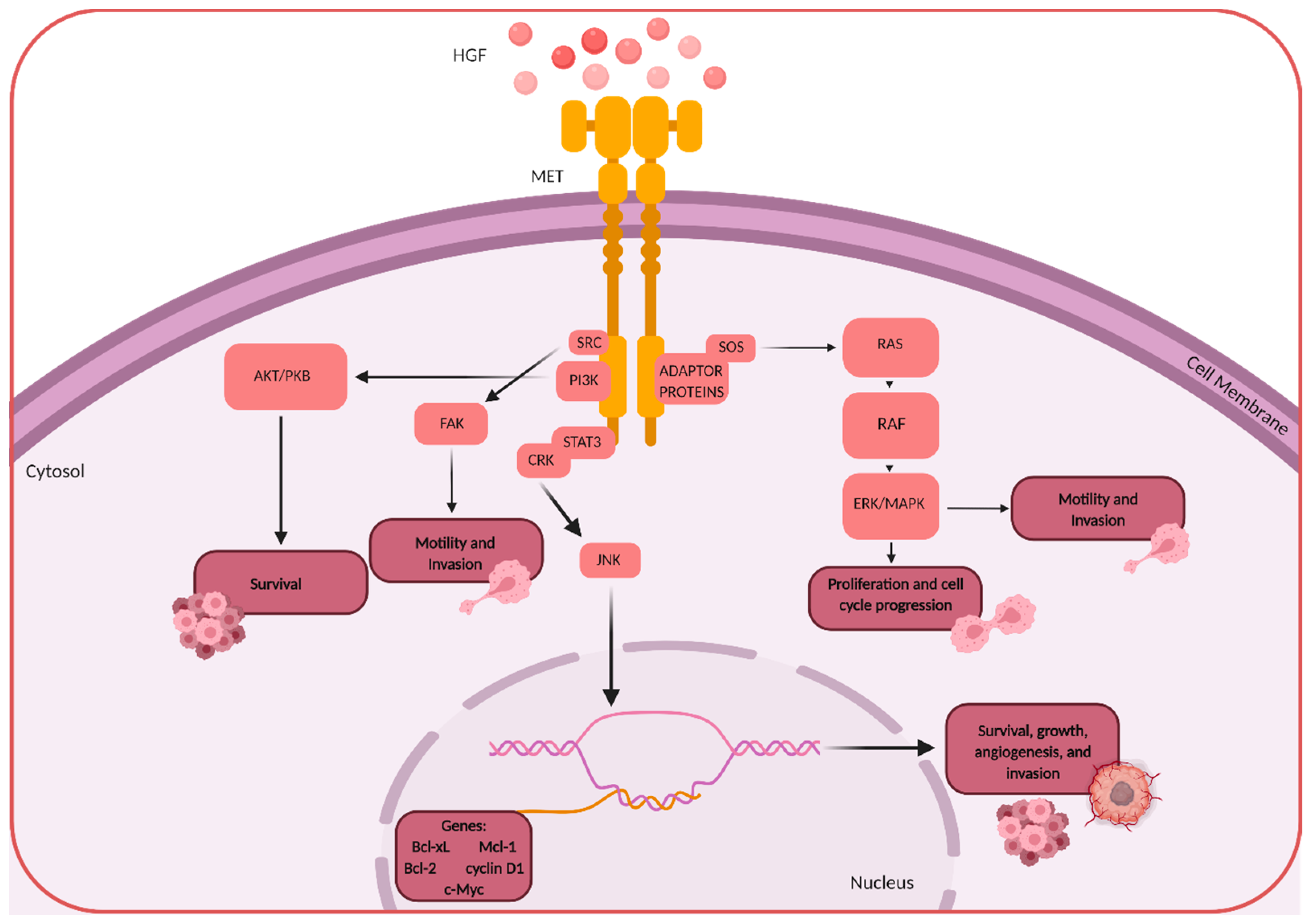{kind=link}
1. HGF/MET Signaling
1.1. Embryonic Development
1.2. Tissue Regeneration and Wound Healing
2. Malignant Brain Tumors
2.1. Primary Malignant Brain Tumors
2.2. Secondary Malignant Brain Tumors (Brain Metastasis)
3. HGF/MET in Brain Tumors
Angiogenesis
4. HGF/MET in Primary Malignant Brain Tumors
4.1. Astrocytomas
4.2. Glioblastomas
4.3. Oligodendrogliomas
4.4. Ependymomas
4.5. Embryonal Central Nervous System Tumors
5. HGF/MET in Brain Metastases
6. HGF/MET Targeted Therapies
6.1. Monoclonal Antibodies
6.2. Small-Molecule Inhibitors
6.3. Delivery Strategies to Cross the BBB
6.3.1. Chemical Stimuli to Create Openings
6.3.2. Physical Stimuli to Create Openings
7. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nakamura, T.; Nawa, K.; Ichihara, A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem. Biophys. Res. Commun. 1984, 122, 1450–1459. [Google Scholar] [PubMed]
- Russell, W.E.; McGowan, J.A.; Bucher, N.L. Partial characterization of a hepatocyte growth factor from rat platelets. J. Cell. Physiol. 1984, 119, 183–192. [Google Scholar] [PubMed]
- Stoker, M.; Gherardi, E.; Perryman, M.; Gray, J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987, 327, 239–242. [Google Scholar] [PubMed]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nat. Cell Biol. 1989, 342, 440–443. [Google Scholar] [CrossRef]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Woude, G.F.V.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Woude, G.F.V. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Woude, G.V. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Bolanos-Garcia, V.M. MET meet adaptors: Functional and structural implications in downstream signalling mediated by the Met receptor. Mol. Cell. Biochem. 2005, 276, 149–157. [Google Scholar] [CrossRef]
- Uehara, Y.; Minowa, O.; Mori, C.; Shiota, K.; Kuno, J.; Noda, T.; Kitamura, N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nat. Cell Biol. 1995, 373, 702–705. [Google Scholar] [CrossRef]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nat. Cell Biol. 1995, 376, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Maina, F.; Klein, R. Hepatocyte growth factor, a versatile signal for developing neurons. Nat. Neurosci. 1999, 2, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Ebens, A.; Brose, K.; Leonardo, E.D.; Hanson, M.G., Jr.; Bladt, F.; Birchmeier, C.; Barres, B.A.; Tessier-Lavigne, M. Hepatocyte Growth Factor/Scatter Factor Is an Axonal Chemoattractant and a Neurotrophic Factor for Spinal Motor Neurons. Neuron 1996, 17, 1157–1172. [Google Scholar] [CrossRef]
- Ohya, W.; Funakoshi, H.; Kurosawa, T.; Nakamura, T. Hepatocyte growth factor (HGF) promotes oligodendrocyte progenitor cell proliferation and inhibits its differentiation during postnatal development in the rat. Brain Res. 2007, 1147, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Gherardi, E. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol. 1998, 8, 404–410. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nakamura, T. Emerging Multipotent Aspects of Hepatocyte Growth Factor. J. Biochem. 1996, 119, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Mizuno, S. The discovery of Hepatocyte Growth Factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B 2010, 86, 588–610. [Google Scholar] [CrossRef]
- Liu, Y.; Tolbert, E.M.; Lin, L.; Thursby, M.A.; Sun, A.M.; Nakamura, T.; Dworkin, L.D. Up-regulation of hepatocyte growth factor receptor: An amplification and targeting mechanism for hepatocyte growth factor action in acute renal failure. Kidney Int. 1999, 55, 442–453. [Google Scholar] [CrossRef]
- Yoshida, S.; Yamaguchi, Y.; Itami, S.; Yoshikawa, K.; Tabata, Y.; Matsumoto, K.; Nakamura, T. Neutralization of Hepatocyte Growth Factor Leads to Retarded Cutaneous Wound Healing Associated with Decreased Neovascularization and Granulation Tissue Formation. J. Investig. Dermatol. 2003, 120, 335–343. [Google Scholar] [CrossRef]
- Nakamura, T.; Sakai, K.; Nakamura, T.; Matsumoto, K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J. Gastroenterol. Hepatol. 2011, 26, 188–202. [Google Scholar] [CrossRef]
- Kosai, K.-I.; Matsumoto, K.; Nagata, S.; Tsujimoto, Y.; Nakamura, T. Abrogation of Fas-Induced Fulminant Hepatic Failure in Mice by Hepatocyte Growth Factor. Biochem. Biophys. Res. Commun. 1998, 244, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Morishita, R.; Hayashi, S.-I.; Matsushita, H.; Nakagami, H.; Moriguchi, A.; Matsumoto, K.; Nakamura, T.; Kaneda, Y.; Ogihara, T. Contribution of Bcl-2, but Not Bcl-xL and Bax, to Antiapoptotic Actions of Hepatocyte Growth Factor in Hypoxia-Conditioned Human Endothelial Cells. Hypertension 2001, 37, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Funakoshi, H.; Nakamura, T. Overexpression of HGF retards disease progression and prolongs life span in a transgenic mouse model of ALS. J. Neurosci. 2002, 22, 6537–6548. [Google Scholar] [CrossRef] [PubMed]
- Kadoyama, K.; Funakoshi, H.; Ohya, W.; Nakamura, T. Hepatocyte growth factor (HGF) attenuates gliosis and motoneuronal degeneration in the brainstem motor nuclei of a transgenic mouse model of ALS. Neurosci. Res. 2007, 59, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Cahill, D.; Turcan, S. Origin of Gliomas. Semin Neurol. 2018, 38, 5–10. [Google Scholar]
- Kelly, P.J. Gliomas: Survival, origin and early detection. Surg. Neurol. Int. 2010, 1, 96. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1andIDH2Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Szerlip, N.J.; Pedraza, A.; Chakravarty, D.; Azim, M.; McGuire, J.; Fang, Y.; Ozawa, T.; Holland, E.C.; Huse, J.T.; Jhanwar, S.; et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl. Acad. Sci. USA 2012, 109, 3041–3046. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef]
- Huber, J.D.; Egleton, R.D.; Davis, T.P. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001, 24, 719–725. [Google Scholar] [CrossRef]
- Dong, X. Current Strategies for Brain Drug Delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Garg, T.; Bhandari, S.; Rath, G.; Goyal, A.K. Current strategies for targeted delivery of bio-active drug molecules in the treatment of brain tumor. J. Drug Target. 2015, 23, 865–887. [Google Scholar] [CrossRef]
- Sanai, N.; Berger, M.S. Surgical oncology for gliomas: The state of the art. Nat. Rev. Clin. Oncol. 2017, 15, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Chambless, L.B.; Kistka, H.M.; Parker, S.L.; Hassam-Malani, L.; McGirt, M.J.; Thompson, R.C.; Mohammad, L. The relative value of postoperative versus preoperative Karnofsky Performance Scale scores as a predictor of survival after surgical resection of glioblastoma multiforme. J. Neuro-Oncol. 2014, 121, 359–364. [Google Scholar] [CrossRef]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Bush, N.A.; Chang, S.M.; Berger, M.S. Current and future strategies for treatment of glioma. Neurosurg. Rev. 2017, 40, 1–14. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar]
- Llaguno, S.A.; Chen, J.; Kwon, C.-H.; Jackson, E.L.; Li, Y.J.; Burns, D.K.; Buylla, A.A.; Parada, L.F. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 2009, 15, 45–56. [Google Scholar] [CrossRef]
- Goffart, N.; Kroonen, J.; Rogister, B. Glioblastoma-initiating cells: Relationship with neural stem cells and the micro-environment. Cancers 2013, 5, 1049–1071. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nat. Cell Biol. 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, A.; Glas, M.; Lal, B.; Ying, M.; Sang, Y.; Xia, S.; Trageser, D.; Guerrero-Cázares, H.; Eberhart, C.G.; et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci. USA 2011, 108, 9951–9956. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Bent, M.V.D.; Perry, A. Oligodendroglioma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 809–827. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nat. Cell Biol. 2017, 553, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; de Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro Oncol. 2015, 16 (Suppl. 10), x1–x36. [Google Scholar] [CrossRef] [PubMed]
- Millard, N.E.; de Braganca, K.C. Medulloblastoma. J. Child Neurol. 2016, 31, 1341–1353. [Google Scholar] [CrossRef]
- Biswas, A.; Kashyap, L.; Kakkar, A.; Sarkar, C.; Julka, P.K. Atypical teratoid/rhabdoid tumors: Challenges and search for solutions. Cancer Manag. Res. 2016, 8, 115–125. [Google Scholar] [CrossRef]
- AbdelBaki, M.S.; Abu Arja, M.H.; Davidson, T.B.; Fangusaro, J.R.; Stanek, J.R.; Dunkel, I.J.; Dhall, G.; Gardner, S.L.; Finlay, J.L. Pineoblastoma in children less than six years of age: The Head Start I, II, and III experience. Pediatr. Blood Cancer 2020, 67, e28252. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Blausen Medical. Medical gallery of Blausen Medical. WikiJournal Med. 2014, 1, 1–79. [Google Scholar]
- Gallego Perez-Larraya, J.; Hildebrand, J. Brain metastases. Handb. Clin. Neurol. 2014, 121, 1143–1157. [Google Scholar] [PubMed]
- Schouten, L.J.; Rutten, J.; Huveneers, H.A.M.; Twijnstra, A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer 2002, 94, 2698–2705. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, L.M.; Posner, J.B.; Posner, J.B. Neurologic Complications of Cancer, 2nd ed.; Contemporary neurology series; Oxford University Press: Oxford, UK; New York, NY, USA, 2009; 634p. [Google Scholar]
- Kebudi, R.; Ayan, I.; Görgün, Ö.; Agaoglu, F.Y.; Vural, S.; Darendeliler, E. Brain metastasis in pediatric extracranial solid tumors: Survey and literature review. J. Neuro-Oncol. 2005, 71, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Nakamura, T.; Kramer, R.H. Hepatocyte growth factor/scatter factor induces tyrosine phosphorylation of focal adhesion kinase (p125FAK) and promotes migration and invasion by oral squamous cell carcinoma cells. J. Biol. Chem. 1994, 269, 31807–31813. [Google Scholar] [PubMed]
- Matsumoto, K.; Nakamura, T. Hepatocyte growth factor and the Met system as a mediator of tumor-stromal interactions. Int. J. Cancer 2006, 119, 477–483. [Google Scholar] [PubMed]
- Abounader, R.; Laterra, J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol. 2005, 7, 436–451. [Google Scholar] [CrossRef]
- Schmidt, L.; Duh, F.-M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Koochekpour, S.; Jeffers, M.; Rulong, S.; Taylor, G.; Klineberg, E.; Hudson, E.A.; Resau, J.H.; Woude, G.F.V. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997, 57, 5391–5398. [Google Scholar]
- Moriyama, T.; Kataoka, H.; Kawano, H.; Yokogami, K.; Nakano, S.; Goya, T.; Uchino, H.; Koono, M.; Wakisaka, S. Comparative analysis of expression of hepatocyte growth factor and its receptor, c-met, in gliomas, meningiomas and schwannomas in humans. Cancer Lett. 1998, 124, 149–155. [Google Scholar] [CrossRef]
- Rosen, E.M.; Laterra, J.; Joseph, A.; Jin, L.; Fuchs, A.; Way, D.; Witte, M.; Weinand, M.; Goldberg, I.D. Scatter factor expression and regulation in human glial tumors. Int. J. Cancer 1996, 67, 248–255. [Google Scholar] [CrossRef]
- Laterra, J.; Nam, M.; Rosen, E.; Rao, J.S.; Lamszus, K.; Goldberg, I.D.; Johnston, P. Scatter factor/hepatocyte growth factor gene transfer enhances glioma growth and angiogenesis in vivo. Lab. Investig. 1997, 76, 565–577. [Google Scholar] [PubMed]
- Laterra, J.; Rosen, E.; Nam, M.; Ranganathan, S.; Fielding, K.; Johnston, P. Scatter Factor/Hepatocyte Growth Factor Expression Enhances Human Glioblastoma Tumorigenicity and Growth. Biochem. Biophys. Res. Commun. 1997, 235, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Abounader, R.; Lal, B.; Luddy, C.; Koe, G.; Davidson, B.; Rosen, E.M.; Laterra, J. In vivo targeting of SF/HGF and c-met expression via U1snRNA/ribozymes inhibits glioma growth and angiogenesis and promotes apoptosis. FASEB J. 2001, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Abounader, R.; Ranganathan, S.; Lal, B.; Fielding, K.; Book, A.; Dietz, H.; Burger, P.; Laterra, J. Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J. Natl. Cancer Inst. 1999, 91, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.A.; Hossain, M.A.; Luddy, C.; Goel, N.; Reznik, T.E.; Laterra, J. Scatter Factor/Hepatocyte Growth Factor Stimulation of Glioblastoma Cell Cycle Progression through G1 Is c-Myc Dependent and Independent of p27 Suppression, Cdk2 Activation, or E2F1-Dependent Transcription. Mol. Cell. Biol. 2002, 22, 2703–2715. [Google Scholar] [CrossRef] [PubMed]
- Bowers, D.C.; Fan, S.; Walter, K.A.; Abounader, R.; Williams, J.A.; Rosen, E.M.; Laterra, J. Scatter factor/hepatocyte growth factor protects against cytotoxic death in human glioblastoma via phosphatidylinositol 3-kinase- and AKT-dependent pathways. Cancer Res. 2000, 60, 4277–4283. [Google Scholar]
- Owusu, B.Y.; Galemmo, R.; Janetka, J.; Klampfer, L. Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment. Cancers 2017, 9, 35. [Google Scholar] [CrossRef]
- Nayak, L.; Reardon, D.A. High-grade Gliomas. Continuum 2017, 23, 1548–1563. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S.; Yamada, Y.; Murayama, K.; Sandika, E.; Sasaki, H.; Yamada, S.; Abe, M.; Hasegawa, M.; Hirose, Y. c-Met Expression Is a Useful Marker for Prognosis Prediction in IDH-Mutant Lower-Grade Gliomas and IDH-Wildtype Glioblastomas. World Neurosurg 2019, 126, e1042–e1049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Farenholtz, K.E.; Yang, Y.; Guessous, F.; Dipierro, C.G.; Calvert, V.S.; Deng, J.; Schiff, D.; Xin, W.; Lee, J.K.; et al. Hepatocyte growth factor sensitizes brain tumors to c-MET kinase inhibition. Clin. Cancer Res. 2013, 19, 1433–1444. [Google Scholar] [CrossRef]
- Kunkel, P.; Müller, S.; Schirmacher, P.; Stavrou, D.; Fillbrandt, R.; Westphal, M.; Lamszus, K. Expression and localization of scatter factor/hepatocyte growth factor in human astrocytomas. Neuro Oncol. 2001, 3, 82–88. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, S.; Oh, Y.-S.; Chang, H.K.; Kim, Y.S.; Hong, S.H.; Kim, J.Y.; Park, Y.-W.; Lee, S.-J.; Song, S.-W.; et al. Humanized Anti-hepatocyte Growth Factor Monoclonal Antibody (YYB-101) Inhibits Ovarian Cancer Progression. Front. Oncol. 2019, 9, 571. [Google Scholar] [CrossRef]
- Seifert, M.; Garbe, M.; Friedrich, B.; Mittelbronn, M.; Klink, B. Comparative transcriptomics reveals similarities and differences between astrocytoma grades. BMC Cancer 2015, 15, 952. [Google Scholar] [CrossRef]
- Collins, V.P. Cellular mechanisms targeted during astrocytoma progression. Cancer Lett. 2002, 188, 1–7. [Google Scholar] [CrossRef]
- Reznik, T.E.; Sang, Y.; Ma, Y.; Abounader, R.; Rosen, E.M.; Xia, S.; Laterra, J. Transcription-Dependent Epidermal Growth Factor Receptor Activation by Hepatocyte Growth Factor. Mol. Cancer Res. 2008, 6, 139–150. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gürsel, D.B.; Connell-Albert, Y.S.; Tuskan, R.G.; Anastassiadis, T.; Walrath, J.C.; Hawes, J.J.; Schaick, J.C.A.-V.; Reilly, K.M. Control of proliferation in astrocytoma cells by the receptor tyrosine kinase/PI3K/AKT signaling axis and the use of PI-103 and TCN as potential anti-astrocytoma therapies. Neuro-Oncology 2011, 13, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma; Codon Publications: Singapore, 2017; pp. 143–153. [Google Scholar]
- Mao, H.; Lebrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated Signaling Pathways in Glioblastoma Multiforme: Molecular Mechanisms and Therapeutic Targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Tabouret, E.; Denicolai, E.; Delfino, C.; Graillon, T.; Boucard, C.; Nanni, I.; Padovani, L.; Figarella-Branger, D.; Chinot, O.L. Changes in PlGF and MET-HGF expressions in paired initial and recurrent glioblastoma. J. Neuro-Oncol. 2016, 130, 431–437. [Google Scholar] [CrossRef]
- Eder, J.P.; Woude, G.F.V.; Boerner, S.A.; Lorusso, P.M.; Maréchal, R.; Mackey, J.R.; Lai, R.; Demetter, P.; Peeters, M.; Polus, M.; et al. Novel Therapeutic Inhibitors of the c-Met Signaling Pathway in Cancer. Clin. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef]
- Cruickshanks, N.; Zhang, Y.; Yuan, F.; Pahuski, M.; Gibert, J.M.; Abounader, R. Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers 2017, 9, 87. [Google Scholar] [CrossRef]
- Cruickshanks, N.; Zhang, Y.; Hine, S.; Gibert, M.; Yuan, F.; Oxford, M.; Grello, C.; Pahuski, M.; Dube, C.; Guessous, F.; et al. Discovery and Therapeutic Exploitation of Mechanisms of Resistance to MET Inhibitors in Glioblastoma. Clin. Cancer Res. 2019, 25, 663–673. [Google Scholar] [CrossRef]
- Jiang, K.; Yao, G.; Hu, L.; Yan, Y.; Liu, J.; Shi, J.; Chang, Y.; Zhang, Y.; Liang, D.; Shen, D.; et al. MOB2 suppresses GBM cell migration and invasion via regulation of FAK/Akt and cAMP/PKA signaling. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Cahill, D.P.; Louis, D.N.; Cairncross, J.G. Molecular background of oligodendroglioma: 1p/19q, IDH, TERT, CIC and FUBP1. CNS Oncol. 2015, 4, 287–294. [Google Scholar] [CrossRef]
- Smits, M. Imaging of oligodendroglioma. Br. J. Radiol. 2016, 89, 20150857. [Google Scholar] [CrossRef]
- Pierscianek, D.; Kim, Y.-H.; Motomura, K.; Mittelbronn, M.; Paulus, W.; Brokinkel, B.; Keyvani, K.; Wrede, K.; Nakazato, Y.; Tanaka, Y.; et al. MET gain in diffuse astrocytomas is associated with poorer outcome. Brain Pathol. 2013, 23, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Baliga, S.; Gandola, L.; Timmermann, B.; Gail, H.; Padovani, L.; Janssens, G.O.; Yock, T.I. Brain tumors: Medulloblastoma, ATRT, ependymoma. Pediatr. Blood Cancer 2020, e28395. [Google Scholar] [CrossRef]
- Wu, J.; Armstrong, T.S.; Gilbert, M.R. Biology and management of ependymomas. Neuro-Oncology 2016, 18, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Sie, M.; Dunnen, W.F.A.D.; Lourens, H.J.; Boer, T.G.J.M.-D.; Scherpen, F.J.G.; Zomerman, W.W.; Kampen, K.R.; Hoving, E.W.; De Bont, E.S.J.M. Growth-Factor-Driven Rescue to Receptor Tyrosine Kinase (RTK) Inhibitors through Akt and Erk Phosphorylation in Pediatric Low Grade Astrocytoma and Ependymoma. PLoS ONE 2015, 10, e0122555. [Google Scholar] [CrossRef]
- Weiner, H.L.; Rothman, M.; Miller, D.C.; Ziff, E.B. Pediatric Brain Tumors Express Multiple Receptor Tyrosine Kinases Including Novel Cell Adhesion Kinases. Pediatr. Neurosurg. 1996, 25, 64–72. [Google Scholar] [CrossRef]
- Odagiri, K.; Omura, M.; Hata, M.; Aida, N.; Niwa, T.; Goto, H.; Ito, S.; Adachi, M.; Yoshida, H.; Yuki, H.; et al. Treatment outcomes and late toxicities in patients with embryonal central nervous system tumors. Radiat. Oncol. 2014, 9, 201. [Google Scholar] [CrossRef][Green Version]
- Bartlett, F.; Kortmann, R.; Saran, F. Medulloblastoma. Clin. Oncol. 2013, 25, 36–45. [Google Scholar] [CrossRef]
- Quinlan, A.; Rizzolo, D. Understanding medulloblastoma. J. Am. Acad. Physician Assist. 2017, 30, 30–36. [Google Scholar] [CrossRef]
- Kongkham, P.N.; Onvani, S.; Smith, C.A.; Rutka, J.T. Inhibition of the MET Receptor Tyrosine Kinase as a Novel Therapeutic Strategy in Medulloblastoma. Transl. Oncol. 2010, 3, 336–343. [Google Scholar] [CrossRef]
- Li, Y.; Lal, B.; Kwon, S.; Fan, X.; Saldanha, U.; Reznik, T.E.; Kuchner, E.B.; Eberhart, C.; Laterra, J.; Abounader, R. The Scatter Factor/Hepatocyte Growth Factor: C-Met Pathway in Human Embryonal Central Nervous System Tumor Malignancy. Cancer Res. 2005, 65, 9355–9362. [Google Scholar] [CrossRef]
- Ding, S.; Merkulova-Rainon, T.; Han, Z.; Tobelem, G. HGF receptor up-regulation contributes to the angiogenic phenotype of human endothelial cells and promotes angiogenesis in vitro. Blood 2003, 101, 4816–4822. [Google Scholar] [CrossRef] [PubMed]
- Provençal, M.; Labbé, D.P.; Veitch, R.; Boivin, D.; Rivard, G.-É.; Sartelet, H.; Robitaille, Y.; Gingras, D.; Béliveau, R. c-Met activation in medulloblastoma induces tissue factor expression and activity: Effects on cell migration. Carcinog. 2009, 30, 1089–1096. [Google Scholar] [CrossRef]
- Bertolini, F.; Spallanzani, A.; Fontana, A.; Depenni, R.; Luppi, G. Brain metastases: An overview. CNS Oncol. 2015, 4, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Gaziel-Sovran, A.; Osman, I.; Hernando, E. In vivo Modeling and Molecular Characterization: A Path Toward Targeted Therapy of Melanoma Brain Metastasis. Front. Oncol. 2013, 3, 127. [Google Scholar] [CrossRef]
- Demkova, L.; Kucerova, L. Role of the HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma. Mol. Cancer 2018, 17, 26. [Google Scholar] [CrossRef]
- Yang, H.; Lee, H.W.; Kim, Y.; Lee, Y.; Choi, Y.-S.; Kim, K.H.; Jin, J.; Lee, J.; Joo, K.M.; Nam, D.-H. Radiosensitization of brain metastasis by targeting c-MET. Lab. Investig. 2013, 93, 344–353. [Google Scholar] [CrossRef]
- Lee, S.J.; Seol, H.J.; Lee, H.W.; Kang, W.Y.; Kang, B.G.; Jin, J.; Jo, M.-Y.; Jin, Y.; Lee, J.-I.; Joo, K.M.; et al. Gene silencing of c-Met leads to brain metastasis inhibitory effects. Clin. Exp. Metastasis 2013, 30, 845–854. [Google Scholar] [CrossRef]
- Mizuno, S.; Nakamura, T. HGF-MET cascade, a key target for inhibiting cancer metastasis: The impact of NK4 discovery on cancer biology and therapeutics. Int. J. Mol. Sci. 2013, 14, 888–919. [Google Scholar] [CrossRef]
- Navis, A.C.; Van Lith, S.A.M.; Van Duijnhoven, S.M.J.; De Pooter, M.; Yetkin-Arik, B.; Wesseling, P.; Hendriks, W.J.; Venselaar, H.; Timmer, M.; Van Cleef, P.; et al. Identification of a novel MET mutation in high-grade glioma resulting in an auto-active intracellular protein. Acta Neuropathol. 2015, 130, 131–144. [Google Scholar] [CrossRef]
- Prat, M.; Oltolina, F.; Basilico, C. Monoclonal Antibodies against the MET/HGF Receptor and Its Ligand: Multitask Tools with Applications from Basic Research to Therapy. Biomedicines 2014, 2, 359–383. [Google Scholar]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients With Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O6-Methylguanine–DNA Methyltransferase Biomarker Analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Adjei, A.A. In the clinic: Ongoing clinical trials evaluating c-MET-inhibiting drugs. Ther. Adv. Med. Oncol. 2011, 3 (Suppl. 1), S37–S50. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.S.; Sweeney, C.J.; Mendelson, D.S.; Eckhardt, S.G.; Anderson, A.; Beaupre, D.M.; Branstetter, D.; Burgess, T.L.; Coxon, A.; Deng, H.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of AMG 102, a Fully Human Hepatocyte Growth Factor-Neutralizing Monoclonal Antibody, in a First-in-Human Study of Patients with Advanced Solid Tumors. Clin. Cancer Res. 2010, 16, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Chekhonin, I.V.; Leopold, A.V.; Gurina, O.I.; Semenova, A.V. Monoclonal antibodies in high-grade gliomas. Ann. Russ. Acad. Med. Sci. 2014, 69, 131–139. [Google Scholar] [CrossRef][Green Version]
- Strickler, J.H.; Lorusso, P.M.; Salgia, R.; Kang, Y.-K.; Yen, C.J.; Lin, C.-C.; Ansell, P.; Motwani, M.; Wong, S.; Yue, H.; et al. Phase I Dose-Escalation and -Expansion Study of Telisotuzumab (ABT-700), an Anti–c-Met Antibody, in Patients with Advanced Solid Tumors. Mol. Cancer Ther. 2020, 19, 1210–1217. [Google Scholar] [CrossRef]
- Rosen, L.S.; Goldman, J.; Algazi, A.P.; Turner, P.K.; Moser, B.; Hu, T.; Wang, X.A.; Tuttle, J.; Wacheck, V.; Wooldridge, J.E.; et al. A First-in-Human Phase I Study of a Bivalent MET Antibody, Emibetuzumab (LY2875358), as Monotherapy and in Combination with Erlotinib in Advanced Cancer. Clin. Cancer Res. 2016, 23, 1910–1919. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Cui, J.J. Targeting receptor tyrosine kinase MET in cancer: Small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453. [Google Scholar] [CrossRef]
- Hughes, P.E.; Rex, K.; Caenepeel, S.; Yang, Y.; Zhang, Y.; Broome, M.A.; Kha, H.T.; Burgess, T.L.; Amore, B.; Kaplan-Lefko, P.J.; et al. In Vitro and In Vivo Activity of AMG 337, a Potent and Selective MET Kinase Inhibitor, in MET-Dependent Cancer Models. Mol. Cancer Ther. 2016, 15, 1568–1579. [Google Scholar] [CrossRef]
- Hu, H.; Mu, Q.; Bao, Z.; Chen, Y.; Liu, Y.; Chen, J.; Wang, K.; Wang, Z.; Nam, Y.; Jiang, B.; et al. Mutational Landscape of Secondary Glioblastoma Guides MET-Targeted Trial in Brain Tumor. Cell 2018, 175, 1665–1678.e18. [Google Scholar] [CrossRef]
- Liu, L.; Norman, M.H.; Lee, M.; Xi, N.; Siegmund, A.; Boezio, A.A.; Booker, S.; Choquette, D.; D’Angelo, N.D.; Germain, J.; et al. Structure-Based Design of Novel Class II c-Met Inhibitors: 2. SAR and Kinase Selectivity Profiles of the Pyrazolone Series. J. Med. Chem. 2012, 55, 1868–1897. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Q.; Yang, G.; Marando, C.; Koblish, H.K.; Hall, L.M.; Fridman, J.S.; Behshad, E.; Wynn, R.; Li, Y.; et al. A Novel Kinase Inhibitor, INCB28060, Blocks c-MET-Dependent Signaling, Neoplastic Activities, and Cross-Talk with EGFR and HER-3. Clin. Cancer Res. 2011, 17, 7127–7138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Feng, Q.; Chen, W.-D.; Wang, Y.-D. HGF/c-MET: A Promising Therapeutic Target in the Digestive System Cancers. Int. J. Mol. Sci. 2018, 19, 3295. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood-brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef]
- Cardoso, F.L.; Brites, D.; Brito, M.A. Looking at the blood-brain barrier: Molecular anatomy and possible investigation approaches. Brain Res. Rev. 2010, 64, 328–363. [Google Scholar] [CrossRef]
- Karyekar, C.S.; Fasano, A.; Raje, S.; Lu, R.; Dowling, T.C.; Eddington, N.D. Zonula Occludens Toxin Increases the Permeability of Molecular Weight Markers and Chemotherapeutic Agents Across the Bovine Brain Microvessel Endothelial Cells. J. Pharm. Sci. 2003, 92, 414–423. [Google Scholar] [CrossRef]
- Abbott, N.J. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell. Mol. Neurobiol. 2000, 20, 131–147. [Google Scholar] [CrossRef]
- Vajkoczy, P.; Menger, M.D. Vascular microenvironment in gliomas. Cancer Treat Res. 2004, 117, 249–262. [Google Scholar]
- Sanovich, E.; Bartus, R.T.; Friden, P.M.; Dean, R.L.; Le, H.Q.; Brightman, M.W. Pathway across blood-brain barrier opened by the bradykinin agonist, RMP-7. Brain Res. 1995, 705, 125–135. [Google Scholar] [CrossRef]
- Saija, A.; Princi, P.; Trombetta, D.; Lanza, M.; De Pasquale, A. Changes in the permeability of the blood-brain barrier following sodium dodecyl sulphate administration in the rat. Exp. Brain Res. 1997, 115, 546–551. [Google Scholar] [CrossRef]
- Hynynen, K. Ultrasound for drug and gene delivery to the brain. Adv. Drug Deliv. Rev. 2008, 60, 1209–1217. [Google Scholar] [PubMed]
- Stam, R. Electromagnetic fields and the blood-brain barrier. Brain Res. Rev. 2010, 65, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Vykhodtseva, N.; McDannold, N.; Hynynen, K. Progress and problems in the application of focused ultrasound for blood-brain barrier disruption. Ultrasonics 2008, 48, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR Imaging–guided Focal Opening of the Blood-Brain Barrier in Rabbits. Radiology 2001, 220, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Al Sawaftah, N.M.; Husseini, G.A. Ultrasound-Mediated Drug Delivery in Cancer Therapy: A Review. J. Nanosci. Nanotechnol. 2020, 20, 7211–7230. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.-B.; Ding, G.-R.; Li, K.-C.; Wang, X.; Zhou, Y.; Zhou, Y.-C.; Li, Y.-R.; Guo, G.-Z. The role of protein kinase C in the opening of blood–brain barrier induced by electromagnetic pulse. Toxicology 2010, 273, 29–34. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mulcahy, E.Q.X.; Colόn, R.R.; Abounader, R. HGF/MET Signaling in Malignant Brain Tumors. Int. J. Mol. Sci. 2020, 21, 7546. https://doi.org/10.3390/ijms21207546
Mulcahy EQX, Colόn RR, Abounader R. HGF/MET Signaling in Malignant Brain Tumors. International Journal of Molecular Sciences. 2020; 21(20):7546. https://doi.org/10.3390/ijms21207546
Chicago/Turabian StyleMulcahy, Elizabeth Qian Xu, Rossymar Rivera Colόn, and Roger Abounader. 2020. "HGF/MET Signaling in Malignant Brain Tumors" International Journal of Molecular Sciences 21, no. 20: 7546. https://doi.org/10.3390/ijms21207546
APA StyleMulcahy, E. Q. X., Colόn, R. R., & Abounader, R. (2020). HGF/MET Signaling in Malignant Brain Tumors. International Journal of Molecular Sciences, 21(20), 7546. https://doi.org/10.3390/ijms21207546