Activation of the VEGF-A/ERK/PLA2 Axis Mediates Early Retinal Endothelial Cell Damage Induced by High Glucose: New Insight from an In Vitro Model of Diabetic Retinopathy

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
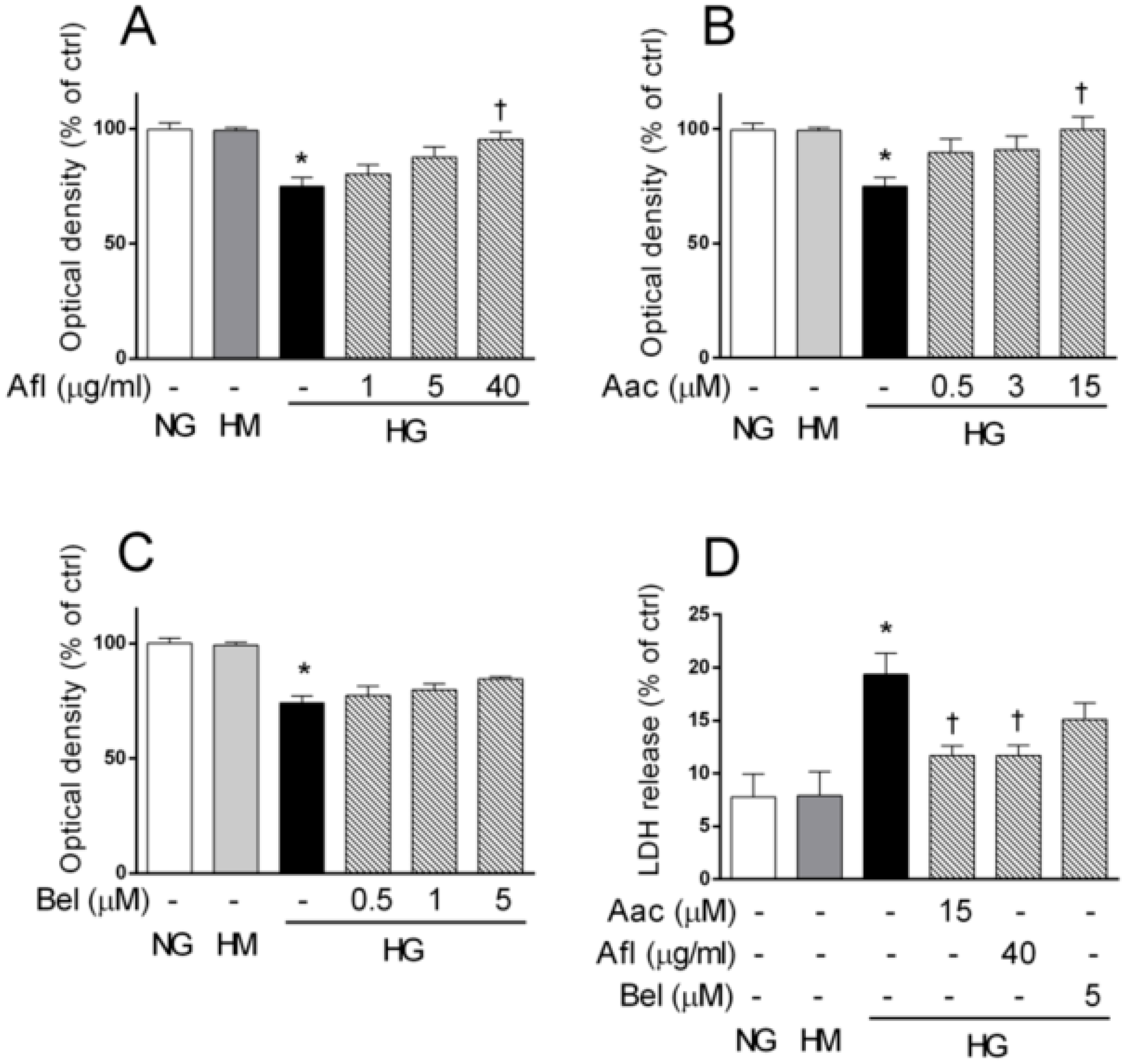
2.1. Effects of Aflibercept and PLA2 Inhibitors on Retinal Endothelial Cell Damage Induced by HG
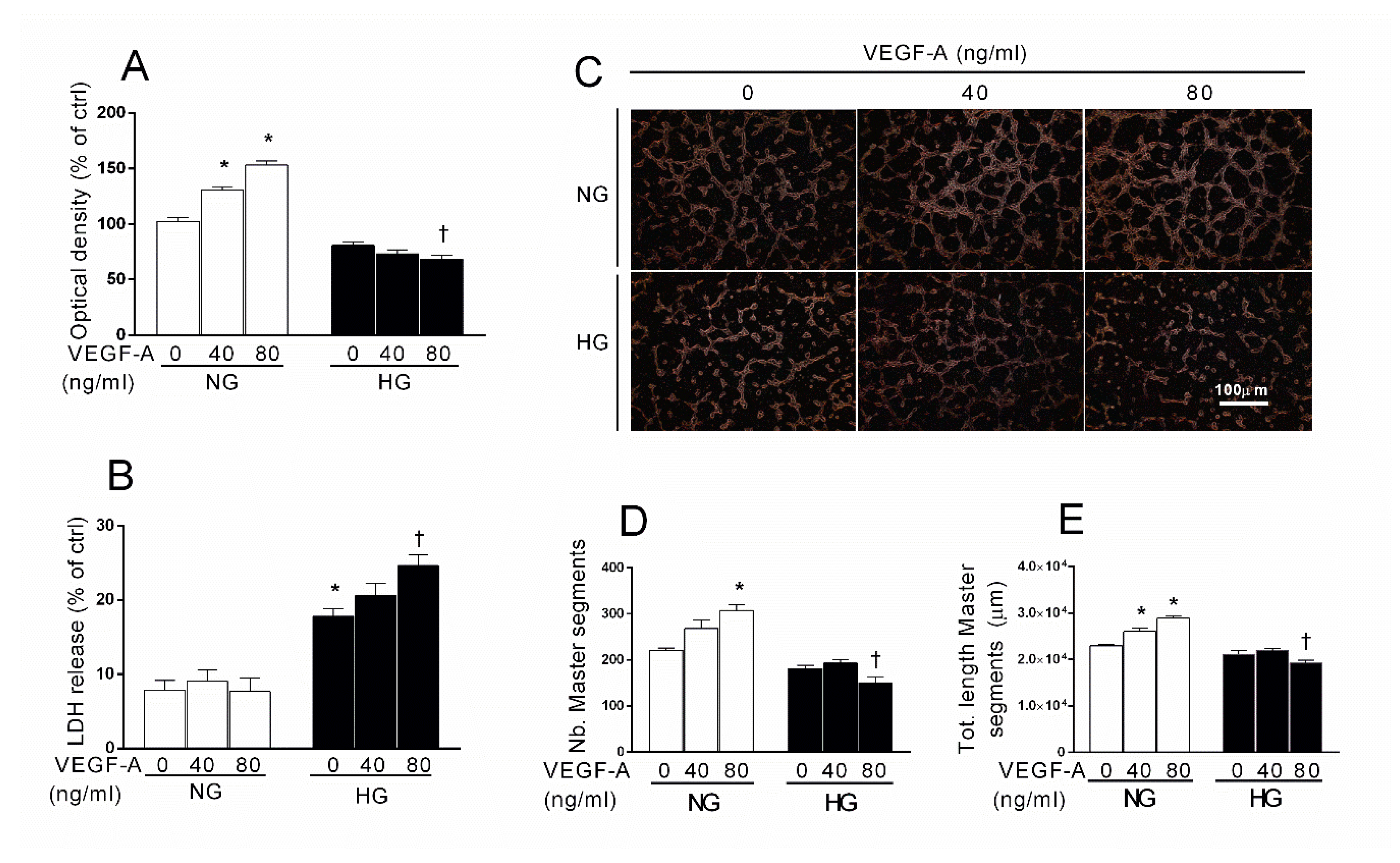
2.2. Effect of Aflibercept and the cPLA2 Inhibitor AACOCF3 on Tube-Like Structure Formation in HRECs Impaired by HG
2.3. Effects of Aflibercept and PLA2 Inhibitors on VEGF-A/PLA2 Axis Activation in HRECs Stimulated by HG
2.4. Effects of Exogenous VEGF-A in HG-Stimulated HREC
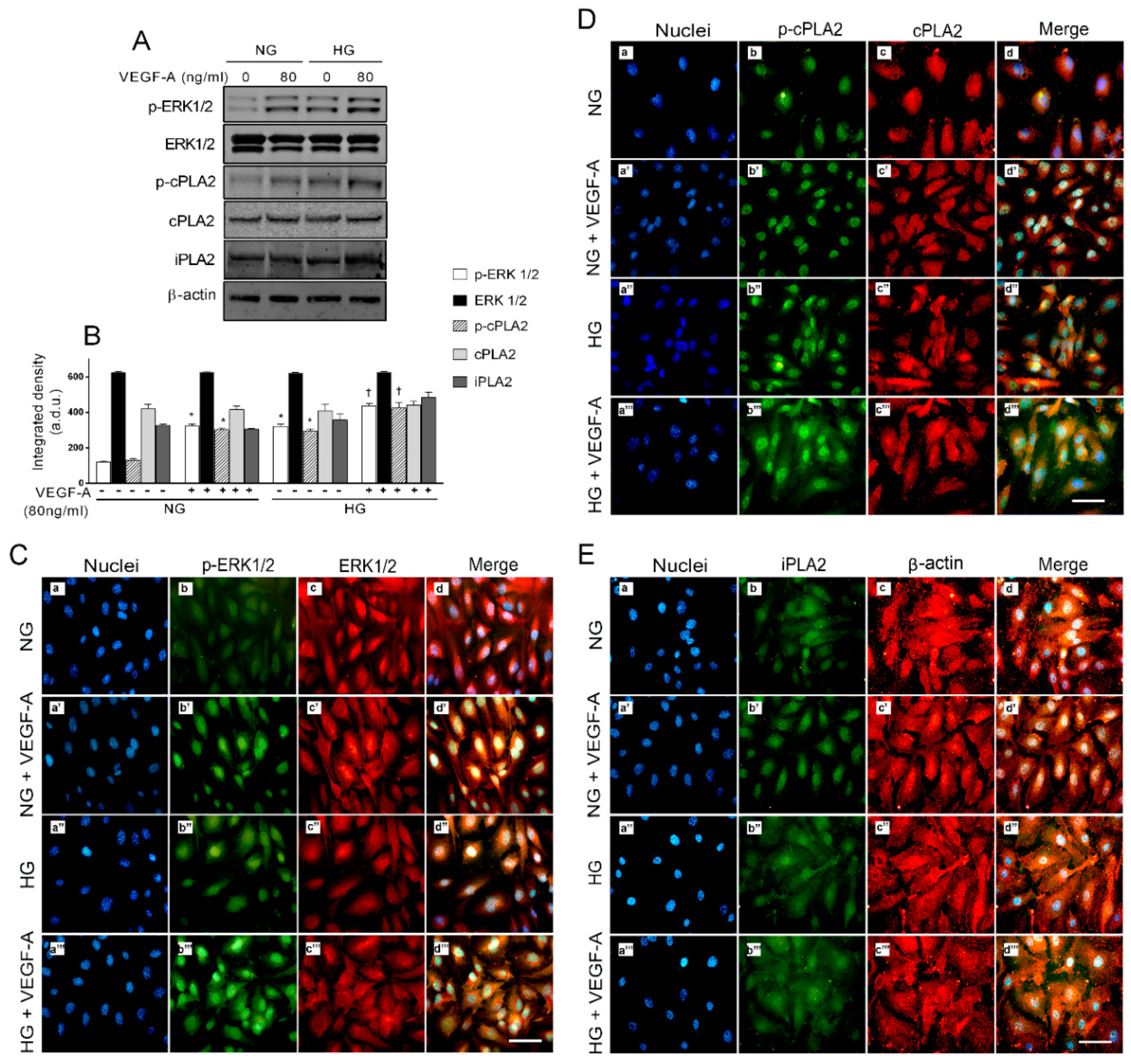
2.5. Activation of ERK1/2/cPLA2 Pathway and iPLA Expression by VEGF-A in Presence of HG in HREC
2.6. Selective PLA2 Silencing Reduces the Detrimental Effects of HG in HRECs
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Cultures and Treatments
4.3. Cell Viability Assay
4.4. Lactate Dehydrogenase (LDH) Release
4.5. Immunocytochemical Analysis
4.6. Tube Formation Assay
4.7. Western Blot Analysis
4.8. Transfection of siRNA
4.9. Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-qPCR)
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ma¨kimattila, S.; Virkama¨ki, A.; Groop, P.-H.; Cockcroft, J.; Utriainen, T.; Fagerudd, J.; Yki-Ja¨rvinen, H. Chronic Hyperglycemia Impairs Endothelial Function and Insulin Sensitivity Via Different Mechanisms in Insulin-Dependent Diabetes Mellitus. Circulation 1996, 94, 1276–1282. [Google Scholar] [CrossRef]
- Chou, J.; Rollins, S.; Fawzi, A.A. Role of endothelial cell and pericyte dysfunction in diabetic retinopathy: Review of techniques in rodent models. In Single Molecule and Single Cell Sequencing; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2014; Volume 801, pp. 669–675. [Google Scholar]
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Chan, P.-S. Oxidative Stress and Diabetic Retinopathy. Exp. Diabetes Res. 2007, 2007, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic Retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Pusparajah, P.; Elee, L.-H.; Kadir, K.E. Molecular Markers of Diabetic Retinopathy: Potential Screening Tool of the Future? Front. Physiol. 2016, 7, 200. [Google Scholar] [CrossRef]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.S.; McHowat, J.; Schnellmann, R.G. Phospholipase A(2)s in cell injury and death. J. Pharmacol. Exp. Ther. 2000, 294, 793–802. [Google Scholar]
- Glaser, K.B. Regulation of Phospholipase A2 Enzymes: Selective Inhibitors and their Pharmacological Potential. Advances in Pharmacology 1995, 32, 31–66. [Google Scholar] [CrossRef]
- Wu, X.; Walker, C.L.; Lu, Q.; Wu, W.; Eddelman, D.B.; Parish, J.M.; Xu, X.-M. RhoA/Rho Kinase Mediates Neuronal Death Through Regulating cPLA2 Activation. Mol. Neurobiol. 2016, 54, 6885–6895. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Hurt-Camejo, E. Phospholipase A2 enzymes and the risk of atherosclerosis. Eur. Hear. J. 2012, 33, 2899–2909. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.Y.; Chuang, D.Y.; Zong, Y.; Jiang, J.; Lee, J.C.M.; Gu, Z.; Simonyi, A. Role of Cytosolic Phospholipase A2 in Oxidative and Inflammatory Signaling Pathways in Different Cell Types in the Central Nervous System. Mol. Neurobiol. 2014, 50, 6–14. [Google Scholar] [CrossRef]
- Taketo, M.M.; Sonoshita, M. Phospholipase A2 and apoptosis. Biochim. Biophys. Acta 2002, 1585, 72–76. [Google Scholar] [CrossRef]
- Chang, J.F.; Yeh, J.C.; Ho, C.T.; Liu, S.H.; Hsieh, C.Y.; Wang, T.M.; Chang, S.W.; Lee, I.T.K.; Huang, Y.; Wang, J.Y.; et al. Targeting ROS and cPLA2/COX2 Expressions Ameliorated Renal Damage in Obese Mice with Endotoxemia. Int. J. Mol. Sci. 2019, 20, 4393. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Jin, X.; Wang, Q.-S.; Wei, S.-H.; Hou, B.-K.; Li, H.-Y.; Zhang, M.-N.; Li, Z.-H. The involvement of high mobility group 1 cytokine and phospholipases A2 in diabetic retinopathy. Lipids Health Dis. 2014, 13, 156. [Google Scholar] [CrossRef]
- Canning, P.; Kenny, B.-A.; Prise, V.; Glenn, J.; Sarker, M.H.; Hudson, N.; Brandt, M.; Lopez, F.J.; Gale, D.; Luthert, P.J.; et al. Lipoprotein-associated phospholipase A2 (Lp-PLA2) as a therapeutic target to prevent retinal vasopermeability during diabetes. Proc. Natl. Acad. Sci. USA 2016, 113, 7213–7218. [Google Scholar] [CrossRef]
- Lupo, G.; Motta, C.; Giurdanella, G.; Anfuso, C.D.; Alberghina, M.; Drago, F.; Salomone, S.; Bucolo, C. Role of phospholipases A2 in diabetic retinopathy: In vitro and in vivo studies. Biochem. Pharmacol. 2013, 86, 1603–1613. [Google Scholar] [CrossRef]
- Giurdanella, G.; Anfuso, C.D.; Olivieri, M.; Lupo, G.; Caporarello, N.; Eandi, C.M.; Drago, F.; Bucolo, C.; Esalomone, S. Aflibercept, bevacizumab and ranibizumab prevent glucose-induced damage in human retinal pericytes in vitro, through a PLA2/COX-2/VEGF-A pathway. Biochem. Pharmacol. 2015, 96, 278–287. [Google Scholar] [CrossRef]
- Balsinde, J.; Balboa, M.A.; Insel, P.A.; Dennis, E.A. Regulation and inhibition of phospholipase A2. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 175–189. [Google Scholar] [CrossRef]
- Murakami, M.; Kudo, I. Phospholipase A2. J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef]
- Leslie, C.C. Cytosolic phospholipase A2: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Turk, J. The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog. Nucleic Acid Res. Mol. Biol. 2001, 67, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Balboa, M.A. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A in activated cells. Cell. Signal. 2005, 17, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Zhang, S.; Barbour, S.E.; Bohrer, A.; Ford, E.L.; Koizumi, A.; Papa, F.R.; Ramanadham, S. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: A role for regulation by SREBP-1. J. Biol. Chem. 2010, 285, 6693–6705. [Google Scholar] [CrossRef]
- Veret, J.; Coant, N.; Berdyshev, E.V.; Skobeleva, A.; Therville, N.; Bailbé, D.; Gorshkova, I.; Natarajan, V.; Portha, B.; Le Stunff, H. Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 β-cells. Biochem. J. 2011, 438, 177–189. [Google Scholar] [CrossRef]
- Boslem, E.; MacIntosh, G.; Preston, A.M.; Bartley, C.; Busch, A.K.; Fuller, M.; Laybutt, D.R.; Meikle, P.J.; Biden, T.J. A lipidomic screen of palmitate-treated MIN6 beta-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem. J. 2011, 435, 267–276. [Google Scholar] [CrossRef]
- Giurdanella, G.; Lazzara, F.; Caporarello, N.; Lupo, G.; Anfuso, C.D.; Eandi, C.M.; Leggio, G.M.; Drago, F.; Bucolo, C.; Salomone, S. Sulodexide prevents activation of the PLA2/COX-2/VEGF inflammatory pathway in human retinal endothelial cells by blocking the effect of AGE/RAGE. Biochem. Pharmacol. 2017, 142, 145–154. [Google Scholar] [CrossRef]
- Askarova, S.; Yang, X.; Sheng, W.; Sun, G.Y.; Lee, J.C. Role of Aβ-receptor for advanced glycation endproducts interaction in oxidative stress and cytosolic phospholipase A₂ activation in astrocytes and cerebral endothelial cells. Neuroscience 2011, 199, 375–385. [Google Scholar] [CrossRef]
- Nicotra, A.; Lupo, G.; Giurdanella, G.; Anfuso, C.D.; Ragusa, N.; Tirolo, C.; Marchetti, B.; Alberghina, M. MAPKs mediate the activation of cytosolic phospholipase A2 by amyloid beta(25-35) peptide in bovine retina pericytes. Biochim. Biophys. Acta 2005, 1733, 172–186. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, D.; Singh, N.K.; Kundumani-Sridharan, V.; Gadiparthi, L.; Rao, C.H.M.; Rao, G.N. Activation of Cytosolic Phospholipase A2 Downstream of the Src-Phospholipase D1 (PLD1)-Protein Kinase C γ (PKC γ) Signaling Axis Is Required for Hypoxia-Induced Pathological Retinal Angiogenesis. J. Biol. Chem. 2011, 286, 22489–22498. [Google Scholar] [PubMed]
- Singh, N.K.; Hansen, D.E.; Kundumani-Sridharan, V.; Rao, G.N. Both Kdr and Flt1 play a vital role in hypoxia-induced Src-PLD1-PKCγ-cPLA2 activation and retinal neovascularization. Blood 2013, 121, 1911–1923. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, S.; Chowdhury, I.H.; Das, S.; Dutta, D.; Saha, A.; Sarkar, R.; Mandal, L.K.; Mukherjee, S.; Bhattacharya, B. Role of NF-κB activation and VEGF gene polymorphisms in VEGF up regulation in non-proliferative and proliferative diabetic retinopathy. Mol. Cell. Biochem. 2015, 405, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Wheeler-Jones, C.; Abu-Ghazaleh, R.; Cospedal, R.; Houliston, R.A.; Martin, J.; Zachary, I. Vascular endothelial growth factor stimulates prostacyclin production and activation of cytosolic phospholipase A2 in endothelial cells via p42/p44 mitogen-activated protein kinase. FEBS Lett. 1997, 420, 28–32. [Google Scholar] [CrossRef]
- Toft-Kehler, A.K.; Andersen, C.; Andreasen, J.R.; Vohra, R.; Junker, N.; Poulsen, K.A.; Kolko, M. Interaction between VEGF and Calcium-Independent Phospholipase A2in Proliferation and Migration of Retinal Pigment Epithelium. Curr. Eye Res. 2012, 37, 500–507. [Google Scholar] [CrossRef]
- Giurdanella, G.; Motta, C.; Muriana, S.; Arena, V.; Anfuso, C.D.; Lupo, G.; Alberghina, M. Cytosolic and calcium-independent phospholipase A2 mediate glioma-enhanced proangiogenic activity of brain endothelial cells. Microvasc. Res. 2011, 81, 1–17. [Google Scholar] [CrossRef]
- Ito, Y.; Iwamoto, Y.; Tanaka, K.; Okuyama, K.; Sugioka, Y. A quantitative assay using basament extracts to study tumor angiogensis in vivo. Int. J. Cancer 1996, 67, 148–152. [Google Scholar] [CrossRef]
- Kaul, K.; Hodgkinson, A.; Tarr, J.M.; Kohner, E.M.; Chibber, R. Is Inflammation a Common Retinal-Renal-Nerve Pathogenic Link in Diabetes? Curr. Diabetes Rev. 2010, 6, 294–303. [Google Scholar] [CrossRef]
- Tikhonenko, M.; Lydic, T.A.; Wang, Y.; Chen, W.; Opreanu, M.; Sochacki, A.; McSorley, K.M.; Renis, R.L.; Kern, T.; Jump, D.B.; et al. Remodeling of Retinal Fatty Acids in an Animal Model of Diabetes: A Decrease in Long-Chain Polyunsaturated Fatty Acids Is Associated with a Decrease in Fatty Acid Elongases Elovl2 and Elovl4. Diabetes 2009, 59, 219–227. [Google Scholar] [CrossRef]
- Barnett, J.M.; Mccollum, G.W.; Penn, J.S. Role of Cytosolic Phospholipase A2 in Retinal Neovascularization. Investig. Opthalmol. Vis. Sci. 2010, 51, 1136–1142. [Google Scholar] [CrossRef]
- Abu El-Asrar, A.M.; Missotten, L.; Geboes, K. Expression of cyclo-oxygenase-2 and downstream enzymes in diabetic fibrovascular epiretinal membranes. Br. J. Ophthalmol. 2008, 92, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Mitsiades, N.; Kirchhof, B.; Koizumi, K.; Döhmen, S.; Adamis, A.P. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-α suppression. FASEB J. 2002, 16, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Ayalasomayajula, S.P.; Kompella, U.B. Celecoxib, a selective cyclooxygenase-2 inhibitor, inhibits retinal vascular endothelial growth factor expression and vascular leakage in a streptozotocin-induced diabetic rat model. Eur. J. Pharmacol. 2003, 458, 283–289. [Google Scholar] [CrossRef]
- Brust, A.-K.; Ulbrich, H.K.; Seigel, G.M.; Pfeiffer, N.; Grus, F.H. Effects of Cyclooxygenase Inhibitors on Apoptotic Neuroretinal Cells. Biomark. Insights 2008, 3, S692. [Google Scholar] [CrossRef] [PubMed]
- Radi, Z.A.; Render, J.A. The pathophysiologic role of cyclo-oxygenases in the eye. J. Ocul. Pharmacol. Ther. 2008, 24, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Lazzara, F.; Fidilio, A.; Platania, C.B.M.; Giurdanella, G.; Salomone, S.; Leggio, G.M.; Tarallo, V.; Cicatiello, V.; De Falco, S.; Eandi, C.M.; et al. Aflibercept regulates retinal inflammation elicited by high glucose via the PlGF/ERK pathway. Biochem. Pharmacol. 2019, 168, 341–351. [Google Scholar] [CrossRef]
- Murakami, T.; Felinski, E.A.; Antonetti, D.A. Occludin Phosphorylation and Ubiquitination Regulate Tight Junction Trafficking and Vascular Endothelial Growth Factor-induced Permeability. J. Biol. Chem. 2009, 284, 21036–21046. [Google Scholar] [CrossRef]
- Harhaj, N.S.; Felinski, E.A.; Wolpert, E.B.; Sundstrom, J.M.; Gardner, T.W.; Antonetti, D.A. VEGF Activation of Protein Kinase C Stimulates Occludin Phosphorylation and Contributes to Endothelial Permeability. Investig. Opthalmol. Vis. Sci. 2006, 47, 5106–5115. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Battah, S.; Ahmed, N.; Karachalias, N.; Agalou, S.; Babaei-Jadidi, R.; Dawnay, A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003, 375, 581–592. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, É. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef]
- Kandarakis, S.A.; Piperi, C.; Topouzis, F.; Papavassiliou, A.G. Emerging role of advanced glycation-end products (AGEs) in the pathobiology of eye diseases. Prog. Retin. Eye Res. 2014, 42, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, C.; Liu, X.; Liao, B.; Pan, X.; Ren, Y.; Fan, M.; Li, M.; He, Z.; Wu, J.; et al. AGEs increased migration and inflammatory responses of adventitial fibroblasts via RAGE, MAPK and NF-κB pathways. Atherosclerosis 2010, 208, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Schiekofer1, S.; Andrassy1, M.; Chen, J.; Rudofsky, G.; Schneider, J.; Wendt, T.; Stefan, N.; Humpert, P.; Fritsche, A.; Stumvoll, M.; et al. Acute Hyperglycemia Causes Intracellular Formation of CML and Activation of ras, p42/44 MAPK, and Nuclear Factor κB in PBMCs. Diabetes 2003, 52, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Sturgis, L.; Haidacher, J.; Zhang, X.N.; Sherwood, S.J.; Bjercke, R.J.; Juhasz, O.; Crow, M.T.; Tilton, R.G.; Denner, L. Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factorkappaB transcriptional activation and cytokine secretion. Diabetes 2001, 50, 1495–1504. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products Sparking the Development of Diabetic Vascular Injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Anfuso, C.D.; Giurdanella, G.; Motta, C.; Muriana, S.; Lupo, G.; Ragusa, N.; Alberghina, M. PKCalpha-MAPK/ERK-phospholipase A2 signaling is required for human melanoma-enhanced brain endothelial cell proliferation and motility. Microvasc. Res. 2009, 78, 338–357. [Google Scholar] [CrossRef]
- Gijón, M.A.; Spencer, D.M.; Siddiqi, A.R.; Bonventre, J.V.; Leslie, C.C. Cytosolic Phospholipase A2Is Required for Macrophage Arachidonic Acid Release by Agonists That Do and Do Not Mobilize Calcium. J. Biol. Chem. 2000, 275, 20146–20156. [Google Scholar] [CrossRef]
- Fatima, S.; Khandekar, Z.; Parmentier, J.H.; Malik, K.U. Cytosolic phospholipase A2 activation by the p38 kinase inhibitor SB203580 in rabbit aortic smooth muscle cells. J. Pharmacol. Exp. Ther. 2001, 298. [Google Scholar]
- Mather, A.; Chen, X.M.; McGinn, S.; Field, M.J.; Summual, S.; Mangiafico, S.; Khang, Y.; Kelly, D.J.; Pollock, C.A. High glucose induced endothelial cell growth inhibition is associated with an increase in TGFbeta1 secretion and inhibition of Ras prenylation via suppression of the mevalonate pathway. Int. J. Biochem. Cell Biol. 2009, 41, 561–569. [Google Scholar] [CrossRef]
- Hernandez, C.; Lecube, A.; Segura, R.M.; Sararols, L.; Simó, R. Nitric oxide and vascular endothelial growth factor concentrations are increased but not related in vitreous fluid of patients with proliferative diabetic retinopathy. Diabet. Med. 2002, 19, 655–660. [Google Scholar] [CrossRef]
- Nikolaou, A.; Kokotou, M.G.; Vasilakaki, S.; Kokotos, G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2019, 1864, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Giurdanella, G.; Montalbano, G.; Gennuso, F.; Brancati, S.; Furno, D.L.; Augello, A.; Bucolo, C.; Drago, F.; Esalomone, S. Isolation, cultivation, and characterization of primary bovine cochlear pericytes: A new in vitro model of stria vascularis. J. Cell. Physiol. 2018, 234, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Anfuso, C.D.; Longo, A.; Distefano, A.; Amorini, A.M.; Salmeri, M.; Zanghì, G.; Giallongo, C.; Giurdanella, G.; Lupo, G. Uveal Melanoma Cells Elicit Retinal Pericyte Phenotypical and Biochemical Changes in an in Vitro Model of Coculture. Int. J. Mol. Sci. 2020, 21, 5557. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giurdanella, G.; Lupo, G.; Gennuso, F.; Conti, F.; Furno, D.L.; Mannino, G.; Anfuso, C.D.; Drago, F.; Salomone, S.; Bucolo, C. Activation of the VEGF-A/ERK/PLA2 Axis Mediates Early Retinal Endothelial Cell Damage Induced by High Glucose: New Insight from an In Vitro Model of Diabetic Retinopathy. Int. J. Mol. Sci. 2020, 21, 7528. https://doi.org/10.3390/ijms21207528
Giurdanella G, Lupo G, Gennuso F, Conti F, Furno DL, Mannino G, Anfuso CD, Drago F, Salomone S, Bucolo C. Activation of the VEGF-A/ERK/PLA2 Axis Mediates Early Retinal Endothelial Cell Damage Induced by High Glucose: New Insight from an In Vitro Model of Diabetic Retinopathy. International Journal of Molecular Sciences. 2020; 21(20):7528. https://doi.org/10.3390/ijms21207528
Chicago/Turabian StyleGiurdanella, Giovanni, Gabriella Lupo, Florinda Gennuso, Federica Conti, Debora Lo Furno, Giuliana Mannino, Carmelina Daniela Anfuso, Filippo Drago, Salvatore Salomone, and Claudio Bucolo. 2020. "Activation of the VEGF-A/ERK/PLA2 Axis Mediates Early Retinal Endothelial Cell Damage Induced by High Glucose: New Insight from an In Vitro Model of Diabetic Retinopathy" International Journal of Molecular Sciences 21, no. 20: 7528. https://doi.org/10.3390/ijms21207528
APA StyleGiurdanella, G., Lupo, G., Gennuso, F., Conti, F., Furno, D. L., Mannino, G., Anfuso, C. D., Drago, F., Salomone, S., & Bucolo, C. (2020). Activation of the VEGF-A/ERK/PLA2 Axis Mediates Early Retinal Endothelial Cell Damage Induced by High Glucose: New Insight from an In Vitro Model of Diabetic Retinopathy. International Journal of Molecular Sciences, 21(20), 7528. https://doi.org/10.3390/ijms21207528