GPR4 Knockout Improves the Neurotoxin-Induced, Caspase-Dependent Mitochondrial Apoptosis of the Dopaminergic Neuronal Cell





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
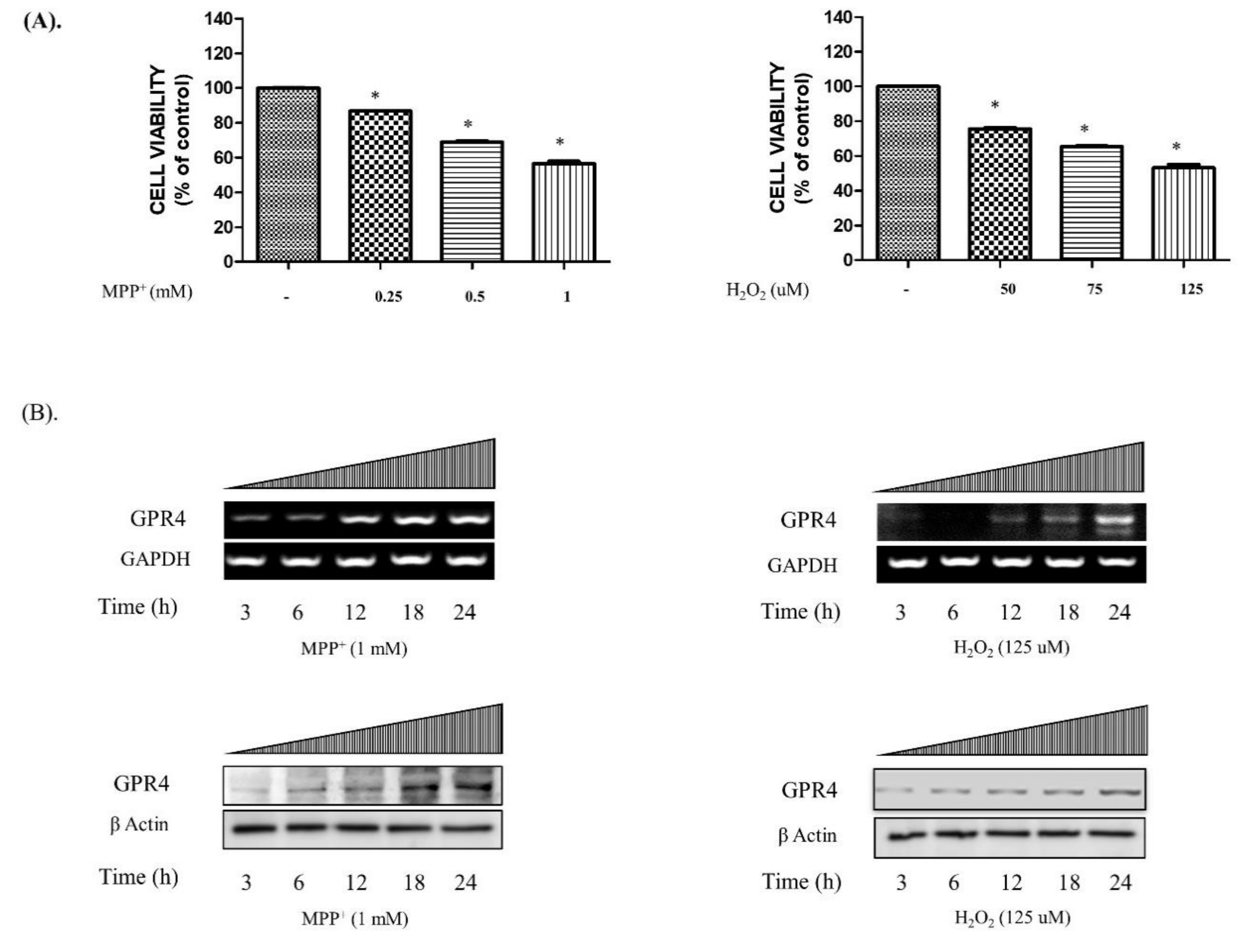
2.1. Expression of GPR4 is Upregulated in Neurotoxin-Stimulated Apoptosis in SH-SY5Y Cells
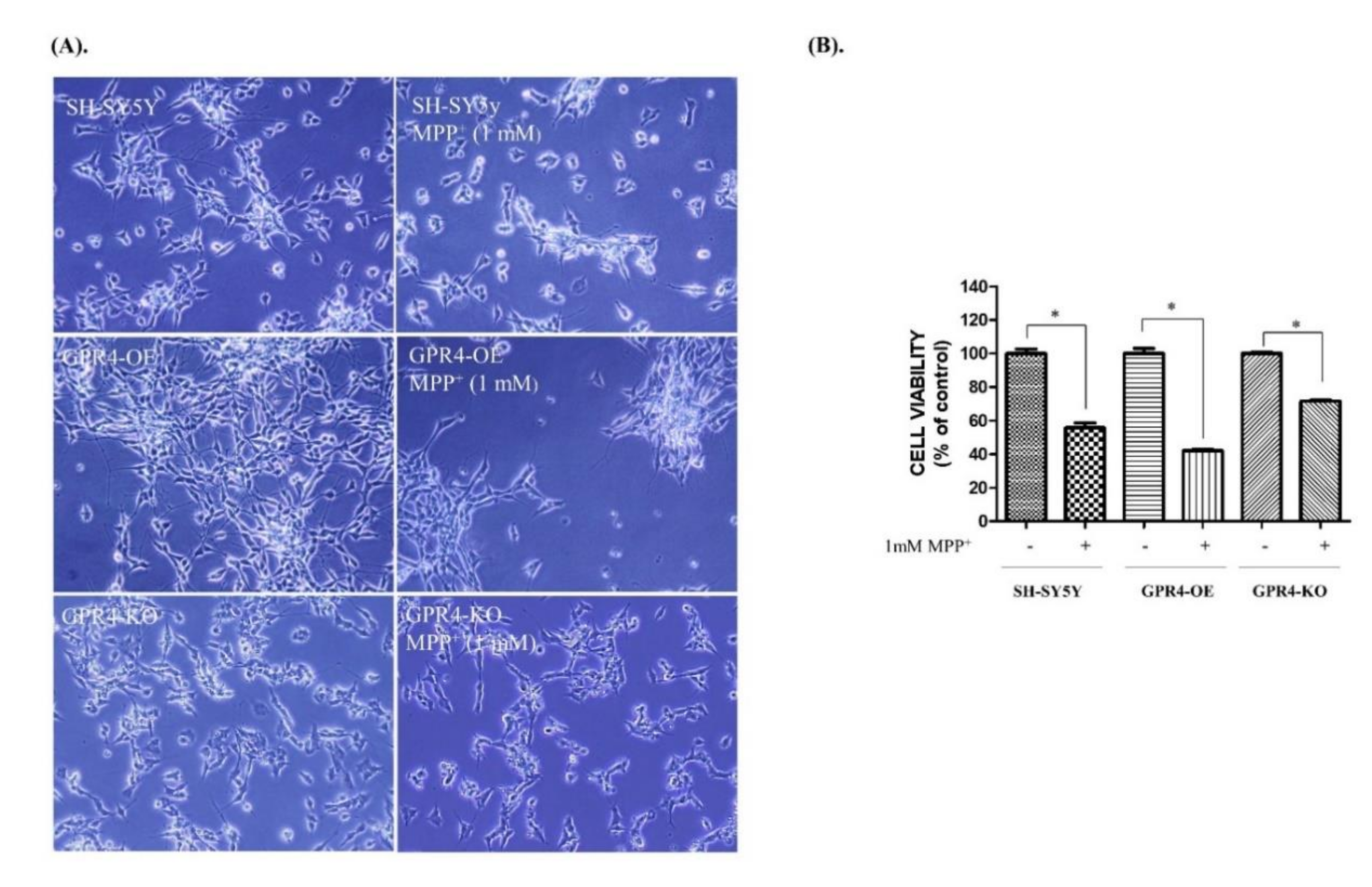
2.2. Knockout of GPR4 Protects SH-SY5Y Cells from Neurotoxin-Stimulated Apoptosis in SH-SY5Y Cells
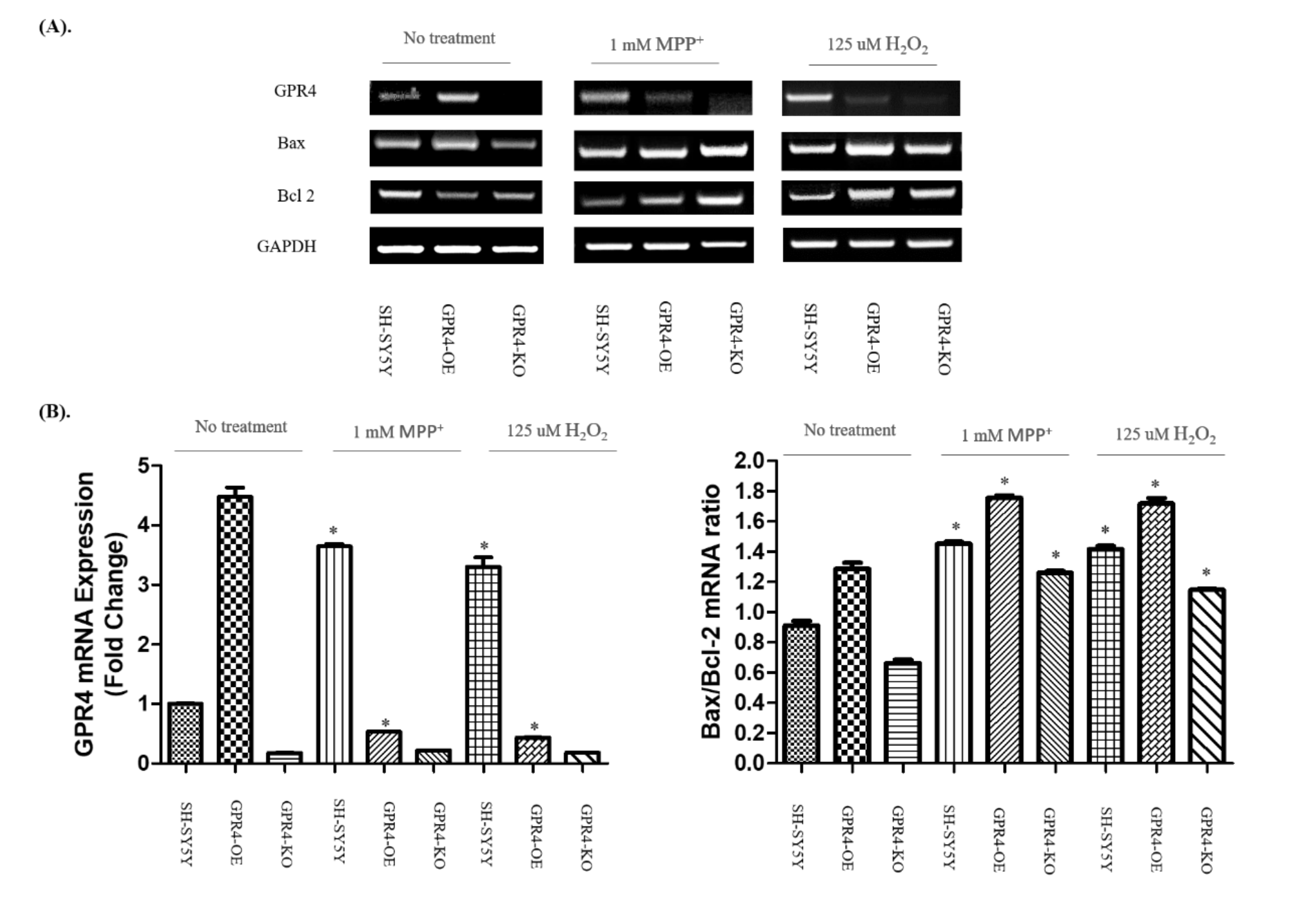
2.3. Knockout of GPR4 Decreases the Bax/Bcl-2 mRNA Ratio during Neurotoxin-Induced Apoptosis in SH-SY5Y Cells
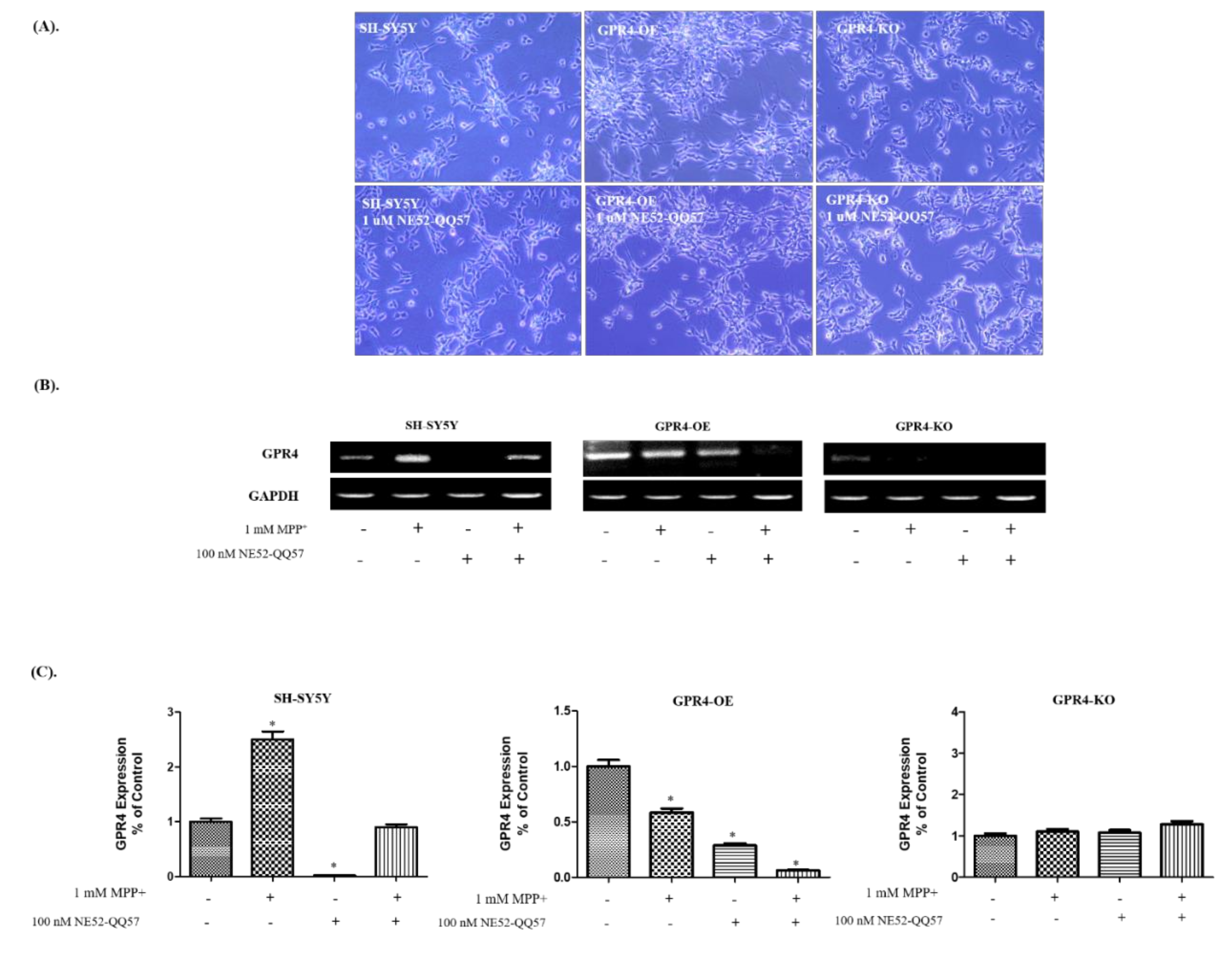
2.4. Effect of a GPR4 Antagonist on the Cellular Morphology and GPR4 mRNA Expression of SH-SY5Y Cells
2.5. Knockout of GPR4 Decreases the Bax/Bcl-2 Protein Ratio and the Cleavage of PARP Expression in Neurotoxin-Stimulated SH-SY5Y Cells
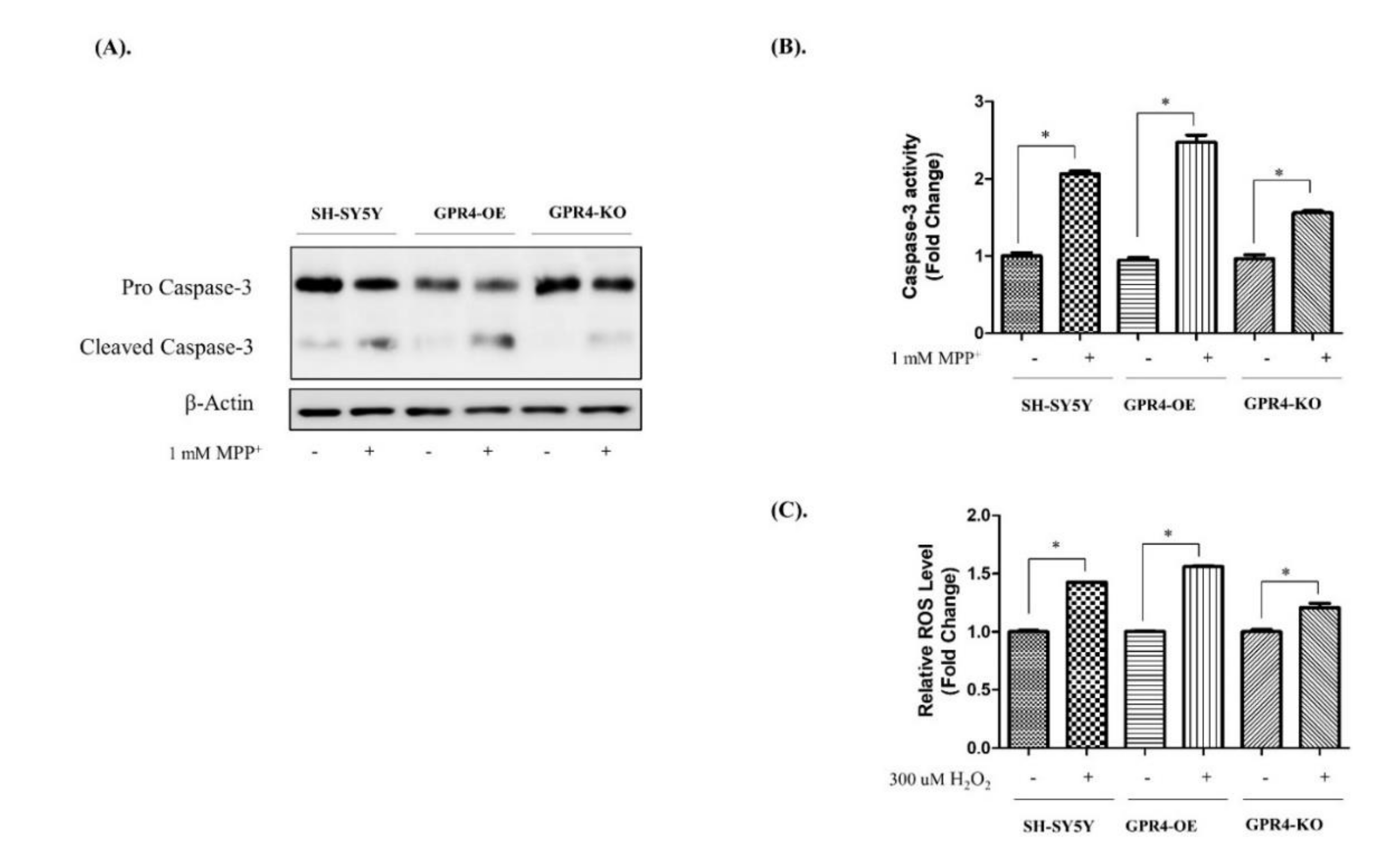
2.6. Knockout of GPR4 Decreases the Caspase-3 Activity and Lowers the ROS Generation in Neurotoxin-Stimulated SH-SY5Y Cells
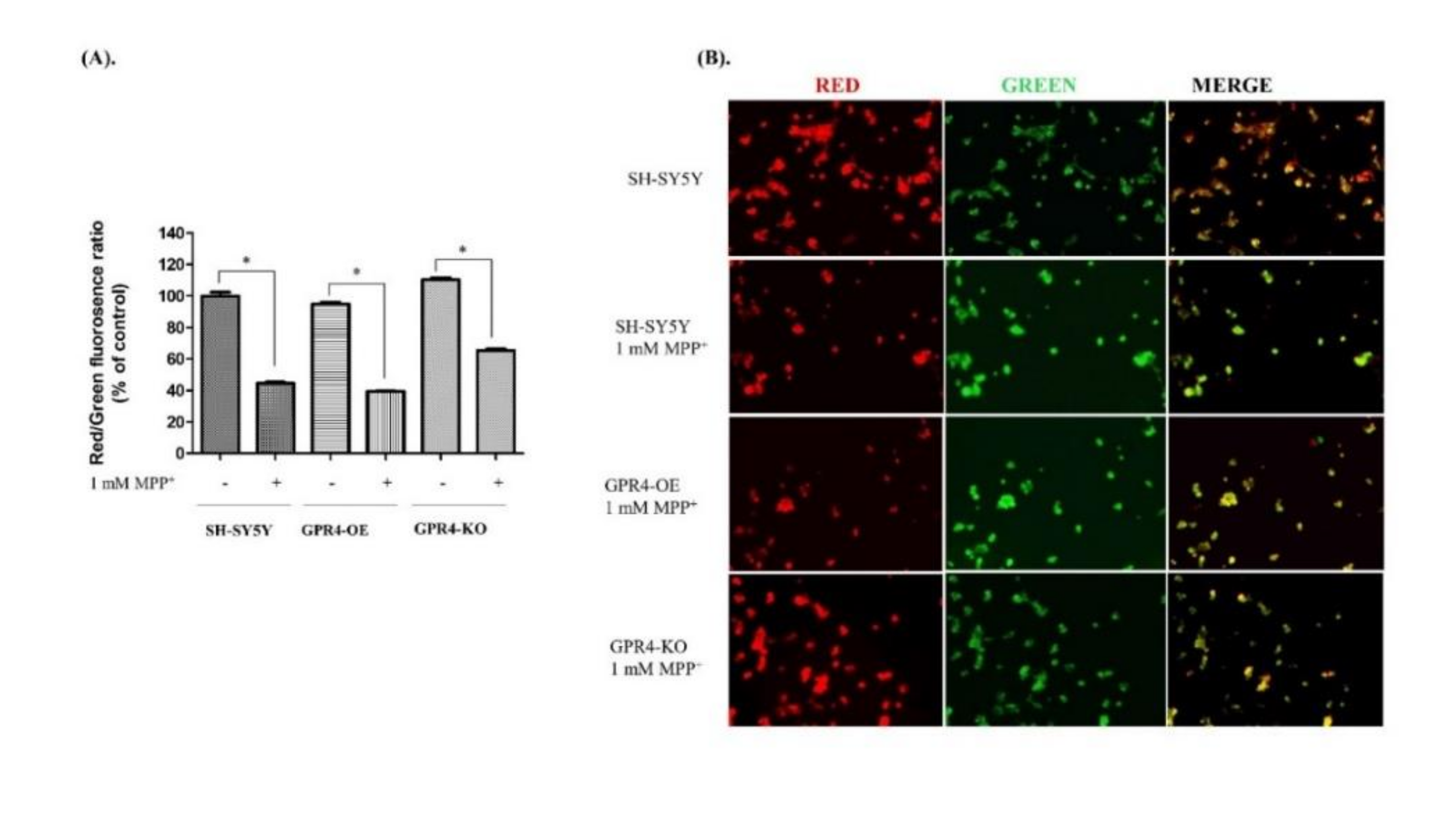
2.7. Knockout of GPR4 Increases the Mitochondrial Membrane Potential (MMP) in Neurotoxin-Stimulated SH-SY5Y Cells
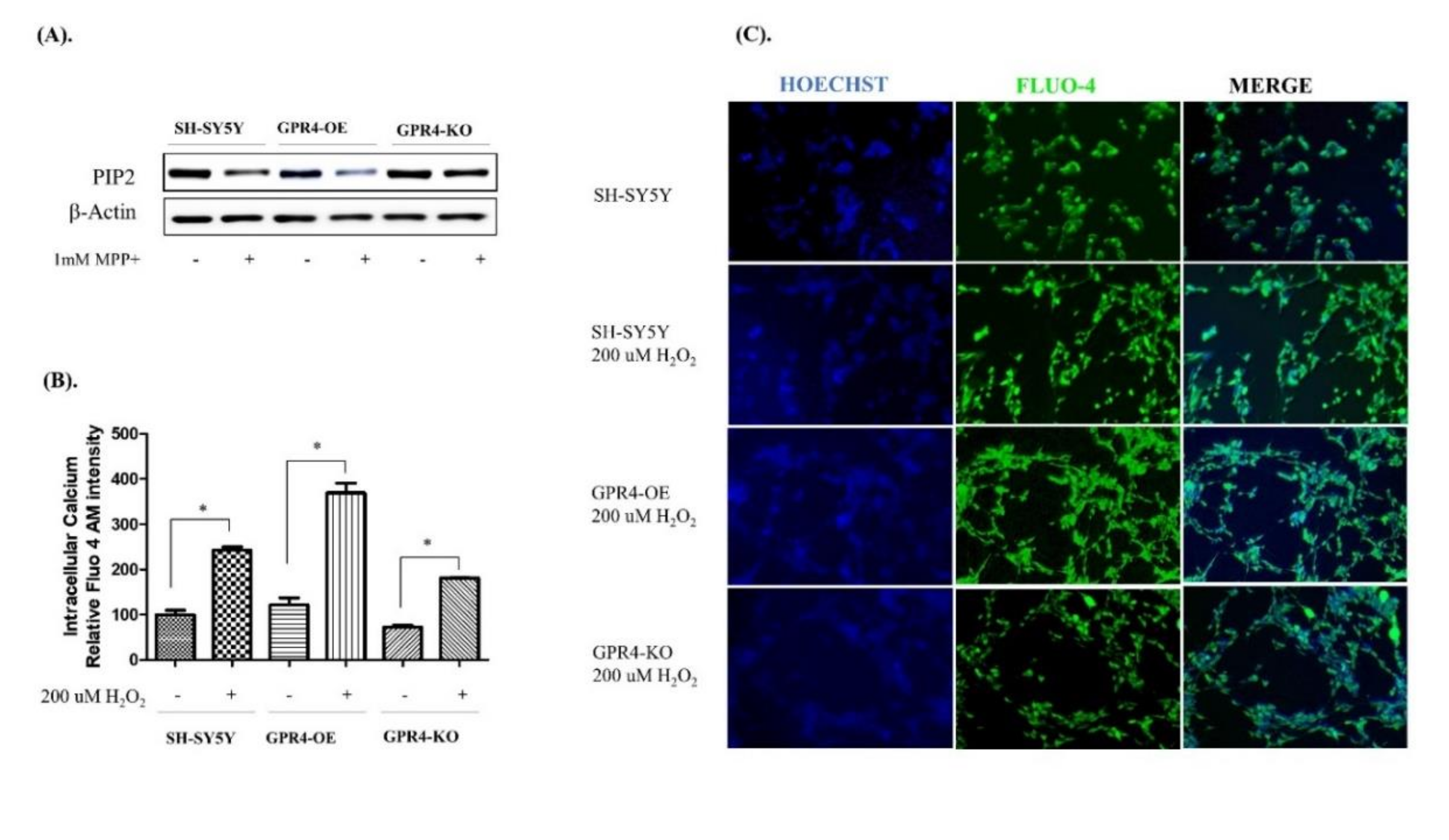
2.8. Knockout of GPR4 Decreases the Intracellular Calcium in Neurotoxin-Stimulated SH-SY5Y Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Transfection
4.3. Measurement of Cell Viability
4.4. Total RNA Isolation for RT-PCR
4.5. Immunoblot Analysis
4.6. Detection of Intracellular ROS
4.7. Assessment of Caspase-3 Activity
4.8. Assessment of Mitochondrial Membrane Potential (MMP)
4.9. Detection of Intracellular Calcium
4.10. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MPTP | 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine |
| MPP+ | 1-Methyl-4-Phenylpyridinium Ion |
| MTT | 3-(3,4-Dimehylthiazol-2-Yl)-2,5-Diphenyl-Tetrazolium Bromide |
| DCF-DA | 7′-Dichlorofluorescein Diacetate |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| ER | Endoplasmic Reticulum |
| FBS | Fetal Bovine Serum |
| HUVEC | Human Umbilical Vein Endothelial Cells |
| MMP | Mitochondrial Membrane Potential |
| Mptp | Mitochondrial Permeability Transition Pore |
| PIP2 | Phosphatidylinositol Biphosphate |
| PARP | Poly (ADP-Ribose) Polymerase |
| SN | Substantia Nigra |
| TDAG8 | T-Cell Death-Associated Gene 8 |
| OGR1 | The Ovarian Cancer G Protein-Coupled Receptor 1 |
References
- Zhang, Q.; Hu, C.; Huang, J.; Liu, W.; Lai, W.; Leng, F.; Tang, Q.; Liu, Y.; Wang, Q.; Zhou, M.; et al. ROCK1 induces dopaminergic nerve cell apoptosis via the activation of Drp1-mediated aberrant mitochondrial fission in Parkinson’s disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–130. [Google Scholar] [CrossRef]
- Ly, J.D.; Grubb, D.R.; Lawen, A.J.A. The mitochondrial membrane potential (Δψ m) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [PubMed]
- Prenek, L.; Boldizsár, F.; Kugyelka, R.; Ugor, E.; Berta, G.; Németh, P.; Berki, T. The regulation of the mitochondrial apoptotic pathway by glucocorticoid receptor in collaboration with Bcl-2 family proteins in developing T cells. Apoptosis 2017, 22, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Gerace, E.; Masi, A.; Resta, F.; Felici, R.; Landucci, E.; Mello, T.; Pellegrini-Giampietro, D.; Mannaioni, G.; Moroni, F. PARP-1 activation causes neuronal death in the hippocampal CA1 region by increasing the expression of Ca2+-permeable AMPA receptors. Neurobiol. Dis. 2014, 70, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Alexander, J.S.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef]
- More, S.V.; Choi, D.-K. Atractylenolide-I Protects Human SH-SY5Y Cells from 1-Methyl-4-Phenylpyridinium-Induced Apoptotic Cell Death. Int. J. Mol. Sci. 2017, 18, 1012. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nat. Cell Biol. 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Guzman, J.N.; Sanchez-Pandila, J.; Schumacker, P.T. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience 2011, 198, 221–231. [Google Scholar]
- Gunter, T.E.; Gunter, K.K. Uptake of Calcium by Mitochondria: Transport and Possible Function. IUBMB Life 2001, 52, 197–204. [Google Scholar] [CrossRef]
- Haupt, S.; Raghu, D.; Haupt, Y. p53 Calls upon CIA (Calcium Induced Apoptosis) to Counter Stress. Front. Oncol. 2015, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between Cytosolic Dopamine, Calcium, and α-Synuclein Causes Selective Death of Substantia Nigra Neurons. Neuron 2009, 62, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.P.; Swerdlow, R.H.; Parker, W.D.; Miller, S.W.; Davis, R.E.; Tuttle, J.B. Altered calcium homeostasis in cells transformed by mitochondria from individuals with Parkinson’s disease. J. Neurochem. 1997, 68, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Singer, T.P.; Ramsay, R.R. Mechanism of the neurotoxicity of MPTP. FEBS Lett. 1990, 274, 1–8. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V. Mechanisms of MPTP toxicity. Mov. Disord. 1998, 13, 35–38. [Google Scholar]
- Dumont, A.; Hehner, S.P.; Hofmann, T.G.; Ueffing, M.; Dröge, W.; Schmitz, M.L. Hydrogen peroxide-induced apoptosis is CD95-independent, requires the release of mitochondria-derived reactive oxygen species and the activation of NF-κB. Oncogene 1999, 18, 747–757. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Uğuz, A.C.; Öz, A.; Nazıroğlu, M. Curcumin inhibits apoptosis by regulating intracellular calcium release, reactive oxygen species and mitochondrial depolarization levels in SH-SY5Y neuronal cells. J. Recept. Signal Transduct. 2016, 36, 395–401. [Google Scholar] [CrossRef]
- Knaryan, V.H.; Samantaray, S.; Park, S.; Azuma, M.; Inoue, J.; Banik, N.L.; Sookyoung, P. SNJ-1945, a calpain inhibitor, protects SH-SY5Y cells against MPP+ and rotenone. J. Neurochem. 2014, 130, 280–290. [Google Scholar] [CrossRef]
- Meyer, T.N.; Gloy, J.; Hug, M.J.; Greger, R.; Schollmeyer, P.; Pavenstädt, H. Hydrogen peroxide increases the intracellular calcium activity in rat mesangial cells in primary culture. Kidney Int. 1996, 49, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.-G.; Vanek, M.; Guerini, D.; Gasser, J.A.; Jones, C.E.; Junker, U.; Hofstetter, H.; Wolf, R.M.; Seuwen, K. Proton-sensing G-protein-coupled receptors. Nat. Cell Biol. 2003, 425, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Kihara, Y.; Shimizu, T. Identification of T Cell Death-associated Gene 8 (TDAG8) as a Novel Acid Sensing G-protein-coupled Receptor. J. Biol. Chem. 2005, 280, 9083–9087. [Google Scholar] [CrossRef] [PubMed]
- Justus, C.R.; Dong, L.; Yang, L.V. Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front. Physiol. 2013, 4, 354. [Google Scholar] [CrossRef]
- Hosford, P.; Mosienko, V.; Kishi, K.; Jurisic, G.; Seuwen, K.; Kinzel, B.; Ludwig, M.; Wells, J.; Christie, I.; Koolen, L.; et al. CNS distribution, signalling properties and central effects of G-protein coupled receptor 4. Neuropharmacology 2018, 138, 381–392. [Google Scholar] [CrossRef]
- Dong, B.; Zhang, X.; Fan, Y.; Cao, S.; Zhang, Y. Acidosis promotes cell apoptosis through the G protein-coupled receptor 4/CCAAT/enhancer-binding protein homologous protein pathway. Oncol. Lett. 2018, 16, 6735–6741. [Google Scholar] [CrossRef]
- Krewson, E.A.; Sanderlin, E.J.; Marie, M.A.; Akhtar, S.N.; Velcicky, J.; Loetscher, P.; Yang, L.V. The Proton-Sensing GPR4 Receptor Regulates Paracellular Gap Formation and Permeability of Vascular Endothelial Cells. iScience 2020, 23, 100848. [Google Scholar] [CrossRef]
- Dong, B.; Zhou, H.; Han, C.; Yao, J.; Xu, L.; Zhang, M.; Fu, Y.; Xia, Q. Ischemia/Reperfusion-Induced CHOP Expression Promotes Apoptosis and Impairs Renal Function Recovery: The Role of Acidosis and GPR4. PLoS ONE 2014, 9, e110944. [Google Scholar] [CrossRef]
- Mortadza, S.S.; Sim, J.A.; Stacey, M.; Jiang, L.-H. Signalling mechanisms mediating Zn2+-induced TRPM2 channel activation and cell death in microglial cells. Sci. Rep. 2017, 7, srep45032. [Google Scholar] [CrossRef]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef]
- Hartmann, A.; Hunot, S.; Michel, P.P.; Muriel, M.-P.; Vyas, S.; Faucheux, B.A.; Mouatt-Prigent, A.; Turmel, H.; Srinivasan, A.; Ruberg, M.; et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2875–2880. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Chung, Y.; Lee, Y.; Lee, Y.; Cho, J.W.; Shin, E.-J.; Kim, H.-C.; Oh, Y.J. Buffering of cytosolic calcium plays a neuroprotective role by preserving the autophagy-lysosome pathway during MPP+-induced neuronal death. Cell Death Discov. 2019, 5, 130. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.; Guo, Q.; Mattson, M.P. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J. Neurosci. Res. 1998, 51, 293–308. [Google Scholar] [CrossRef]
- Lynch, K.; Fernández, G.; Pappalardo, A.; Peluso, J.J. Basic Fibroblast Growth Factor Inhibits Apoptosis of Spontaneously Immortalized Granulosa Cells by Regulating Intracellular Free Calcium Levels through a Protein Kinase Cδ-Dependent Pathway. Endocrinol. 2000, 141, 4209–4217. [Google Scholar] [CrossRef]
- Kiselyov, K.; Shin, D.M.; Muallem, S. Signalling specificity in GPCR-dependent Ca2+ signalling. Cell. Signal. 2003, 15, 243–253. [Google Scholar] [CrossRef]
- Frick, K.K.; Krieger, N.S.; Nehrke, K.; Bushinsky, A.D. Metabolic Acidosis Increases Intracellular Calcium in Bone Cells Through Activation of the Proton Receptor OGR1. J. Bone Miner. Res. 2009, 24, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Tobo, M.; Tomura, H.; Mogi, C.; Wang, J.-Q.; Liu, J.-P.; Komachi, M.; Damirin, A.; Kimura, T.; Murata, N.; Kurose, H.; et al. Previously postulated “ligand-independent” signaling of GPR4 is mediated through proton-sensing mechanisms. Cell. Signal. 2007, 19, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.N.; Velic, A.; Soliz, J.; Shi, Y.; Li, K.; Wang, S.; Weaver, J.L.; Sen, J.; Abbott, S.B.; Lazarenko, R.M.; et al. Regulation of breathing by CO2 requires the proton-activated receptor GPR4 in retrotrapezoid nucleus neurons. Science 2015, 348, 1255–1260. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Fall, C.P.; Bennett, J.P. Characterization and time course of MPP+ -induced apoptosis in human SH-SY5Y neuroblastoma cells. J. Neurosci. Res. 1999, 55, 620–628. [Google Scholar] [CrossRef]
- Kim, I.-S.; Ganesan, P.; Choi, D.-K. Cx43 Mediates Resistance against MPP+-Induced Apoptosis in SH-SY5Y Neuroblastoma Cells via Modulating the Mitochondrial Apoptosis Pathway. Int. J. Mol. Sci. 2016, 17, 1819. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S.J.N. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem. Int. 2016, 99, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-P.; Nakakura, T.; Tomura, H.; Tobo, M.; Mogi, C.; Wang, J.-Q.; He, X.-D.; Takano, M.; Damirin, A.; Komachi, M.; et al. Each one of certain histidine residues in G-protein-coupled receptor GPR4 is critical for extracellular proton-induced stimulation of multiple G-protein-signaling pathways. Pharmacol. Res. 2010, 61, 499–505. [Google Scholar] [CrossRef]
- Karthivashan, G.; Park, S.-Y.; Kweon, M.-H.; Kim, J.; Haque, E.; Cho, D.-Y.; Kim, I.-S.; Cho, E.-A.; Ganesan, P.; Choi, D.-K. Ameliorative potential of desalted Salicornia europaea L. extract in multifaceted Alzheimer’s-like scopolamine-induced amnesic mice model. Sci. Rep. 2018, 8, 7174. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, M.E.; Akther, M.; Azam, S.; Choi, D.-K.; Kim, I.-S. GPR4 Knockout Improves the Neurotoxin-Induced, Caspase-Dependent Mitochondrial Apoptosis of the Dopaminergic Neuronal Cell. Int. J. Mol. Sci. 2020, 21, 7517. https://doi.org/10.3390/ijms21207517
Haque ME, Akther M, Azam S, Choi D-K, Kim I-S. GPR4 Knockout Improves the Neurotoxin-Induced, Caspase-Dependent Mitochondrial Apoptosis of the Dopaminergic Neuronal Cell. International Journal of Molecular Sciences. 2020; 21(20):7517. https://doi.org/10.3390/ijms21207517
Chicago/Turabian StyleHaque, Md Ezazul, Mahbuba Akther, Shofiul Azam, Dong-Kug Choi, and In-Su Kim. 2020. "GPR4 Knockout Improves the Neurotoxin-Induced, Caspase-Dependent Mitochondrial Apoptosis of the Dopaminergic Neuronal Cell" International Journal of Molecular Sciences 21, no. 20: 7517. https://doi.org/10.3390/ijms21207517
APA StyleHaque, M. E., Akther, M., Azam, S., Choi, D.-K., & Kim, I.-S. (2020). GPR4 Knockout Improves the Neurotoxin-Induced, Caspase-Dependent Mitochondrial Apoptosis of the Dopaminergic Neuronal Cell. International Journal of Molecular Sciences, 21(20), 7517. https://doi.org/10.3390/ijms21207517