Chiral Cyclobutane-Containing Cell-Penetrating Peptides as Selective Vectors for Anti-Leishmania Drug Delivery Systems

 , , , ,
, , , ,  and
and 
Abstract
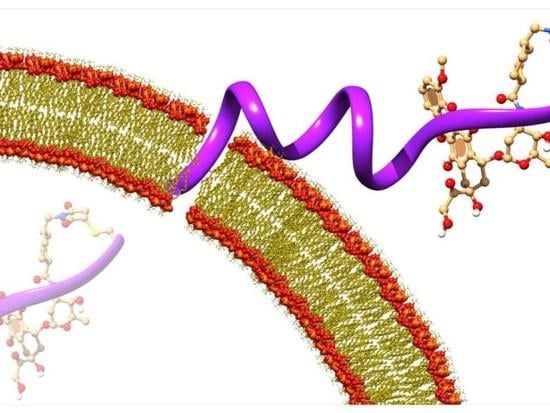
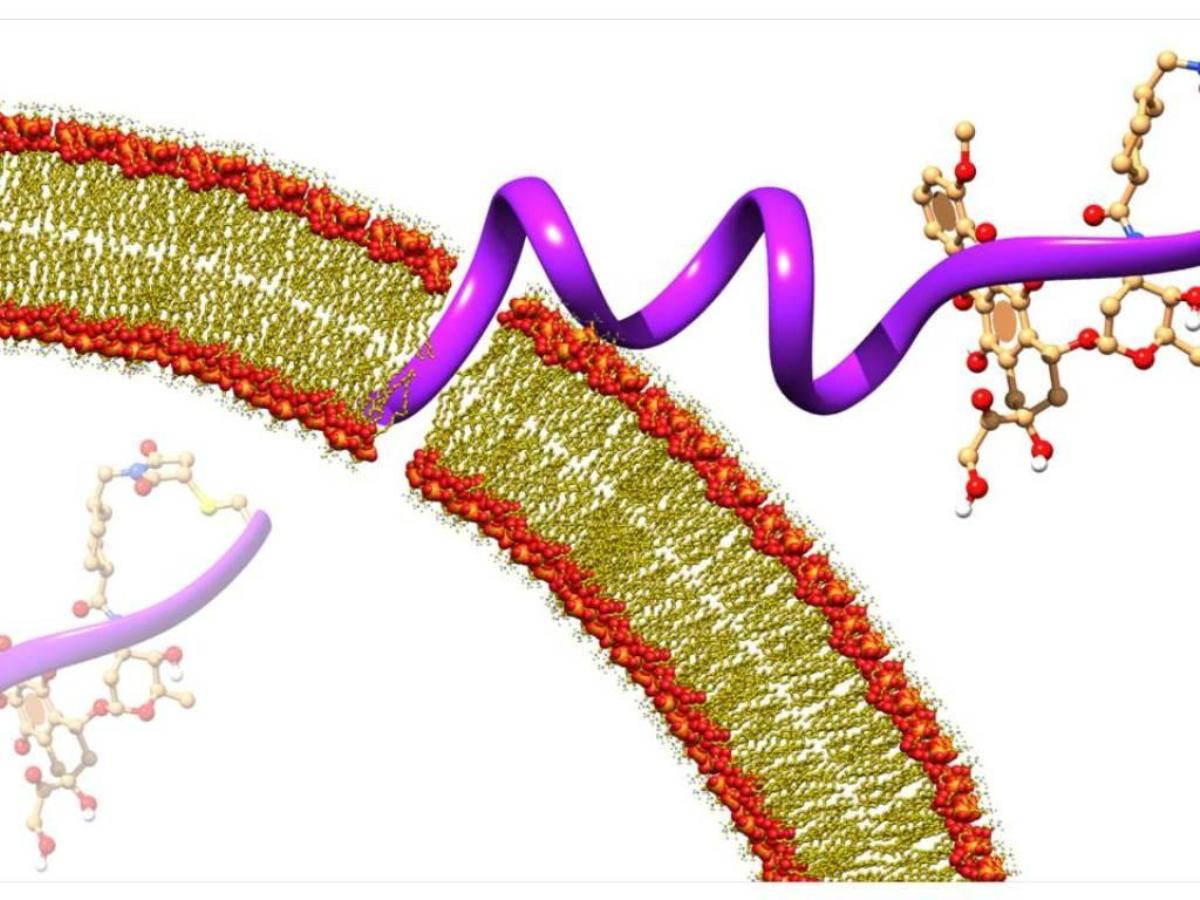{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
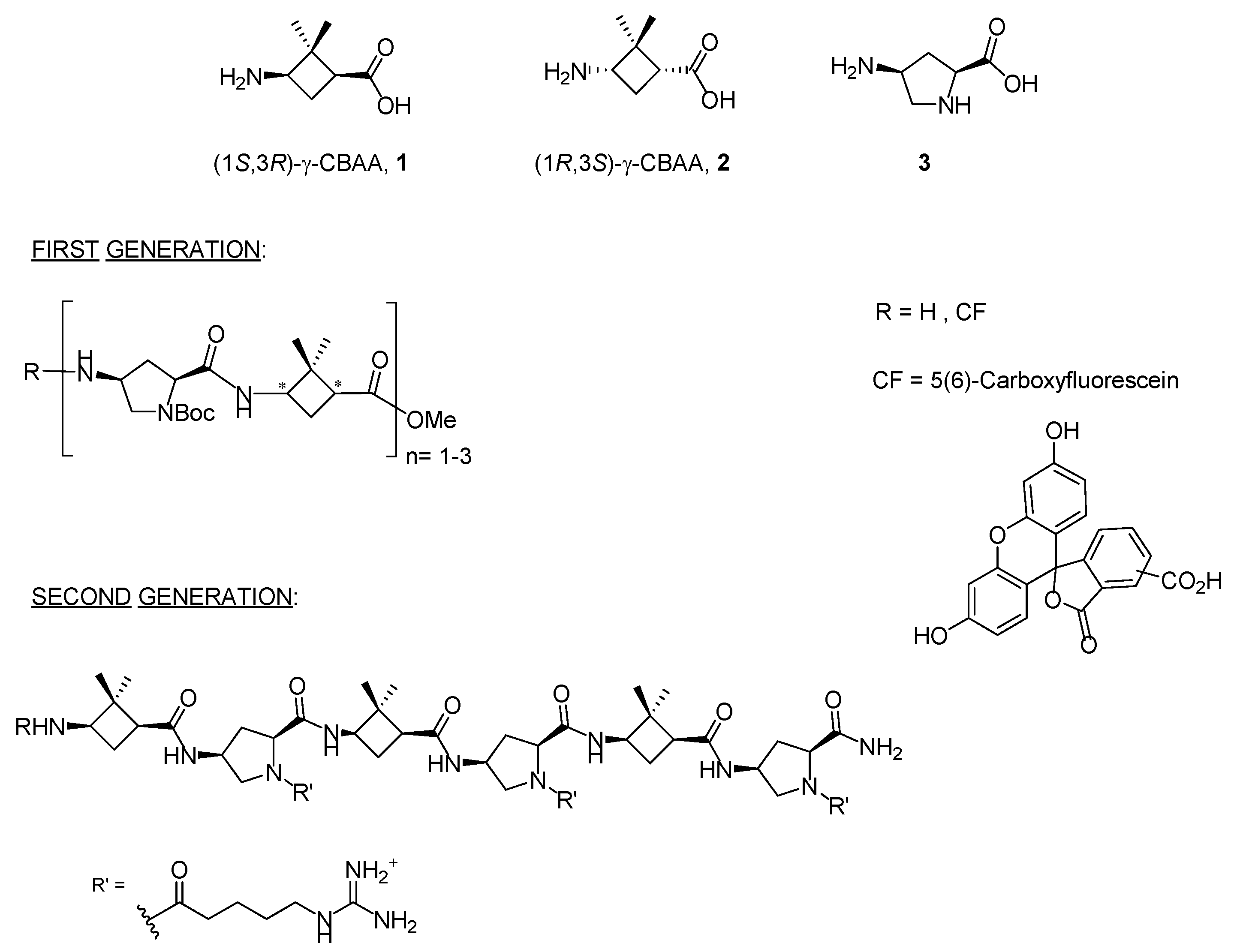
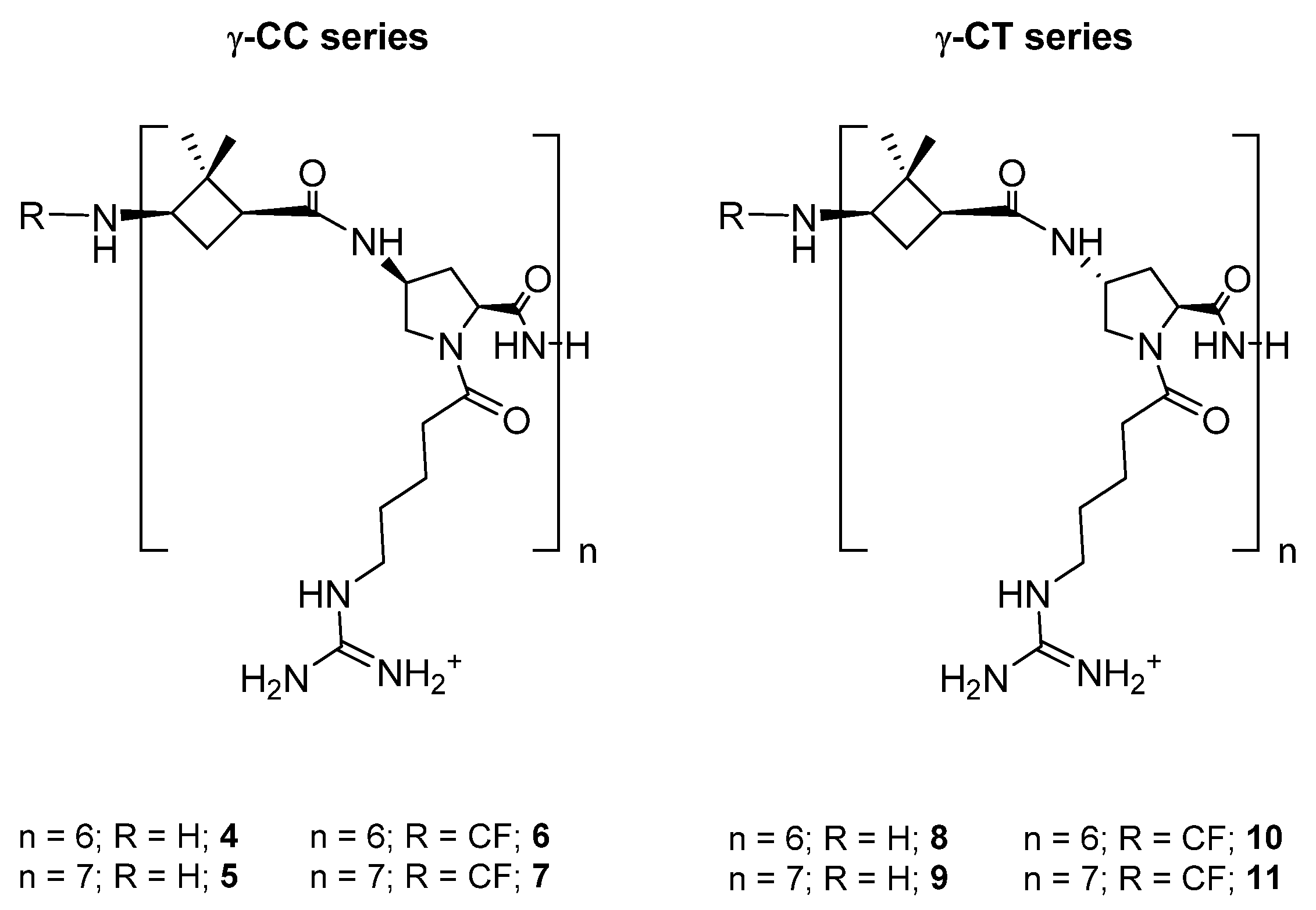
2.1. Design and Synthesis of Peptides and Conjugates
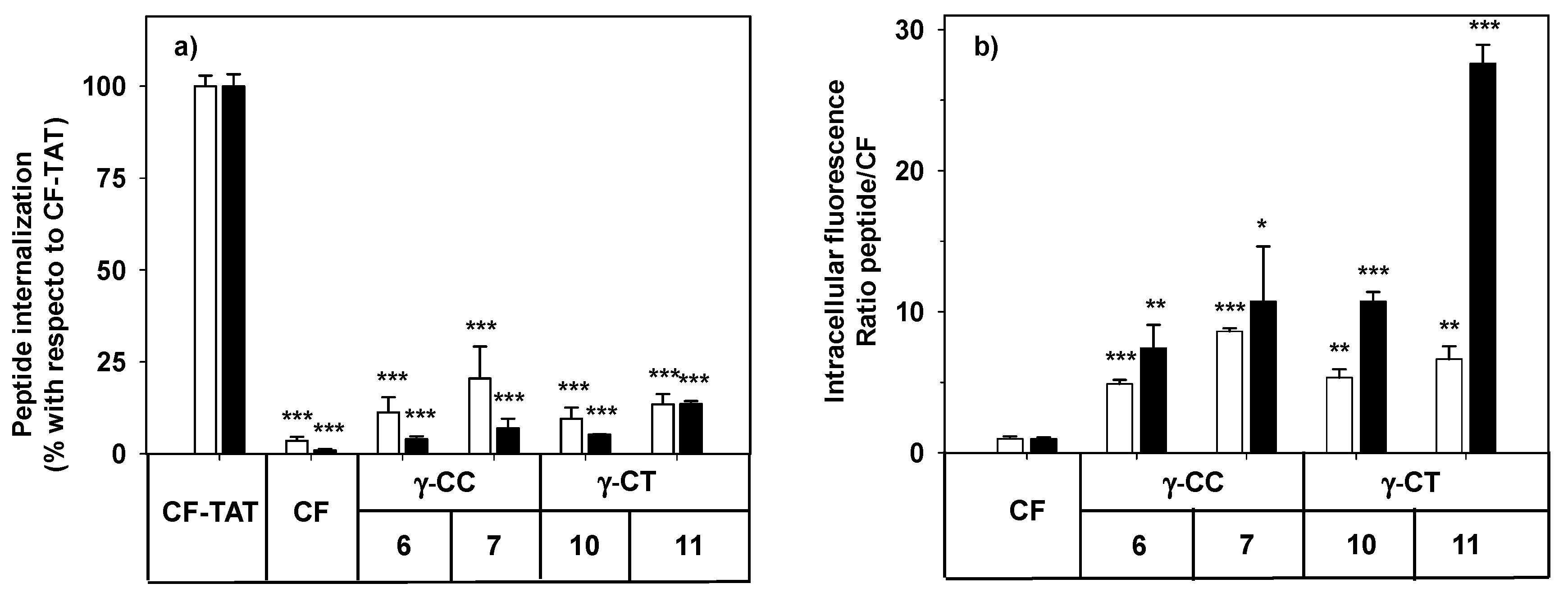
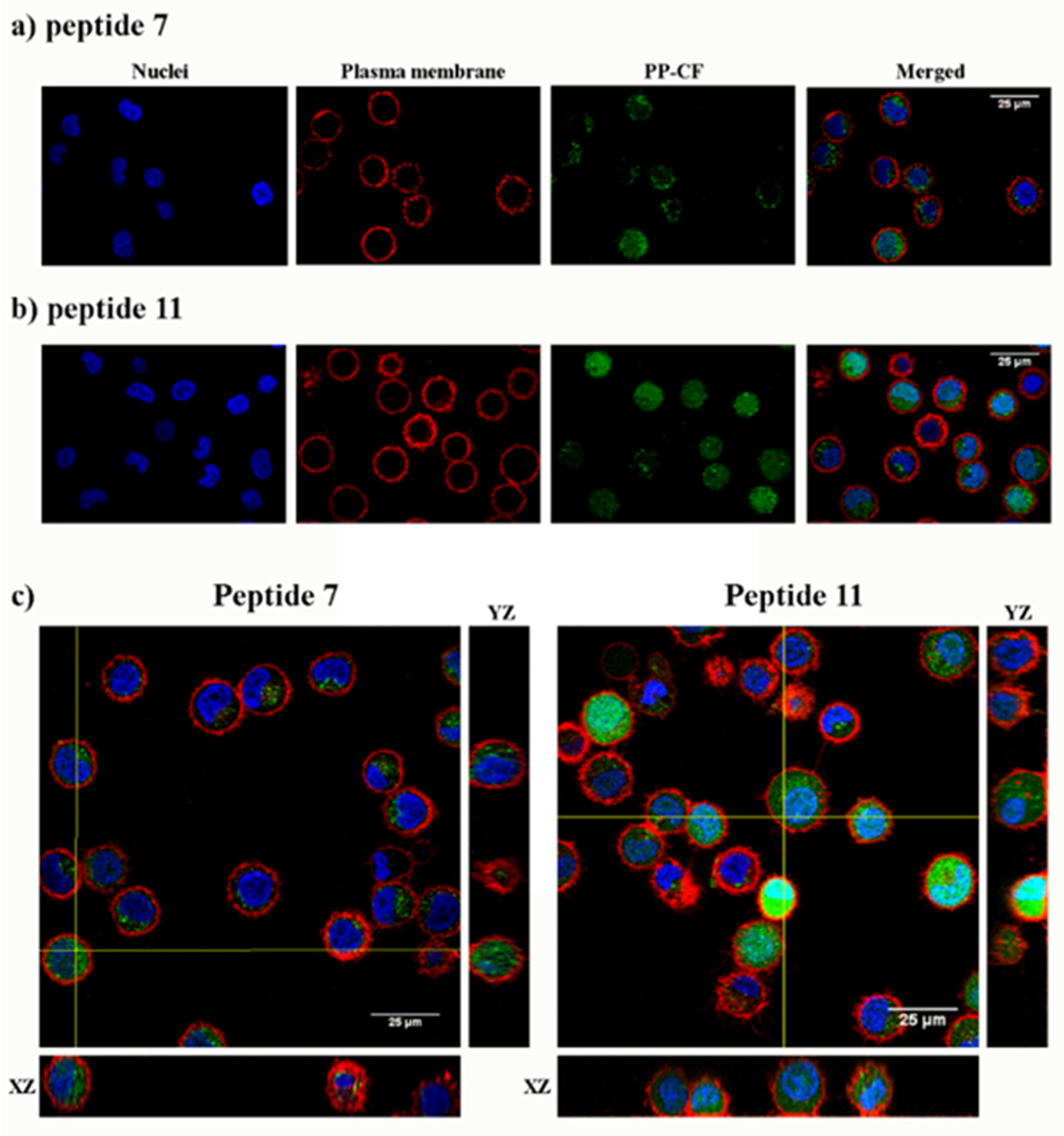
2.2. Cytotoxicity and Cellular Uptake in HeLa Cells
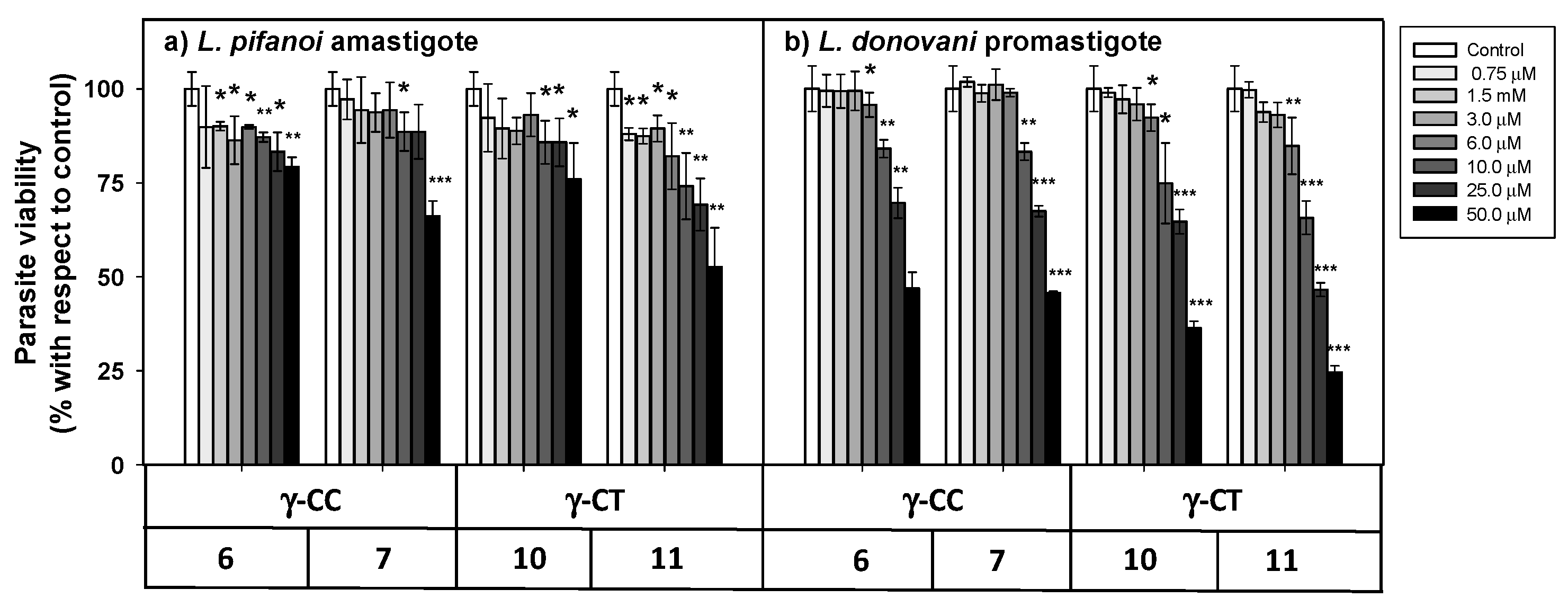
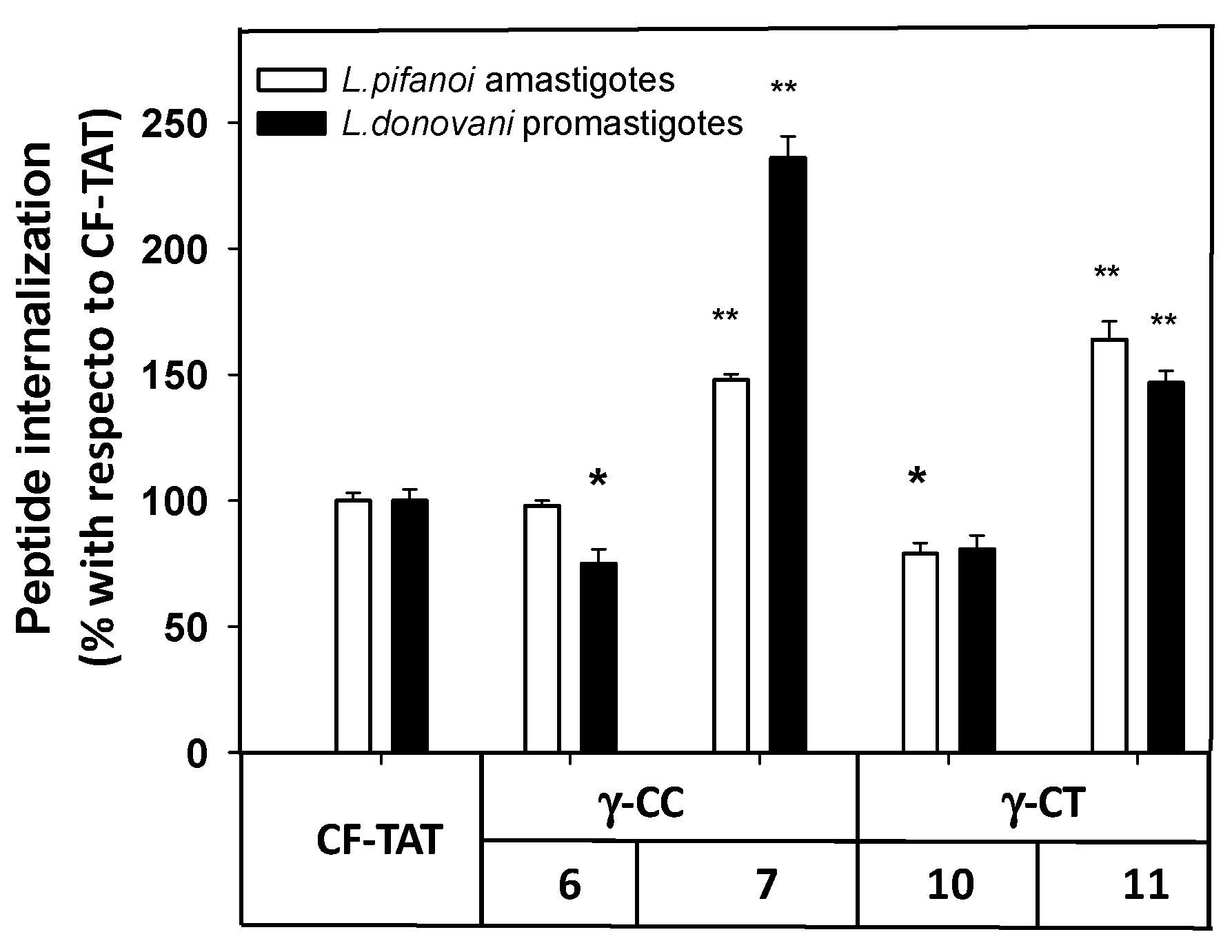
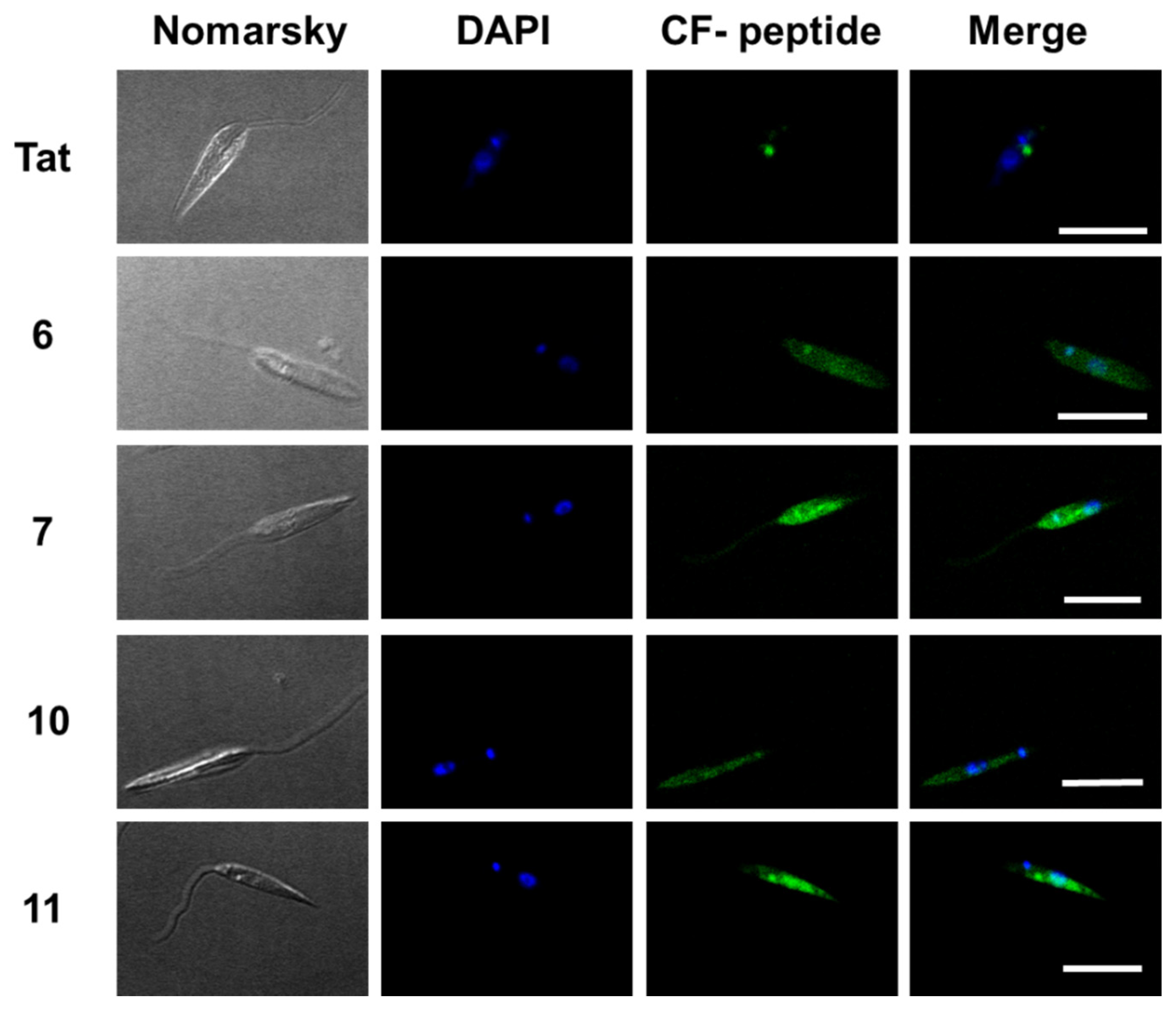
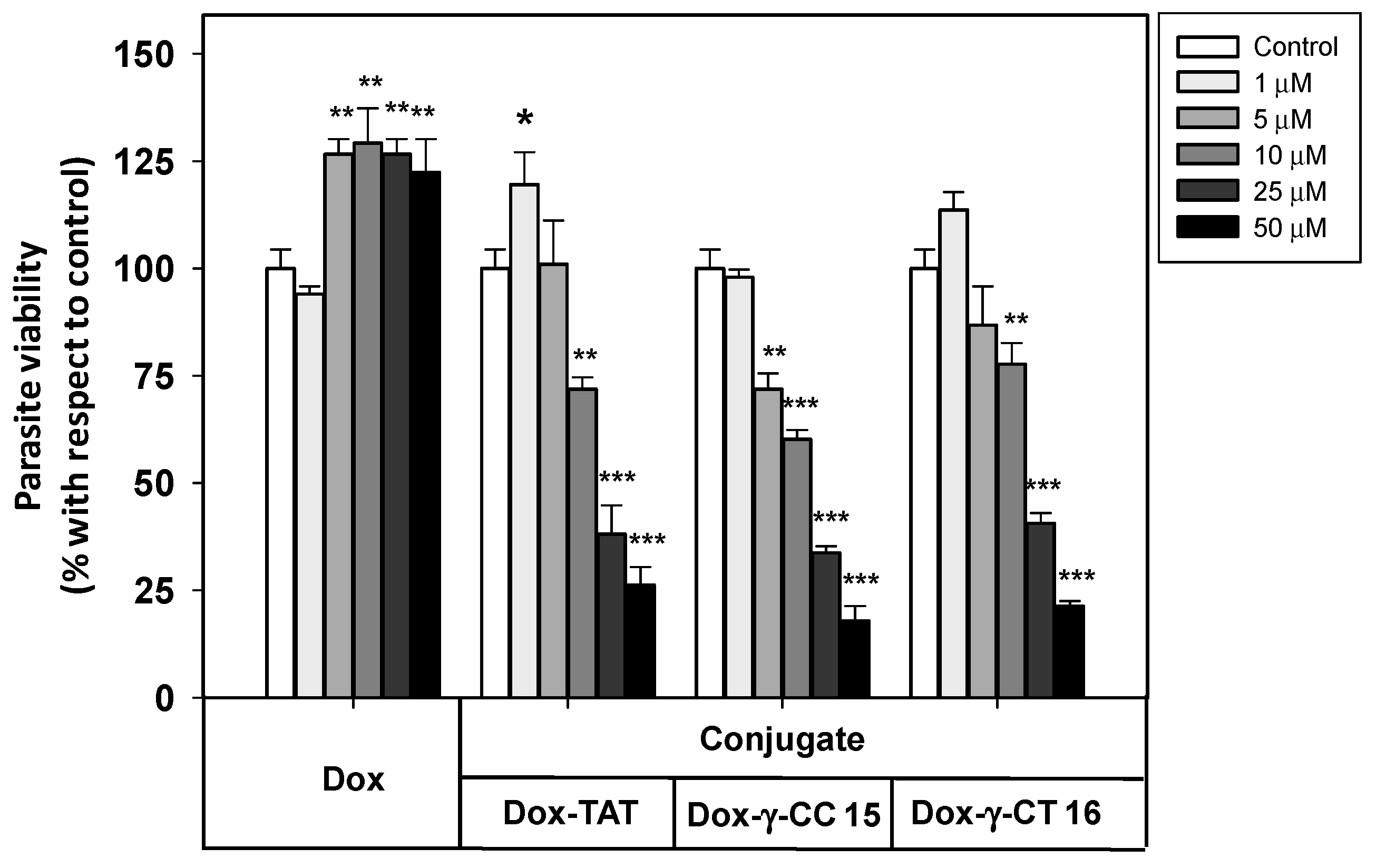
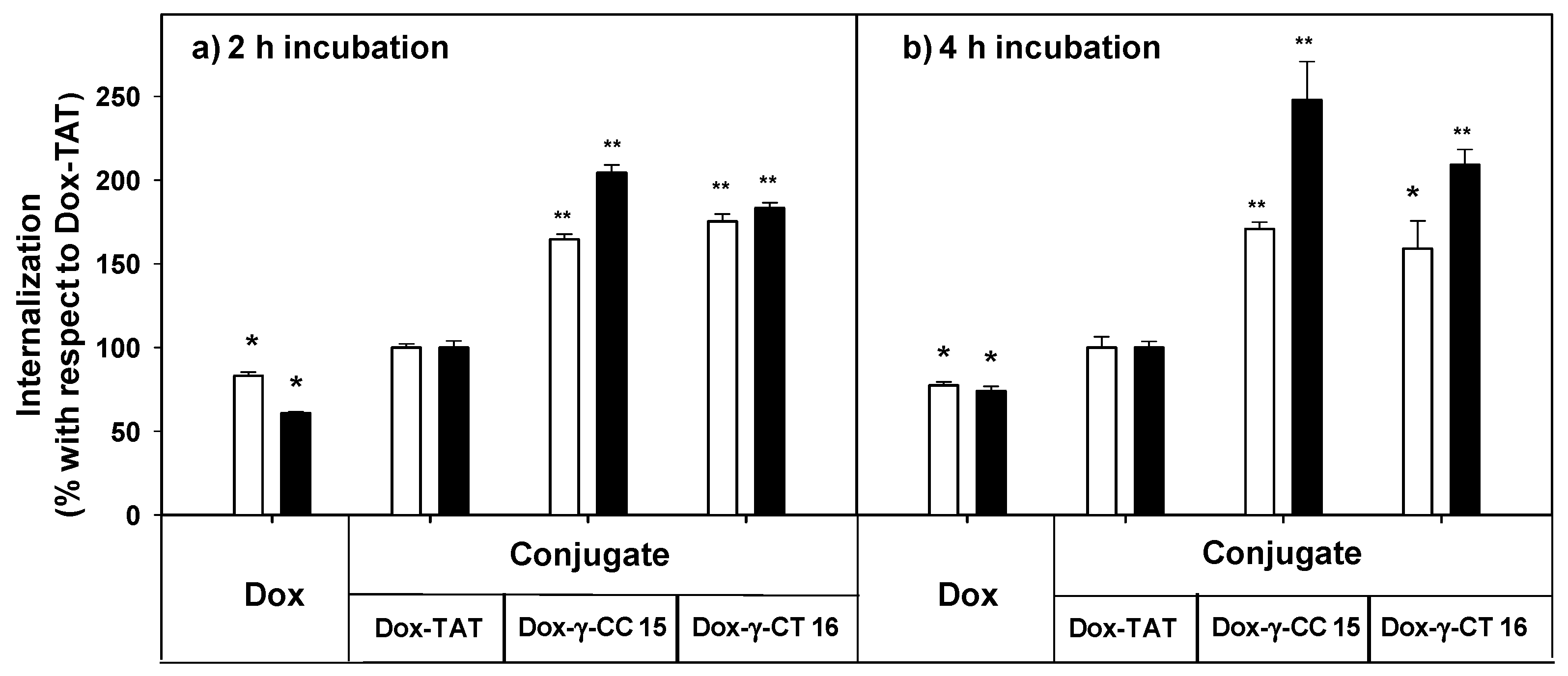
2.3. Uptake, Microbicidal Activity and Intracellular Location of Peptides on Leishmania Parasites
2.4. CD Spectroscopy
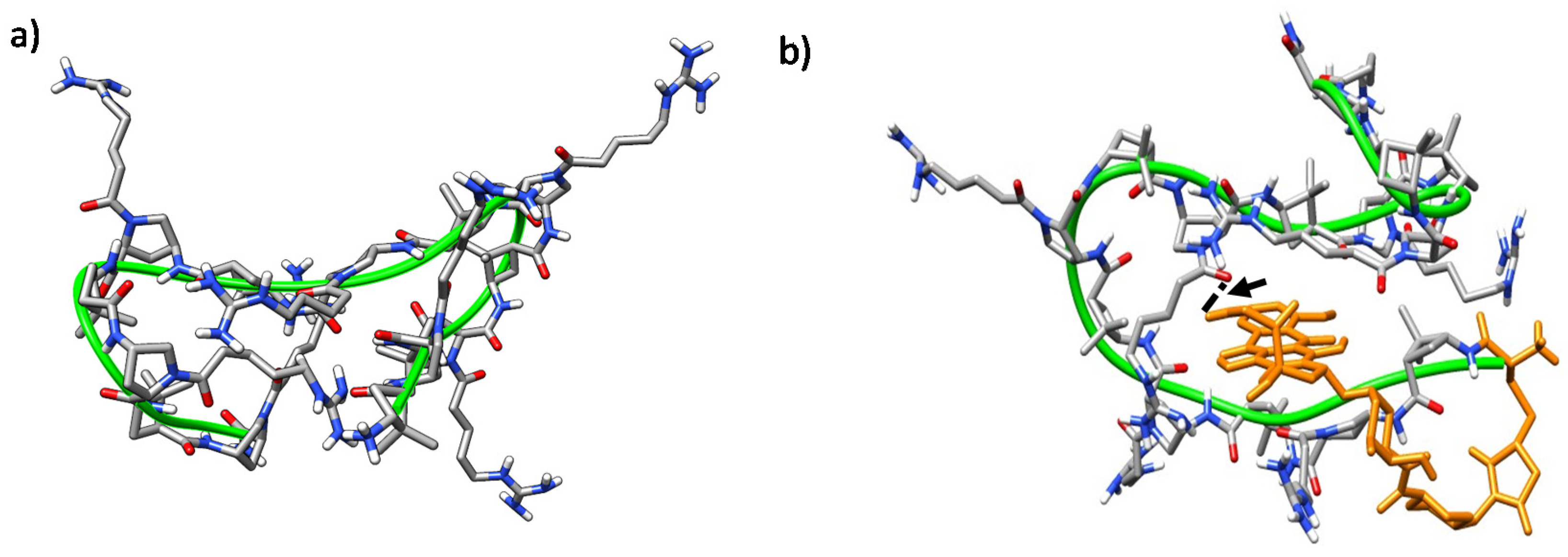
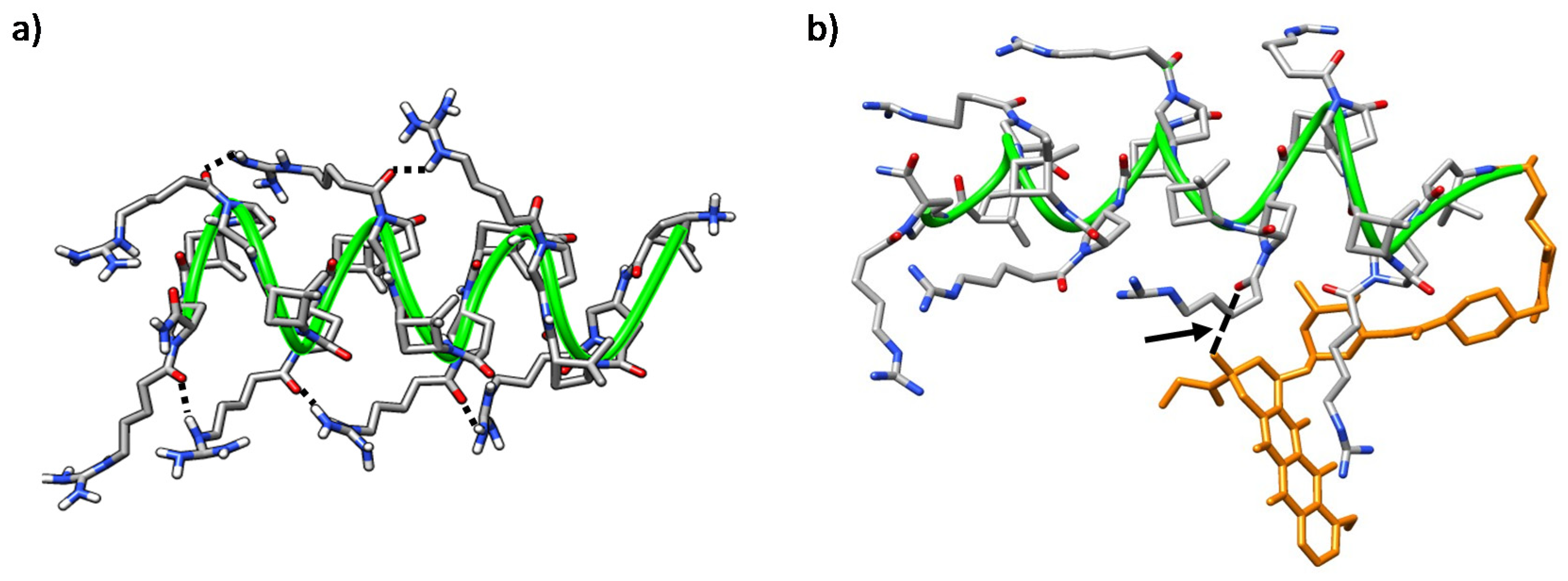
2.5. Molecular Modeling
3. Materials and Methods
3.1. Synthesis of the γ-CC Peptides (4–7), γ-CT Peptides (8–11) and TAT48–57 Peptides
3.2. Synthesis of Cysteine Elongated Peptides (12) and (13)
3.3. Synthesis of Doxorubicin-MCC (14)
3.4. Synthesis of Conjugates Dox-γ-CC (15) and Dox-γ-CT (16)
3.5. Peptide and Conjugate Purification
3.6. Peptide Characterization
3.6.1. Peptide γ-CC (4)
3.6.2. Peptide γ-CC (5)
3.6.3. Peptide CF-γ-CC (6)
3.6.4. Peptide CF-γ-CC (7)
3.6.5. Peptide γ-CT (8)
3.6.6. Peptide γ-CT (9)
3.6.7. Peptide CF-γ-CT (10)
3.6.8. Peptide CF-γ-CT (11)
3.6.9. TAT48–57
3.6.10. TAT48–57-CF
3.6.11. Peptide (12)
3.6.12. Peptide (13)
3.6.13. TAT48–57-Cys
3.6.14. Conjugate Dox- γ-CC (15)
3.6.15. Conjugate Dox- γ-CT (16)
3.6.16. Dox-TAT48–57
3.7. Cellular Viability, Internalization, and Localization Experiments with HeLa Cells
3.7.1. HeLa Cells Culture
3.7.2. Cellular Viability
3.7.3. Peptide Internalization
3.8. Cellular Viability, Internalization and Localization Experiments with Leishmania Parasites
3.8.1. Cellular Viability
3.8.2. Peptide Uptake for Leishmania donovani Promastigotes
3.8.3. Intracellular Localization of the Internalized Peptides in Leishmania donovani Promastigotes
3.9. Circular Dichroism (CD) Spectroscopy Details
3.10. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| Alloc | Allyloxycarbonyl |
| CBAA | Cyclobutane Amino Acid |
| CF | 5(6)-Carboxyfluorescein |
| CPP | Cell Penetrating Peptide |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| DCM | Dichloromethane |
| DDS | Drug Delivery Systems |
| DFT | Density Functional Theory |
| DIPEA | Diisopropylethylamine |
| DMF | Dimethylformamide |
| DMSO | Dimethyl Sulfoxide |
| Dox | Doxorubicin |
| EDTA | Ethylenediaminetetraacetic Acid |
| ESI | Electrospray Ionization |
| FBS | Fetal Bovine Serum |
| Glc | Glucose |
| HBSS | Hanks Buffered Saline Solution |
| Fmoc | Fluorenylmethyloxycarbonyl |
| HPLC | High-Performance Liquid Chromatography |
| LPG | Lipophosphoglycan |
| MCC | 4-(N-maleimidomethyl)cyclohexane-1-carboxylate |
| MD | Molecular Dynamics |
| MEM | Minimum Essential Medium |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide |
| NMR | Nuclear Magnetic Resonance |
| PBS | Phosphate-Buffered Saline |
| PCA | Principal Component Analysis |
| PES | Potential Energy Surface |
| PI | Propidium Iodide |
| RESP | Restrained Electrostatic Potential |
| RMSD | Root-Mean-Square Deviation |
| RMSF | Root-Mean-Square Fluctuation |
| RP | Reverse Phase |
| SD | Standard Deviation |
| SM | Supplementary Materials |
| SMCC | Succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate |
| SPPS | Solid Phase Peptide Synthesis |
References
- Hossen, S.; Hossain, M.; Basher, M.K.; Mia, M.N.H.; Rahman, M.T.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, F.; Xiong, F.; Gu, N. The smart drug delivery system and its clinical potential. Theranostics 2016, 6, 1306–1323. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Heitz, V.; Sour, A.; Bolze, F.; Kessler, P.; Flamigni, L.; Ventura, B.; Bonnet, C.S.; Toth, E. A Theranostic agent combining a two-photon-absorbing photosensitizer for photodynamic therapy and a gadolinium(III) complex for MRI detection. Chem. Eur. J. 2016, 22, 2775–2786. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, Ü. Cell-penetrating Peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Langel, Ü. Cell-Penetrating Peptides in Processes and Applications; CRC Press Pharmacology and Toxicology Series: Boca Raton, FL, USA, 2002. [Google Scholar]
- Lundberg, P.; Langel, Ü. A brief introduction to cell-penetrating peptides. J. Mol. Recognit. 2003, 16, 227–233. [Google Scholar] [CrossRef]
- Vivès, E.; Schmidt, J.; Pèlegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 2008, 1786, 126–138. [Google Scholar] [CrossRef]
- Koren, E.; Torchillin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, Ü. Cell-penetrating peptides: Design, synthesis, and applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, J.; Xu, D. Cell penetrating peptides as noninvasive transmembrane vectors for the development of new functional drug delivery systems. J. Control. Release 2016, 229, 130–139. [Google Scholar] [CrossRef]
- Dissanayake, S.; Denny, W.A.; Gamage, S.; Sarojini, V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J. Control. Release 2017, 250, 62–76. [Google Scholar] [CrossRef]
- Fominaya, J.; Bravo, J.; Rebollo, A. Strategies to stabilize cell penetrating peptides for in vivo applications. Ther. Del. 2015, 6, 1171–1194. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Dobitz, S.; Aronoff, M.R.; Wennemers, H. Oligoprolines as molecular entities for controlling distance in biological and material sciences. Acc. Chem. Res. 2017, 50, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Potocky, T.B.; Menon, A.K.; Gellman, S.H. Effects of conformational stability and geometry of guanidinium display on cell entry by β-peptides. J. Am. Chem. Soc. 2005, 127, 3686–3687. [Google Scholar] [CrossRef]
- Nagel, Y.A.; Raschle, P.S.; Wennemers, H. Effect of preorganized charge-display on the cell-penetrating properties of cationic peptides. Angew. Chem. 2017, 56, 122–126. [Google Scholar] [CrossRef]
- Tian, Y.; Zeng, X.; Li, J.; Jiang, Y.; Zhao, H.; Wang, D.; Huang, X.; Li, Z. Achieving enhanced cell penetration of short conformationally constrained peptides through amphiphilicity tuning. Chem. Sci. 2017, 8, 7576–7581. [Google Scholar] [CrossRef]
- Lättig-Tünnemann, G.; Prinz, M.; Hoffmann, D.; Behlke, J.; Palm-Apergi, C.; Morano, I.; Herce, H.D.; Cardoso, M.C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, 2, 453. [Google Scholar] [CrossRef]
- Nischan, N.; Herce, H.D.; Natale, F.; Bohlke, N.; Budisa, N.; Cardoso, M.C.; Hackenberger, C.P.R. Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem. 2015, 54, 1950–1953. [Google Scholar] [CrossRef]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef]
- Gutiérrez-Abad, R.; Carbajo, D.; Nolis, P.; Acosta-Silva, C.; Cobos, J.A.; Illa, O.; Royo, M.; Ortuño, R.M. Synthesis and structural study of highly constrained hybrid cyclobutane-proline γ,γ-peptides. Amino Acids 2011, 41, 673–686. [Google Scholar] [CrossRef]
- Gorrea, E.; Carbajo, D.; Gutiérrez-Abad, R.; Illa, O.; Branchadell, V.; Royo, M.; Ortuño, R.M. Searching for new cell-penetrating agents: Hybrid cyclobutane–proline γ,γ-peptides. Org. Biomol. Chem. 2012, 10, 4050–4057. [Google Scholar] [CrossRef]
- Farrera-Sinfreu, J.; Giralt, E.; Castel, S.; Albericio, F.; Royo, M. Cell-penetrating cis-γ-amino-l-proline-derived peptides. J. Am. Chem. Soc. 2005, 127, 9459–9468. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.; Augusto, M.T.; Felício, M.R.; Hollmann, A.; Franco, O.L.; Gonçalves, S.; Santos, N.C. Designing improved active peptides for therapeutic approaches against infectious diseases. Biotechnol. Adv. 2018, 36, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Rivas, L.; Nácher-Vázquez, M.; Andreu, D. The Physical Matrix of the Plasma Membrane as a Target: The Charm of Drugs with Low Specificity. In Drug Discovery for Leishmaniasis; Rivas, L., Gil, C., Eds.; The Royal Society of Chemistry: London, UK, 2018; pp. 248–281. [Google Scholar]
- Agbowuro, A.A.; Huston, W.M.; Gamble, A.B.; Tyndall, J.D.A. Proteases and protease inhibitors in infectious diseases. Med. Res. Rev. 2018, 38, 1295–1331. [Google Scholar] [CrossRef]
- World Health Organization. Available online: http://www.who.int/leishmaniasis/en/ (accessed on 7 September 2020).
- Chakravarty, J.; Sundar, S. Drug resistance in leishmaniasis. J. Glob. Infect. Dis. 2010, 2, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Nagle, A.S.; Khare, S.; Kumar, A.B.; Supek, F.; Buchynskyy, A.; Mathison, C.J.N.; Chennamaneni, N.K.; Pendem, N.; Buckner, F.S.; Gelb, M.H.; et al. Recent developments in drug discovery for leishmaniasis and human african trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. [Google Scholar] [CrossRef] [PubMed]
- Hefnawy, A.; Berg, M.; Dujardin, J.-C.; De Muylder, G. Exploiting knowledge on Leishmania drug resistance to support the quest for new drugs. Trends Parasitol. 2017, 33, 162–174. [Google Scholar] [CrossRef]
- Paromomycin SULFATE. Available online: https://www.webmd.com/drugs/2/drug-5160/paromomycin-oral/details#side-effects (accessed on 4 September 2020).
- Drugs and Supplements Miltefosine (Oral Route). Available online: htpps://www.mayoclinic.org/drugs-supplements/miltefosine-oral-route/side-effects/drg-20095231 (accessed on 4 September 2020).
- Naderer, T.; Vince, J.E.; McConville, M.J. Surface determinants of Leishmania parasites and their role in infectivity in the mammalian host. Curr. Mol. Med. 2004, 4, 649–665. [Google Scholar] [CrossRef]
- Walrant, A.; Cardon, S.; Burlina, F.; Sagan, S. Membrane crossing and membranotropic activity of cell-penetrating peptides: Dangerous liaisons? Acc. Chem. Res. 2017, 50, 2968–2975. [Google Scholar] [CrossRef]
- Morgan, G.W.; Hall, B.S.; Denny, P.W.; Carrington, M.; Field, M.C. The Kinetoplastida endocytic apparatus. Part I: A dynamic system for nutrition and evasion of host defences. Trends Parasitol. 2002, 18, 491–496. [Google Scholar] [CrossRef]
- Forestier, C.L.; Gao, Q.; Boons, G.J. Leishmania lipophosphoglycan: How to establish structure-activity relationships for this highly complex and multifunctional glycoconjugate? Front. Cell. Infect. Microbiol. 2015, 4, 193. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Atayde, V.D.; Isnard, A.; Hassani, K.; Shio, M.T. Leishmania virulence factors: Focus on the metalloprotease GP63. Microbes Infect. 2012, 14, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Luque-Ortega, J.R.; Rivas, L. Characterization of the Leishmanicidal Activity of Antimicrobial Peptides. In Antimicrobial Peptides. Methods in Molecular Biology (Methods and Protocols); Giuliani, A., Rinaldi, A., Eds.; Humana Press: Totowa, NJ, USA, 2010; Volume 618, pp. 393–420. [Google Scholar]
- Silva, T.; Abengózar, M.A.; Fernández-Reyes, M.; Andreu, D.; Nazmi, K.; Bolscher, J.G.M.; Bastos, M.; Rivas, L. Enhanced leishmanicidal activity of cryptopeptide chimeras from the active N1 domain of bovine lactoferrin. Amino Acids 2012, 43, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Santaquiteria, M.; Sánchez-Murcia, P.A.; Toro, M.A.; de Lucio, H.; Gutiérrez, K.J.; de Castro, S.; Carneiro, F.A.C.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.-J.; et al. First example of peptides targeting the dimer interface of Leishmania infantum trypanothione reductase with potent in vitro antileishmanial activity. Eur. J. Med. Chem. 2017, 135, 49–59. [Google Scholar] [CrossRef]
- Keller, A.-A.; Mussbach, F.; Breitling, R.; Hemmerich, P.; Schaefer, B.; Lorkowski, S.; Reissmann, S. Relationships between cargo, cell penetrating peptides and cell type for uptake of non-covalent complexes into live cells. Pharmaceuticals 2013, 6, 184–203. [Google Scholar] [CrossRef]
- Keller, A.A.; Breitling, R.; Hemmerich, P.; Kappe, K.; Braun, M.; Wittig, B.; Schaefer, B.; Lorkowski, S.; Reissmann, S. Transduction of proteins into Leishmania tarentolae by formation of non-covalent complexes with cell-penetrating peptides. J. Cell. Biochem. 2014, 115, 243–252. [Google Scholar] [CrossRef]
- Keller, A.A.; Scheiding, B.; Breitling, R.; Licht, A.; Hemmerich, P.; Lorkowski, S.; Reissmann, S. Transduction and transfection of difficult-to-transfect cells: Systematic attempts for the transfection of protozoa Leishmania. J. Cell. Biochem. 2019, 120, 14–27. [Google Scholar] [CrossRef]
- Kóczán, G.; Ghose, A.C.; Mookerjee, A.; Hudecz, F. Methotrexate conjugate with branched polypeptide influences Leishmania donovani infection in vitro and in experimental animals. Bioconjug. Chem. 2002, 13, 518–524. [Google Scholar] [CrossRef]
- Luque-Ortega, J.R.; de la Torre, B.G.; Hornillos, V.; Bart, J.-M.; Rueda, C.; Navarro, M.; Amat-Guerri, F.; Ulises Acuña, A.; Andreu, D.; Rivas, L. Defeating Leishmania resistance to miltefosine (hexadecylphosphocholine) by peptide-mediated drug smuggling: A proof of mechanism for trypanosomatid chemotherapy. J. Control. Release 2012, 161, 835–842. [Google Scholar] [CrossRef]
- Torre, B.G.; Hornillos, V.; Luque-Ortega, J.R.; Abengozar, M.A.; Amat-Guerri, F.; Ulises Acuna, A.; Rivas, L.; Andreu, D. A BODIPY-embedding miltefosine analog linked to cell-penetrating TAT(48-60) peptide favors intracellular delivery and visualization of the antiparasitic drug. Amino Acids 2014, 46, 1047–1058. [Google Scholar] [CrossRef]
- de Lucio, H.; Gamo, A.M.; Ruiz-Santaquiteria, M.; de Castro, S.; Sánchez-Murcia, P.A.; Toro, M.A.; Gutiérrez, K.J.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.-J.; et al. Improved proteolytic stability and potent activity against Leishmania infantum trypanothione reductase of α/β-peptide foldamers conjugated to cell-penetrating peptides. Eur. J. Med. Chem. 2017, 140, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Defaus, S.; Gallo, M.; Abengózar, M.A.; Rivas, L.; Andreu, D. A synthetic strategy for conjugation of paromomycin to cell-penetrating TAT(48-60) for delivery and visualization into Leishmania parasites. Int. J. Pept. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.; Quiang, Z.; Chu, M.; Shi, D.; Ren, J. Polymeric drug delivery system with actively targeted cell penetration and nuclear targeting for cancer therapy. ACS Appl. Bio Mater. 2019, 2, 1724–1731. [Google Scholar] [CrossRef]
- Sett, R.; Basu, N.; Ghosh, A.K.; Das, P.K. Potential of doxorubicin as an antileishmanial agent. J. Parasitol. 1992, 78, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Das, L.; Kole, L.; Karmakar, S.; Datta, N.; Das, P.K. Targeting of parasite-specific immunoliposome-encapsulated doxorubicin in the treatment of experimental visceral leishmaniasis. J. Infect. Dis. 2004, 189, 1024–1034. [Google Scholar] [CrossRef]
- Sundar, S.; Chakravarty, J. Investigational drugs for visceral leishmaniosis. Expert Opin. Investig. Drugs 2015, 24, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus TAT trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the TAT protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 TAT protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef]
- Mosnann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Futaki, S. Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv. Drug. Deliv. Rev. 2005, 57, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, T.A.; Timmers, M.M.; Olson, E.S.; Jiang, T.; Tsien, R.Y. Systemic in vivo distribution of activatable cell penetrating peptides is superior to cell penetrating peptides. Integr. Biol. 2009, 1, 371–381. [Google Scholar] [CrossRef] [PubMed]
- WHO Technical report series. In Proceedings of the Control of the leishmaniases: Report of a Meeting of the WHO Expert Commitee on the Control of Leishmaniases, Geneva, Switzerland, 22–26 March 2010.
- Seebach, D.; Brenner, M.; Rueping, M.; Jaun, B. γ2-, γ 3-, and γ 2,3,4-Amino acids, coupling to γ-hexapeptides: CD spectra, NMR solution and X-ray Crystal structures of γ -peptides. Chem. Eur. J. 2002, 8, 573–584. [Google Scholar] [CrossRef]
- Deshayes, S.; Plénat, T.; Aldrian-Herrada, G.; Divita, G.; Le Grimellec, C.; Heitz, F. Primary amphipathic cell-penetrating peptides: structural requirements and interactions with model membranes. Biochemistry 2004, 43, 7698–7706. [Google Scholar] [CrossRef]
- Ziegler, A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv. Drug Deliv. Rev. 2008, 60, 580–597. [Google Scholar] [CrossRef]
- Zaro, J.L.; Vekich, J.E.; Tran, T.; Shen, W.-C. Nuclear localization of cell-penetrating peptides is dependent on endocytosis rather than cytosolic delivery in CHO cells. Mol. Pharm. 2009, 6, 337–344. [Google Scholar] [CrossRef]
- Jiao, C.Y.; Delaroche, D.; Durlina, F.; Alves, I.D.; Chassaing, G.; Sagan, S. Translocation and endocytosis for cell-penetrating peptide internalization. J. Biol. Chem. 2009, 284, 33957–33965. [Google Scholar] [CrossRef]
- Pae, J.; Liivamägi, L.; Lubenets, D.; Arukuusk, P.; Langel, Ü.; Pooga, M. Glycosaminoglycans are required for translocation of amphipathic cell-penetrating peptides across membranes. Biochim. Biophys. Acta 2016, 1858, 1860–1867. [Google Scholar] [CrossRef]
- Farrera-Sinfreu, J.; Zaccaro, L.; Vidal, D.; Salvatella, X.; Giralt, E.; Pons, M.; Albericio, F.; Royo, M. A new class of foldamers based on cis-γ-amino-l-proline. J. Am. Chem. Soc. 2004, 126, 6048–6057. [Google Scholar] [CrossRef]
- Nakase, I.; Takeuchi, T.; Tanaka, G.; Futaki, S. Methodological and cellular aspects that govern the internalization mechanisms of arginine-rich cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Izawa, H.; Kinai, M.; Ifuku, S.; Morimoto, M.; Saimoto, H. Guanidinylated chitosan inspired by arginine-rich cell-penetrating peptides. Int. J. Biol. Macromol. 2019, 125, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Vazdar, M.; Heyda, J.; Mason, P.E.; Tesei, G.; Allolio, C.; Lund, M.; Jungwirth, P. Arginine “magic”: Guanidinium Like-Charge Ion Pairing from Aqueous Salts to Cell Penetrating Peptides. Acc. Chem. Res. 2018, 51, 1455–1464. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.; McGibbon, R.; Zhao, Y.; Beauchamp, K.; Wang, L.; Simmonett, A.; Harrigan, M.; Stern, C.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- Rodríguez-Guerra Pedregal, J.; Alonso-Cotchico, L.; Velasco-Carneros, L.; Mareéchal, J.-D. OMM Protocol: A command line application to launch molecular dynamics simulations with openMM. ChemRxiv 2018. [Google Scholar] [CrossRef]
- Sciortino, G.; Sánchez-Aparicio, J.-E.; Rodríguez-Guerra Pedregal, J.; Garribba, E.; Maréchal, J.-D. Computational insight into the interaction of oxaliplatin with insulin. Metallomics 2019, 11, 765–773. [Google Scholar] [CrossRef]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Illa, O.; Olivares, J.-A.; Gaztelumendi, N.; Martínez-Castro, L.; Ospina, J.; Abengozar, M.-Á.; Sciortino, G.; Maréchal, J.-D.; Nogués, C.; Royo, M.; et al. Chiral Cyclobutane-Containing Cell-Penetrating Peptides as Selective Vectors for Anti-Leishmania Drug Delivery Systems. Int. J. Mol. Sci. 2020, 21, 7502. https://doi.org/10.3390/ijms21207502
Illa O, Olivares J-A, Gaztelumendi N, Martínez-Castro L, Ospina J, Abengozar M-Á, Sciortino G, Maréchal J-D, Nogués C, Royo M, et al. Chiral Cyclobutane-Containing Cell-Penetrating Peptides as Selective Vectors for Anti-Leishmania Drug Delivery Systems. International Journal of Molecular Sciences. 2020; 21(20):7502. https://doi.org/10.3390/ijms21207502
Chicago/Turabian StyleIlla, Ona, José-Antonio Olivares, Nerea Gaztelumendi, Laura Martínez-Castro, Jimena Ospina, María-Ángeles Abengozar, Giuseppe Sciortino, Jean-Didier Maréchal, Carme Nogués, Míriam Royo, and et al. 2020. "Chiral Cyclobutane-Containing Cell-Penetrating Peptides as Selective Vectors for Anti-Leishmania Drug Delivery Systems" International Journal of Molecular Sciences 21, no. 20: 7502. https://doi.org/10.3390/ijms21207502
APA StyleIlla, O., Olivares, J.-A., Gaztelumendi, N., Martínez-Castro, L., Ospina, J., Abengozar, M.-Á., Sciortino, G., Maréchal, J.-D., Nogués, C., Royo, M., Rivas, L., & Ortuño, R. M. (2020). Chiral Cyclobutane-Containing Cell-Penetrating Peptides as Selective Vectors for Anti-Leishmania Drug Delivery Systems. International Journal of Molecular Sciences, 21(20), 7502. https://doi.org/10.3390/ijms21207502