Toward Drug-Like Multispecific Antibodies by Design

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
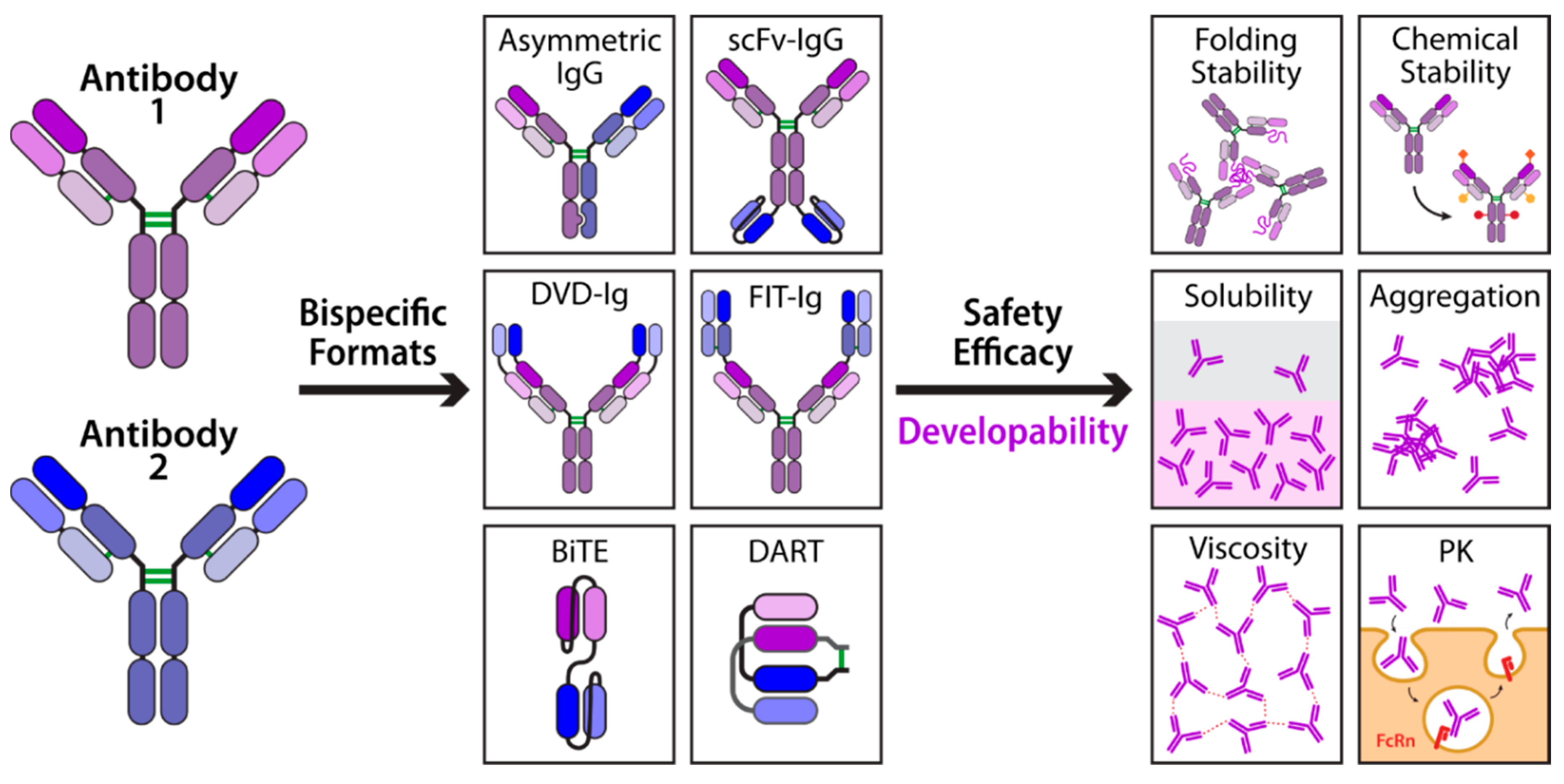
1. Introduction
2. Physical and Chemical Stability
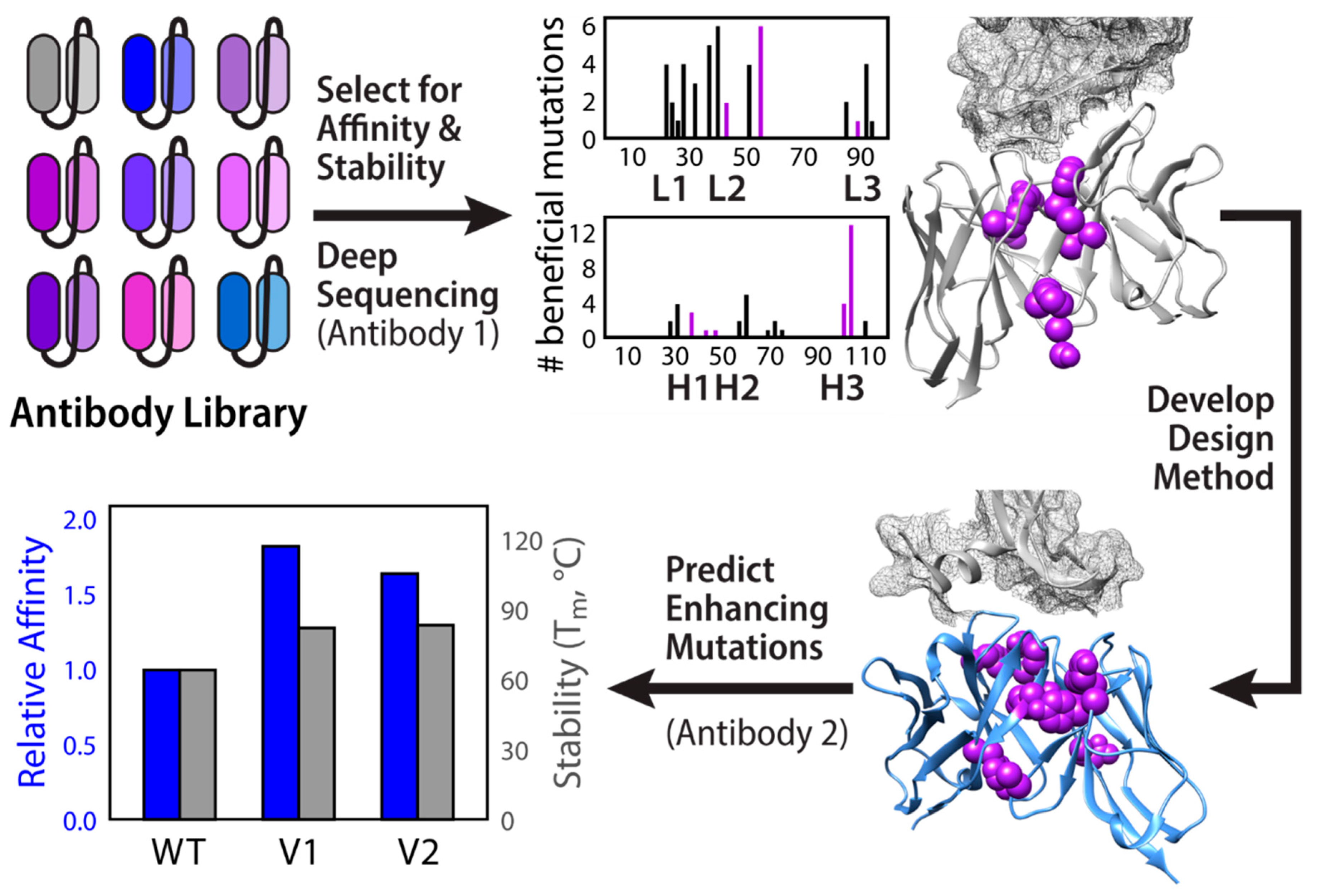
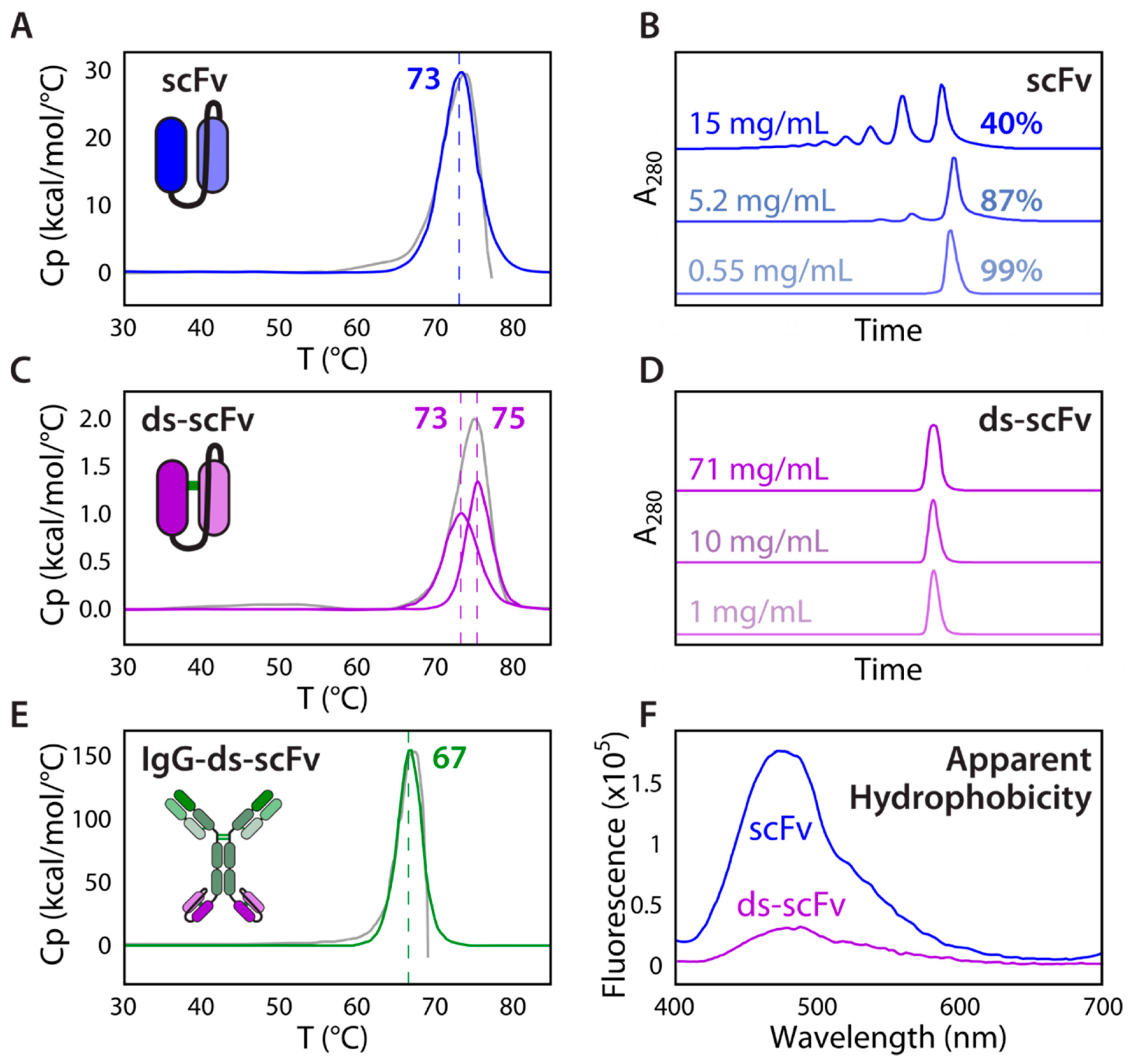
2.1. Folding and Assembly
2.2. Aggregation
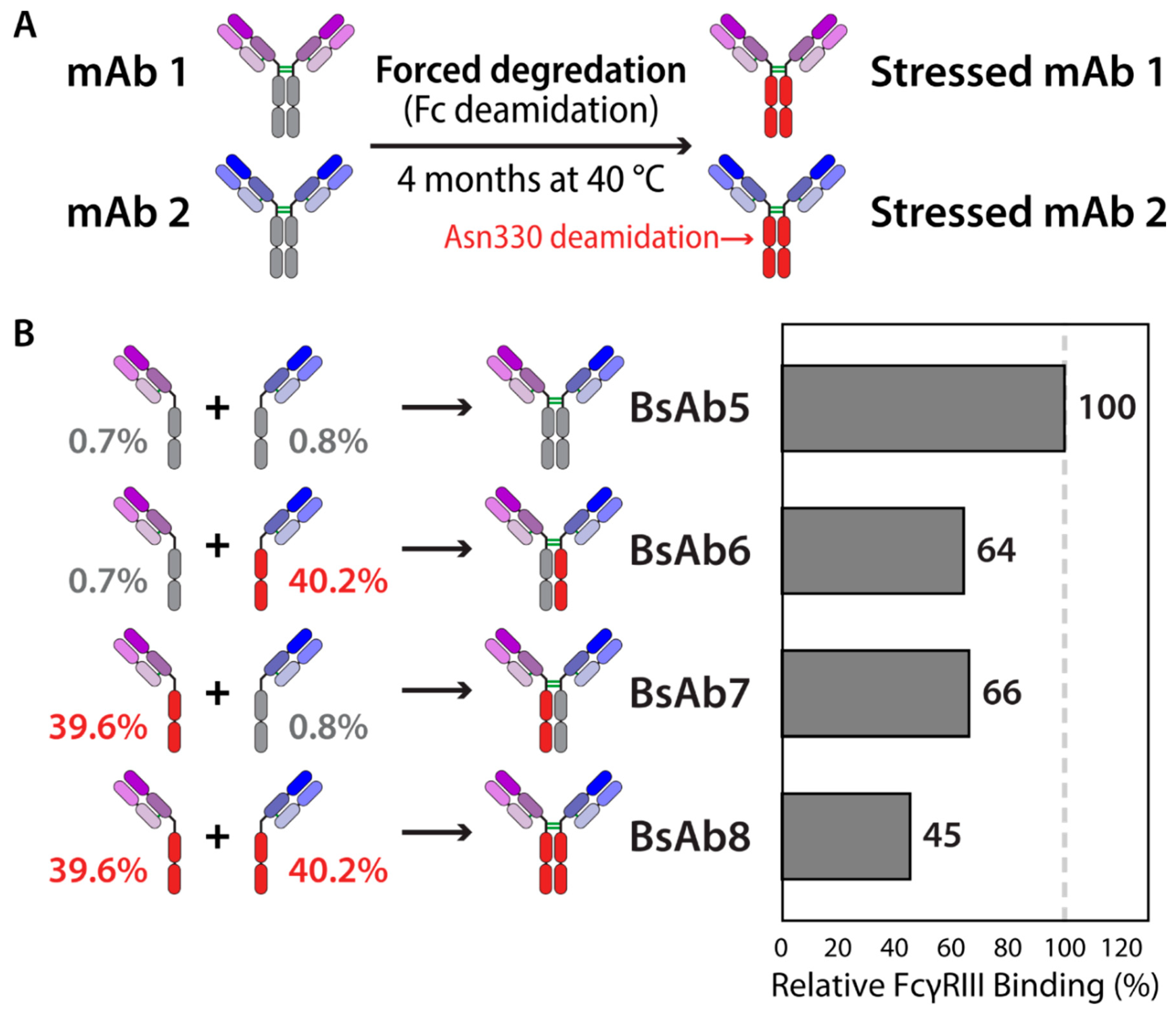
2.3. Chemical Stability
3. Self-Association and High Concentration Properties
3.1. Self-Association and Solubility
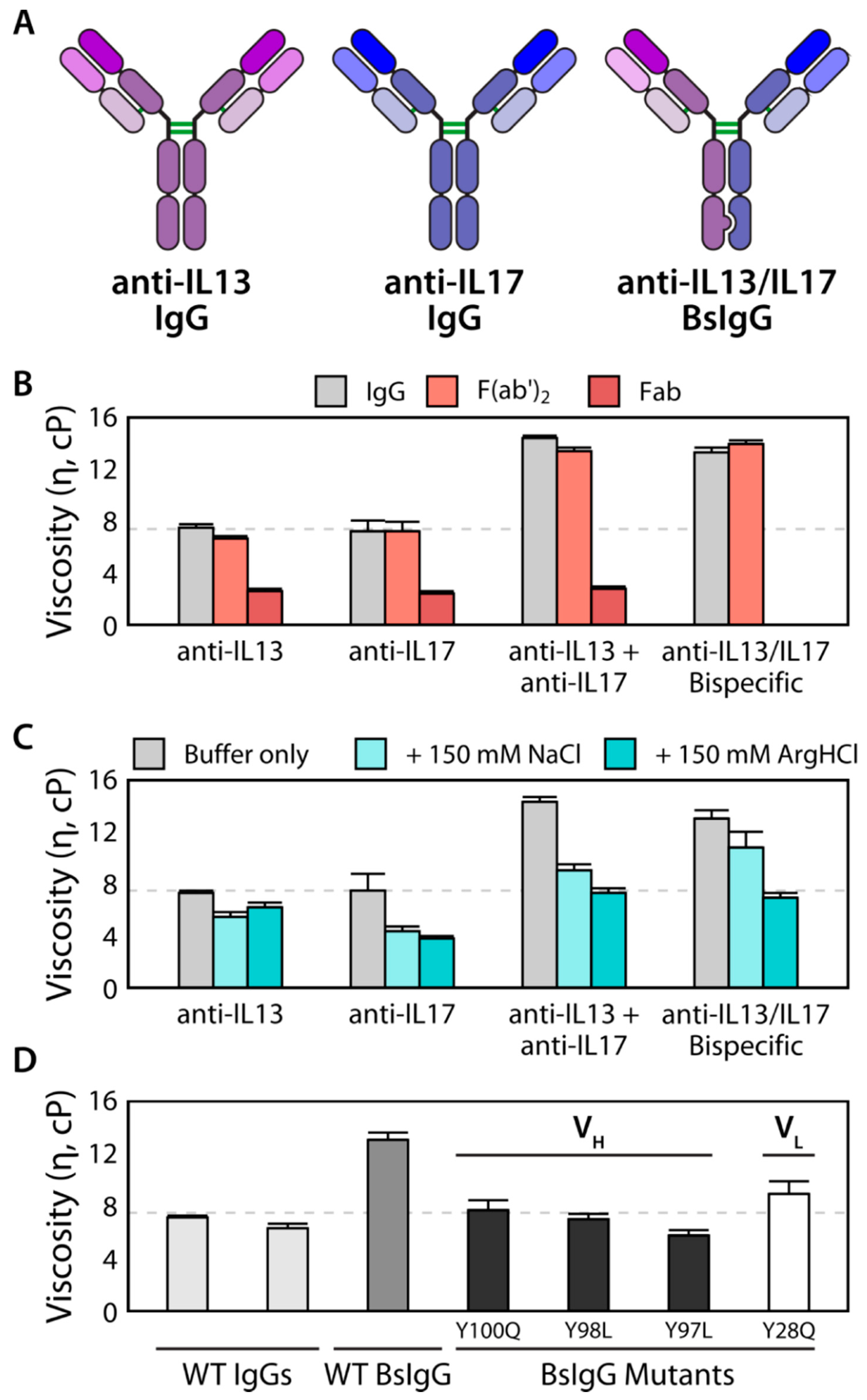
3.2. Viscosity
4. Polyspecificty and In Vivo Properties
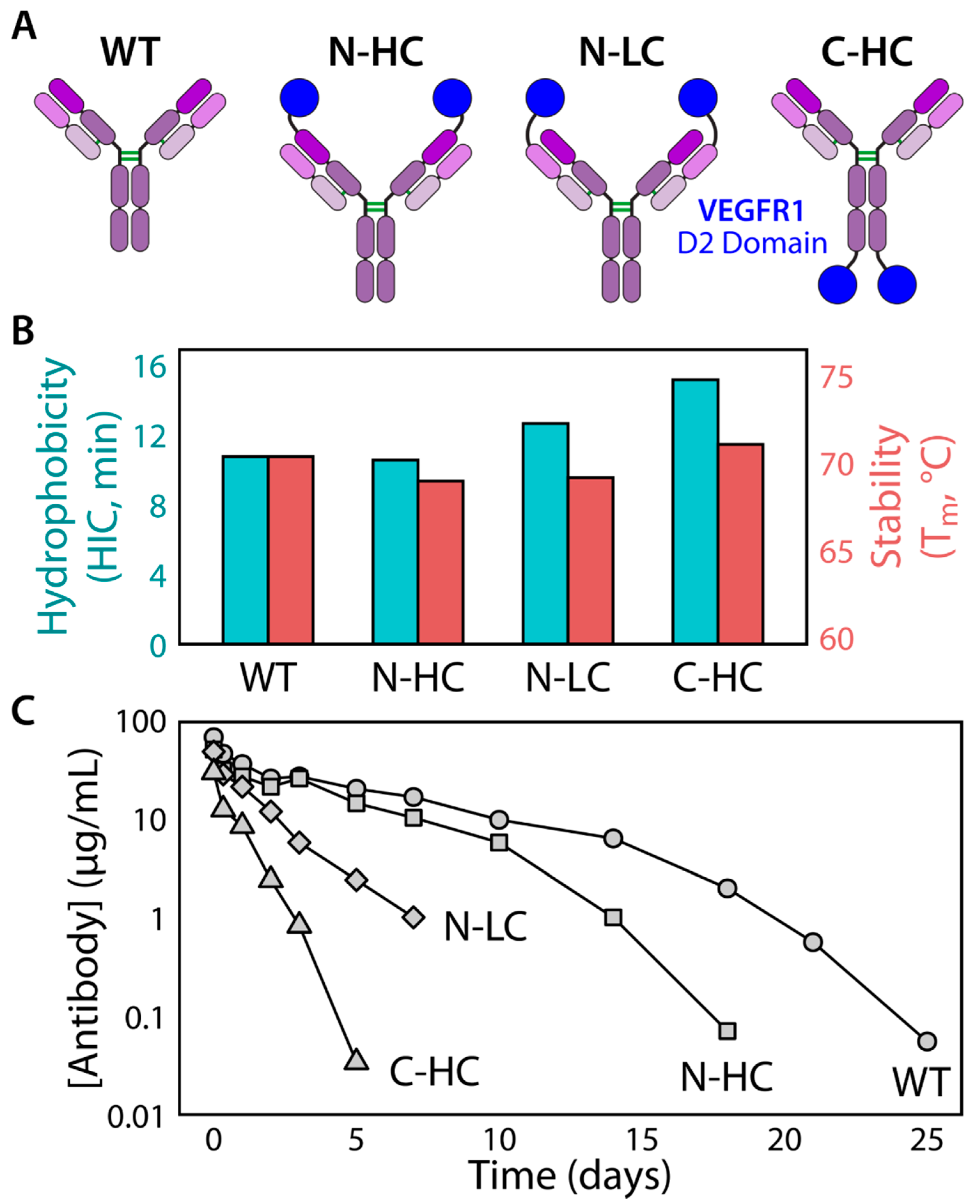
4.1. Polyspecificity and Pharmacokinetics
4.2. Immunogenicity
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. mAbs 2020, 12, 1703531. [Google Scholar] [CrossRef]
- Trabolsi, A.; Arumov, A.; Schatz, J.H. T Cell–Activating Bispecific Antibodies in Cancer Therapy. J. Immunol. 2019, 203, 585–592. [Google Scholar] [CrossRef]
- Maher, J.; Adami, A.A. Antitumor Immunity: Easy as 1, 2, 3 with Monoclonal Bispecific Trifunctional Antibodies? Cancer Res. 2013, 73, 5613–5617. [Google Scholar] [CrossRef]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical development and future directions. mAbs 2010, 2, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Lenting, P.J.; Denis, C.V.; Christophe, O.D. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: How does it actually compare to factor VIII? Blood 2017, 130, 2463–2468. [Google Scholar] [CrossRef] [PubMed]
- Shima, M.; Hanabusa, H.; Taki, M.; Matsushita, T.; Sato, T.; Fukutake, K.; Fukazawa, N.; Yoneyama, K.; Yoshida, H.; Nogami, K. Factor VIII–Mimetic Function of Humanized Bispecific Antibody in Hemophilia A. N. Engl. J. Med. 2016, 374, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 2020, 580, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor Regression in Cancer Patients by Very Low Doses of a T Cell-Engaging Antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Einsele, H.; Borghaei, H.; Orlowski, R.Z.; Subklewe, M.; Roboz, G.J.; Zugmaier, G.; Kufer, P.; Iskander, K.; Kantarjian, H. The BiTE (bispecific T-cell engager) platform: Development and future potential of a targeted immuno-oncology therapy across tumor types. Cancer 2020, 126, 3192–3201. [Google Scholar] [CrossRef]
- Löffler, A.; Kufer, P.; Lutterbüse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmüller, G.; Dörken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef]
- Oberg, H.H.; Kellner, C.; Gonnermann, D.; Sebens, S.; Bauerschlag, D.; Gramatzki, M.; Kabelitz, D.; Peipp, M.; Wesch, D. Tribody [(HER2)2xCD16] Is More Effective Than Trastuzumab in Enhancing γδ T Cell and Natural Killer Cell Cytotoxicity Against HER2-Expressing Cancer Cells. Front. Immunol. 2018, 9, 814. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, J.U.; Felices, M.; Todhunter, D.; Taras, E.; Miller, J.S.; Vallera, D.A. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget 2016, 7, 73830–73844. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, J.U.; Gleason, M.K.; Dougherty, P.R.; Miller, J.S.; Vallera, D.A. Heterodimeric Bispecific Single Chain Variable Fragments (scFv) Killer Engagers (BiKEs) Enhance NK-cell Activity Against CD133+ Colorectal Cancer Cells. Target. Oncol. 2016, 11, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef]
- Husain, B.; Ellerman, D. Expanding the Boundaries of Biotherapeutics with Bispecific Antibodies. BioDrugs 2018, 32, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Saxena, A.; Sidhu, S.S.; Wu, D. Fc Engineering for Developing Therapeutic Bispecific Antibodies and Novel Scaffolds. Front. Immunol. 2017, 8, 38. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef]
- Tiller, K.E.; Tessier, P.M. Advances in Antibody Design. Annu. Rev. Biomed. Eng. 2015, 17, 191–216. [Google Scholar] [CrossRef]
- Rouet, R.; Lowe, D.; Christ, D. Stability engineering of the human antibody repertoire. FEBS Lett. 2014, 588, 269–277. [Google Scholar] [CrossRef]
- McConnell, A.D.; Spasojevich, V.; Macomber, J.L.; Krapf, I.P.; Chen, A.; Sheffer, J.C.; Berkebile, A.; Horlick, R.A.; Neben, S.; King, D.J.; et al. An integrated approach to extreme thermostabilization and affinity maturation of an antibody. Protein Eng. Des. Sel. 2013, 26, 151–164. [Google Scholar] [CrossRef] [PubMed]
- McConnell, A.D.; Zhang, X.; Macomber, J.L.; Chau, B.; Sheffer, J.C.; Rahmanian, S.; Hare, E.; Spasojevic, V.; Horlick, R.A.; King, D.J.; et al. A general approach to antibody thermostabilization. mAbs 2014, 6, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Ghayur, T. Rationale and development of multispecific antibody drugs. Expert Rev. Clin. Pharmacol. 2010, 3, 491–508. [Google Scholar] [CrossRef] [PubMed]
- Hagihara, Y.; Saerens, D. Engineering disulfide bonds within an antibody. Biochim. Biophys. Acta 2014, 1844, 2016–2023. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.; Liu, J.L.; Zabetakis, D.; Lee, A.B.; Anderson, G.P.; Goldman, E.R. Improving the biophysical properties of anti-ricin single-domain antibodies. Biotechnol. Rep. 2015, 6, 27–35. [Google Scholar] [CrossRef]
- Anderson, G.P.; Liu, J.H.; Zabetakis, D.; Liu, J.L.; Goldman, E.R. Thermal stabilization of anti-α-cobratoxin single domain antibodies. Toxicon 2017, 129, 68–73. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kandalaft, H.; Hussack, G.; Raphael, S.; Ding, W.; Kelly, J.F.; Henry, K.A.; Tanha, J. Evaluation of a noncanonical Cys40-Cys55 disulfide linkage for stabilization of single-domain antibodies. Protein Sci. 2019, 28, 881–888. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kandalaft, H.; Ding, W.; Ryan, S.; Van Faassen, H.; Hirama, T.; Foote, S.J.; MacKenzie, C.R.; Tanha, J. Disulfide linkage engineering for improving biophysical properties of human VH domains. Protein Eng. Des. Sel. 2012, 25, 581–590. [Google Scholar] [CrossRef][Green Version]
- Hussack, G.; Hirama, T.; Ding, W.; MacKenzie, R.; Tanha, J. Engineered Single-Domain Antibodies with High Protease Resistance and Thermal Stability. PLoS ONE 2011, 6, e28218. [Google Scholar] [CrossRef]
- Hagihara, Y.; Mine, S.; Uegaki, K. Stabilization of an Immunoglobulin Fold Domain by an Engineered Disulfide Bond at the Buried Hydrophobic Region. J. Biol. Chem. 2007, 282, 36489–36495. [Google Scholar] [CrossRef]
- Zabetakis, D.; Olson, M.A.; Anderson, G.P.; Legler, P.M.; Goldman, E.R. Evaluation of Disulfide Bond Position to Enhance the Thermal Stability of a Highly Stable Single Domain Antibody. PLoS ONE 2014, 9, e115405. [Google Scholar] [CrossRef]
- Bhatta, P.; Humphreys, D.T. Relative Contribution of Framework and CDR Regions in Antibody Variable Domains to Multimerisation of Fv- and scFv-Containing Bispecific Antibodies. Antibodies 2018, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Goldman, E.R.; Zabetakis, D.; Walper, S.A.; Turner, K.; Shriver-Lake, L.C.; Anderson, G.P. Enhanced production of a single domain antibody with an engineered stabilizing extra disulfide bond. Microb. Cell Factories 2015, 14, 158. [Google Scholar] [CrossRef] [PubMed]
- Schlapschy, M.; Grimm, S.; Skerra, A. A system for concomitant overexpression of four periplasmic folding catalysts to improve secretory protein production in Escherichia coli. Protein Eng. Des. Sel. 2006, 19, 385–390. [Google Scholar] [CrossRef]
- Shriver-Lake, L.C.; Goldman, E.R.; Zabetakis, D.; Anderson, G.P. Improved production of single domain antibodies with two disulfide bonds by co-expression of chaperone proteins in the Escherichia coli periplasm. J. Immunol. Methods 2017, 443, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Plückthun, A. Improving in vivo folding and stability of a single-chain Fv antibody fragment by loop grafting. Protein Eng. 1997, 10, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Honegger, A.; Malebranche, A.D.; Röthlisberger, D.; Plückthun, A. The influence of the framework core residues on the biophysical properties of immunoglobulin heavy chain variable domains. Protein Eng. Des. Sel. 2009, 22, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Queen, C.; Schneider, W.P.; Selick, H.E.; Payne, P.W.; Landolfi, N.F.; Duncan, J.F.; Avdalovic, N.M.; Levitt, M.; Junghans, R.P.; Waldmann, T.A. A humanized antibody that binds to the interleukin 2 receptor. Proc. Natl. Acad. Sci. USA 1989, 86, 10029–10033. [Google Scholar] [CrossRef]
- Julian, M.C.; Li, L.-J.; Garde, S.; Wilen, R.; Tessier, P.M. Efficient affinity maturation of antibody variable domains requires co-selection of compensatory mutations to maintain thermodynamic stability. Sci. Rep. 2017, 7, 45259. [Google Scholar] [CrossRef]
- Lehmann, A.; Wixted, J.H.F.; Shapovalov, M.V.; Roder, H.; Dunbrack, R.L.; Robinson, M.K. Stability engineering of anti-EGFR scFv antibodies by rational design of a lambda-to-kappa swap of the VL framework using a structure-guided approach. mAbs 2015, 7, 1058–1071. [Google Scholar] [CrossRef]
- Shusta, E.V.; Holler, P.D.; Kieke, M.C.; Kranz, D.M.; Wittrup, K. Directed evolution of a stable scaffold for T-cell receptor engineering. Nat. Biotechnol. 2000, 18, 754–759. [Google Scholar] [CrossRef]
- Orr, B.; Carr, L.; Wittrup, K.; Roy, E.; Kranz, D.M. Rapid Method for Measuring ScFv Thermal Stability by Yeast Surface Display. Biotechnol. Prog. 2003, 19, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Franklin, E.; Cunningham, O.; Fennell, B. Parallel Evolution of Antibody Affinity and Thermal Stability for Optimal Biotherapeutic Development. Adv. Struct. Safe. Stud. 2018, 457–477. [Google Scholar] [CrossRef]
- Traxlmayr, M.W.; Obinger, C. Directed evolution of proteins for increased stability and expression using yeast display. Arch. Biochem. Biophys. 2012, 526, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Traxlmayr, M.W.; Shusta, E.V. Directed Evolution of Protein Thermal Stability Using Yeast Surface Display. Methods Mol. Biol. 2017, 1575, 45–65. [Google Scholar] [CrossRef]
- Xu, L.; Kohli, N.; Rennard, R.; Jiao, Y.; Razlog, M.; Zhang, K.; Baum, J.; Johnson, B.; Tang, J.; Schoeberl, B.; et al. Rapid optimization and prototyping for therapeutic antibody-like molecules. mAbs 2013, 5, 237–254. [Google Scholar] [CrossRef]
- Jia, Y.; Ren, P.; Duan, S.; Zeng, P.; Xie, D.; Zeng, F. An optimized yeast display strategy for efficient scFv antibody selection using ribosomal skipping system and thermo resistant yeast. Biotechnol. Lett. 2019, 41, 1067–1076. [Google Scholar] [CrossRef]
- Scott, M.J.; Lee, J.A.; Wake, M.S.; Batt, K.V.; Wattam, T.A.; Hiles, I.D.; Batuwangala, T.D.; Ashman, C.I.; Steward, M. ‘In-Format’ screening of a novel bispecific antibody format reveals significant potency improvements relative to unformatted molecules. mAbs 2016, 9, 85–93. [Google Scholar] [CrossRef]
- Kim, D.Y.; Hussack, G.; Kandalaft, H.; Tanha, J. Mutational approaches to improve the biophysical properties of human single-domain antibodies. Biochim. Biophys. Acta 2014, 1844, 1983–2001. [Google Scholar] [CrossRef]
- Geddie, M.L.; Kohli, N.; Kirpotin, D.B.; Razlog, M.; Jiao, Y.; Kornaga, T.; Rennard, R.; Xu, L.; Schoerberl, B.; Marks, J.D.; et al. Improving the developability of an anti-EphA2 single-chain variable fragment for nanoparticle targeting. mAbs 2017, 9, 58–67. [Google Scholar] [CrossRef]
- Liu, J.L.; Shriver-Lake, L.C.; Anderson, G.P.; Zabetakis, D.; Goldman, E.R. Selection, characterization, and thermal stabilization of llama single domain antibodies towards Ebola virus glycoprotein. Microb. Cell Factories 2017, 16, 223. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.; Flock, T.; Soler, N.; Zaiss, M.; Vincke, C.; Sterckx, Y.G.-J.; Kastelic, D.; Muyldermans, S.; Hoheisel, J.D. Exploiting sequence and stability information for directing nanobody stability engineering. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2196–2205. [Google Scholar] [CrossRef]
- Baran, D.; Pszolla, M.G.; Lapidoth, G.D.; Norn, C.; Dym, O.; Unger, T.; Albeck, S.; Tyka, M.D.; Fleishman, S.J. Principles for computational design of binding antibodies. Proc. Natl. Acad. Sci. USA 2017, 114, 10900–10905. [Google Scholar] [CrossRef] [PubMed]
- Adolf-Bryfogle, J.; Kalyuzhniy, O.; Kubitz, M.; Weitzner, B.D.; Hu, X.; Adachi, Y.; Schief, W.R.; Dunbrack, R.L. RosettaAntibodyDesign (RAbD): A general framework for computational antibody design. PLoS Comput. Biol. 2018, 14, e1006112. [Google Scholar] [CrossRef] [PubMed]
- Pérez, A.-M.W.; Sormanni, P.; Andersen, J.S.; Sakhnini, L.I.; León, I.R.; Bjelke, J.R.; Gajhede, A.J.; De Maria, L.; Otzen, D.E.; Vendruscolo, M.; et al. In vitro and in silico assessment of the developability of a designed monoclonal antibody library. mAbs 2019, 11, 388–400. [Google Scholar] [CrossRef]
- Soler, M.A.; Medagli, B.; Semrau, M.S.; Storici, P.; Bajc, G.; De Marco, A.; Laio, A.; Fortuna, S. A consensus protocol for the in silico optimisation of antibody fragments. Chem. Commun. 2019, 55, 14043–14046. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Der, B.S.; Karamitros, C.S.; Li, W.; Marshall, N.M.; Lungu, O.I.; Miklos, A.E.; Xu, J.; Kang, T.H.; Lee, C.; et al. Computer-based engineering of thermostabilized antibody fragments. AIChE J. 2020, 66. [Google Scholar] [CrossRef]
- Kuroda, D.; Tsumoto, K. Engineering Stability, Viscosity, and Immunogenicity of Antibodies by Computational Design. J. Pharm. Sci. 2020, 109, 1631–1651. [Google Scholar] [CrossRef]
- Lapidoth, G.D.; Baran, D.; Pszolla, G.M.; Norn, C.; Alon, A.; Tyka, M.D.; Fleishman, S.J. AbDesign: An algorithm for combinatorial backbone design guided by natural conformations and sequences. Proteins 2015, 83, 1385–1406. [Google Scholar] [CrossRef]
- Warszawski, S.; Katz, A.B.; Lipsh, R.; Khmelnitsky, L.; Ben Nissan, G.; Javitt, G.; Dym, O.; Unger, T.; Knop, O.; Albeck, S.; et al. Optimizing antibody affinity and stability by the automated design of the variable light-heavy chain interfaces. PLoS Comput. Biol. 2019, 15, e1007207. [Google Scholar] [CrossRef]
- Fischer, N.; Elson, G.; Magistrelli, G.; Dheilly, E.; Fouque, N.; Laurendon, A.; Gueneau, F.; Ravn, U.; Depoisier, J.-F.; Moine, V.; et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat. Commun. 2015, 6, 6113. [Google Scholar] [CrossRef]
- Amaral, M.; Hölper, S.; Lange, C.; Jung, J.; Sjuts, H.; Weil, S.; Fischer, M.; Radoevic, K.; Rao, E. Engineered Technologies and Bioanalysis of multispecific Antibody Formats. J. Appl. Bioanal. 2020, 6, 26–51. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Meesters, J.I.; Priem, P.; De Jong, R.N.; Bremer, E.T.J.V.D.; Van Kampen, M.D.; Gerritsen, A.F.; Schuurman, J.; Parren, P.W.H.I. Controlled Fab-arm exchange for the generation of stable bispecific IgG1. Nat. Protoc. 2014, 9, 2450–2463. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Meesters, J.I.; De Goeij, B.E.C.G.; Bremer, E.T.J.V.D.; Neijssen, J.; Van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145–5150. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef] [PubMed]
- De Nardis, C.; Hendriks, L.J.A.; Poirier, E.; Arvinte, T.; Gros, P.; Bakker, A.B.H.; De Kruif, J. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J. Biol. Chem. 2017, 292, 14706–14717. [Google Scholar] [CrossRef] [PubMed]
- Krah, S.; Schröter, C.; Eller, C.; Rhiel, L.; Rasche, N.; Beck, J.; Sellmann, C.; Günther, R.; Toleikis, L.; Hock, B.; et al. Generation of human bispecific common light chain antibodies by combining animal immunization and yeast display. Protein Eng. Des. Sel. 2017, 30, 291–301. [Google Scholar] [CrossRef]
- Shiraiwa, H.; Narita, A.; Kamata-Sakurai, M.; Ishiguro, T.; Sano, Y.; Hironiwa, N.; Tsushima, T.; Segawa, H.; Tsunenari, T.; Ikeda, Y.; et al. Engineering a bispecific antibody with a common light chain: Identification and optimization of an anti-CD3 epsilon and anti-GPC3 bispecific antibody, ERY974. Methods 2019, 154, 10–20. [Google Scholar] [CrossRef]
- Lewis, S.M.; Wu, X.; Pustilnik, A.; Sereno, A.; Huang, F.; Rick, H.L.; Guntas, G.; Leaver-Fay, A.; Smith, E.M.; Ho, C.; et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat. Biotechnol. 2014, 32, 191–198. [Google Scholar] [CrossRef]
- Liu, Z.; Leng, E.C.; Gunasekaran, K.; Pentony, M.; Shen, M.; Howard, M.; Stoops, J.; Manchulenko, K.; Razinkov, V.; Liu, H.; et al. A Novel Antibody Engineering Strategy for Making Monovalent Bispecific Heterodimeric IgG Antibodies by Electrostatic Steering Mechanism. J. Biol. Chem. 2015, 290, 7535–7562. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Cai, H.; Jin, Y.; Wang, P.; Zhang, Q.; Lin, Y.; Wang, W.; Cheng, J.; Zeng, N.; Xu, T.; et al. Structural basis of a novel heterodimeric Fc for bispecific antibody production. Oncotarget 2017, 8, 51037–51049. [Google Scholar] [CrossRef] [PubMed]
- Froning, K.J.; Leaver-Fay, A.; Wu, X.; Phan, S.; Gao, L.; Huang, F.; Pustilnik, A.; Bacica, M.; Houlihan, K.; Chai, Q.; et al. Computational design of a specific heavy chain/κ light chain interface for expressing fully IgG bispecific antibodies. Protein Sci. 2017, 26, 2021–2038. [Google Scholar] [CrossRef]
- Dillon, M.; Yin, Y.; Zhou, J.; Mccarty, L.; Ellerman, D.; Slaga, D.; Junttila, T.T.; Han, G.; Sandoval, W.; Ovacik, M.A.; et al. Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. mAbs 2017, 9, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.K.; Phung, W.; Han, G.; Yin, Y.; Kim, I.; Sandoval, W.; Carter, P.J. Elucidating heavy/light chain pairing preferences to facilitate the assembly of bispecific IgG in single cells. mAbs 2019, 11, 1254–1265. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves first bispecific. Nat. Rev. Drug Discov. 2015, 14, 7. [Google Scholar] [CrossRef]
- Johnson, S.; Burke, S.; Huang, L.; Gorlatov, S.; Li, H.; Wang, W.; Zhang, W.; Tuaillon, N.; Rainey, J.; Barat, B.; et al. Effector Cell Recruitment with Novel Fv-based Dual-affinity Re-targeting Protein Leads to Potent Tumor Cytolysis and In Vivo B-cell Depletion. J. Mol. Biol. 2010, 399, 436–449. [Google Scholar] [CrossRef]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef]
- Rader, C. DARTs take aim at BiTEs. Blood 2011, 117, 4403–4404. [Google Scholar] [CrossRef]
- Wu, X.; Sereno, A.J.; Huang, F.; Lewis, S.M.; Lieu, R.L.; Weldon, C.; Torres, C.; Fine, C.; Batt, M.A.; Fitchett, J.R.; et al. Fab-based bispecific antibody formats with robust biophysical properties and biological activity. mAbs 2015, 7, 470–482. [Google Scholar] [CrossRef]
- Gong, S.; Ren, F.; Wu, D.; Wu, X.; Wu, C. Fabs-in-tandem immunoglobulin is a novel and versatile bispecific design for engaging multiple therapeutic targets. mAbs 2017, 9, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.-R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Haga, N.; Asano, R.; Nakazawa, H.; Hattori, T.; Takeda, D.; Sugiyama, A.; Kurotani, R.; Kumagai, I.; Umetsu, M.; et al. Generation of camelid VHH bispecific constructs via in-cell intein-mediated protein trans-splicing. Prot. Eng. Des. Sel. 2017, 30, 15–21. [Google Scholar]
- Hemmi, S.; Asano, R.; Kimura, K.; Umetsu, M.; Nakanishi, T.; Kumagai, I.; Makabe, K. Construction of a circularly connected VHH bispecific antibody (cyclobody) for the desirable positioning of antigen-binding sites. Biochem. Biophys. Res. Commun. 2020, 523, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Chen, J.; Ding, K.; Zong, H.; Xie, Y.; Jiang, H.; Zhang, B.; Lu, H.; Yin, W.; Gilly, J.; et al. Efficient generation of bispecific IgG antibodies by split intein mediated protein trans-splicing system. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Han, L.; Zong, H.; Zhou, Y.; Pan, Z.; Chen, J.; Ding, K.; Xie, Y.; Jiang, H.; Zhang, B.; Lu, H.; et al. Naturally split intein Npu DnaE mediated rapid generation of bispecific IgG antibodies. Methods 2019, 154, 32–37. [Google Scholar] [CrossRef]
- Hofmann, T.; Schmidt, J.; Ciesielski, E.; Becker, S.; Rysiok, T.; Schütte, M.; Toleikis, L.; Kolmar, H.; Doerner, A. Intein mediated high throughput screening for bispecific antibodies. mAbs 2020, 12, 1731938. [Google Scholar] [CrossRef]
- Moussa, E.M.; Panchal, J.P.; Moorthy, B.S.; Blum, J.S.; Joubert, M.K.; Narhi, L.O.; Topp, E. Immunogenicity of Therapeutic Protein Aggregates. J. Pharm. Sci. 2016, 105, 417–430. [Google Scholar] [CrossRef]
- Ratanji, K.D.; Derrick, J.P.; Dearman, R.J.; Kimber, I. Immunogenicity of therapeutic proteins: Influence of aggregation. J. Immunotoxicol. 2014, 11, 99–109. [Google Scholar] [CrossRef]
- Perchiacca, J.M.; Bhattacharya, M.; Tessier, P.M. Mutational analysis of domain antibodies reveals aggregation hotspots within and near the complementarity determining regions. Proteins Struct. Funct. Bioinform. 2011, 79, 2637–2647. [Google Scholar] [CrossRef]
- Perchiacca, J.M.; Ladiwala, A.R.A.; Bhattacharya, M.; Tessier, P.M. Aggregation-resistant domain antibodies engineered with charged mutations near the edges of the complementarity-determining regions. Protein Eng. Des. Sel. 2012, 25, 591–602. [Google Scholar] [CrossRef]
- Dudgeon, K.; Rouet, R.; Kokmeijer, I.; Schofield, P.R.; Stolp, J.; Langley, D.B.; Stock, D.; Christ, D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 10879–10884. [Google Scholar] [CrossRef]
- Perchiacca, J.M.; Lee, C.C.; Tessier, P.M. Optimal charged mutations in the complementarity-determining regions that prevent domain antibody aggregation are dependent on the antibody scaffold. Protein Eng. Des. Sel. 2014, 27, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Van Der Kant, R.; Karow-Zwick, A.R.; Van Durme, J.; Blech, M.; Gallardo, R.; Seeliger, D.; Aßfalg, K.; Baatsen, P.; Compernolle, G.; Gils, A.; et al. Prediction and Reduction of the Aggregation of Monoclonal Antibodies. J. Mol. Biol. 2017, 429, 1244–1261. [Google Scholar] [CrossRef]
- Chaudhuri, R.; Cheng, Y.; Middaugh, C.R.; Volkin, D. High-Throughput Biophysical Analysis of Protein Therapeutics to Examine Interrelationships Between Aggregate Formation and Conformational Stability. AAPS J. 2014, 16, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Prediction of Aggregation Prone Regions of Therapeutic Proteins. J. Phys. Chem. B 2010, 114, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J. Therapeutic protein aggregation: Mechanisms, design, and control. Trends Biotechnol. 2014, 32, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Perchiacca, J.M.; Tessier, P.M. Toward aggregation-resistant antibodies by design. Trends Biotechnol. 2013, 31, 612–620. [Google Scholar] [CrossRef]
- Schaefer, Z.P.; Bailey, L.J.; Kossiakoff, A.A. A polar ring endows improved specificity to an antibody fragment. Protein Sci. 2016, 25, 1290–1298. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J.M. Protein-protein interactions: A review of protein dimer structures. Prog. Biophys. Mol. Biol. 1995, 63, 31–65. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sakhnini, L.I.; Greisen, P.J.; Wiberg, C.; Bozoky, Z.; Lund, S.; Perez, A.-M.W.; Karkov, H.S.; Huus, K.; Hansen, J.-J.; Bülow, L.; et al. Improving the Developability of an Antigen Binding Fragment by Aspartate Substitutions. Biochemistry 2019, 58, 2750–2759. [Google Scholar] [CrossRef] [PubMed]
- Brummitt, R.K.; Nesta, D.P.; Chang, L.; Chase, S.F.; Laue, T.M.; Roberts, C.J. Nonnative Aggregation of an IgG1 Antibody in Acidic Conditions: Part 1. Unfolding, Colloidal Interactions, and Formation of High-Molecular-Weight Aggregates. J. Pharm. Sci. 2011, 100, 2087–2103. [Google Scholar] [CrossRef] [PubMed]
- Austerberry, J.; Dajani, R.; Panova, S.; Roberts, D.; Golovanov, A.P.; Pluen, A.; Van Der Walle, C.F.; Uddin, S.; Warwicker, J.; Derrick, J.P.; et al. The effect of charge mutations on the stability and aggregation of a human single chain Fv fragment. Eur. J. Pharm. Biopharm. 2017, 115, 18–30. [Google Scholar] [CrossRef]
- Austerberry, J.I.; Thistlethwaite, A.; Fisher, K.; Golovanov, A.P.; Pluen, A.; Esfandiary, R.; Van Der Walle, C.F.; Warwicker, J.; Derrick, J.P.; Curtis, R. Arginine to Lysine Mutations Increase the Aggregation Stability of a Single-Chain Variable Fragment through Unfolded-State Interactions. Biochemistry 2019, 58, 3413–3421. [Google Scholar] [CrossRef]
- Shan, L.; Mody, N.; Sormani, P.; Rosenthal, K.L.; Damschroder, M.M.; Esfandiary, R.; Sormanni, P. Developability Assessment of Engineered Monoclonal Antibody Variants with a Complex Self-Association Behavior Using Complementary Analytical and in Silico Tools. Mol. Pharm. 2018, 15, 5697–5710. [Google Scholar] [CrossRef]
- Hsu, H.-J.; Lee, K.H.; Jian, J.-W.; Chang, H.-J.; Yu, C.-M.; Lee, Y.-C.; Chen, I.-C.; Peng, H.-P.; Wu, C.Y.; Huang, Y.-F.; et al. Antibody Variable Domain Interface and Framework Sequence Requirements for Stability and Function by High-Throughput Experiments. Structure 2014, 22, 22–34. [Google Scholar] [CrossRef]
- Wörn, A.; Plückthun, A. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 2001, 305, 989–1010. [Google Scholar] [CrossRef]
- Jung, S.; Honegger, A.; Plückthun, A. Selection for improved protein stability by phage display 1 1Edited by J. A. Wells. J. Mol. Biol. 1999, 294, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Wörn, A.; Plückthun, A. Different Equilibrium Stability Behavior of ScFv Fragments: Identification, Classification, and Improvement by Protein Engineering. Biochemistry 1999, 38, 8739–8750. [Google Scholar] [CrossRef] [PubMed]
- Wörn, A.; Plückthun, A. Mutual Stabilization of VLand VHin Single-Chain Antibody Fragments, Investigated with Mutants Engineered for Stability. Biochemistry 1998, 37, 13120–13127. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, S.; Kobashigawa, Y.; Fukuda, N.; Teramoto, M.; Toyota, Y.; Liu, C.; Ikeguchi, Y.; Sato, T.; Sato, Y.; Kimura, H.; et al. Cyclization of Single-Chain Fv Antibodies Markedly Suppressed Their Characteristic Aggregation Mediated by Inter-Chain VH-VL Interactions. Molecules 2019, 24, 2620. [Google Scholar] [CrossRef]
- Miller, B.R.; Glaser, S.M.; Demarest, S.J. Rapid Screening Platform for Stabilization of scFvs in Escherichia coli. Methods Mol. Biol. 2009, 525, 279–289, xiv. [Google Scholar]
- Jain, T.; Sun, T.; Durand, S.; Hall, A.; Houston, N.R.; Nett, J.H.; Sharkey, B.; Bobrowicz, B.; Caffry, I.; Yu, Y.; et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 2017, 114, 944–949. [Google Scholar] [CrossRef]
- Bailly, M.; Mieczkowski, C.; Juan, V.; Metwally, E.; Tomazela, D.; Baker, J.; Uchida, M.; Kofman, E.; Raoufi, F.; Motlagh, S.; et al. Predicting Antibody Developability Profiles Through Early Stage Discovery Screening. mAbs 2020, 12, 1743053. [Google Scholar] [CrossRef]
- Clarke, S.C.; Ma, B.; Trinklein, N.D.; Schellenberger, U.; Osborn, M.J.; Ouisse, L.-H.; Boudreau, A.; Davison, L.M.; Harris, K.E.; Ugamraj, H.S.; et al. Multispecific Antibody Development Platform Based on Human Heavy Chain Antibodies. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Lauer, T.M.; Agrawal, N.J.; Chennamsetty, N.; Egodage, K.; Helk, B.; Trout, B.L. Developability Index: A Rapid in Silico Tool for the Screening of Antibody Aggregation Propensity. J. Pharm. Sci. 2012, 101, 102–115. [Google Scholar] [CrossRef]
- Obrezanova, O.; Arnell, A.; De La Cuesta, R.G.; Berthelot, M.E.; Gallagher, T.R.; Zurdo, J.; Stallwood, Y. Aggregation risk prediction for antibodies and its application to biotherapeutic development. mAbs 2015, 7, 352–363. [Google Scholar] [CrossRef]
- Zhang, C.; Samad, M.; Yu, H.; Chakroun, N.; Hilton, D.; Dalby, P.A. Computational Design to Reduce Conformational Flexibility and Aggregation Rates of an Antibody Fab Fragment. Mol. Pharm. 2018, 15, 3079–3092. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Das, T.K.; Singh, S.K.; Kumar, S. Potential aggregation prone regions in biotherapeutics: A survey of commercial monoclonal antibodies. mAbs 2009, 1, 254–267. [Google Scholar] [CrossRef]
- Wang, X.; Singh, S.K.; Kumar, S. Potential Aggregation-Prone Regions in Complementarity-Determining Regions of Antibodies and Their Contribution Towards Antigen Recognition: A Computational Analysis. Pharm. Res. 2010, 27, 1512–1529. [Google Scholar] [CrossRef] [PubMed]
- Christ, D.; Famm, K.; Winter, G. Repertoires of aggregation-resistant human antibody domains. Protein Eng. Des. Sel. 2007, 20, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-J.; Luo, J.; O’Neil, K.T.; Kang, J.; Lacy, E.R.; Canziani, G.; Baker, A.; Huang, M.; Tang, Q.M.; Raju, T.; et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng. Des. Sel. 2010, 23, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Julian, M.C.; Tiller, K.E.; Meng, F.; Duconge, S.E.; Akter, R.; Raleigh, D.P.; Tessier, P.M. Design and Optimization of Anti-amyloid Domain Antibodies Specific for β-Amyloid and Islet Amyloid Polypeptide. J. Biol. Chem. 2016, 291, 2858–2873. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.G.; Rajendra, Y.; Hougland, M.D.; Boyles, J.S.; Barnard, G.C. Polymer-mediated flocculation of transient CHO cultures as a simple, high throughput method to facilitate antibody discovery. Biotechnol. Prog. 2017, 33, 1393–1400. [Google Scholar] [CrossRef]
- Evans, A.R.; Capaldi, M.T.; Goparaju, G.; Colter, D.; Shi, F.F.; Aubert, S.; Li, L.-C.; Mo, J.; Lewis, M.J.; Hu, P.; et al. Using bispecific antibodies in forced degradation studies to analyze the structure-function relationships of symmetrically and asymmetrically modified antibodies. mAbs 2019, 11, 1101–1112. [Google Scholar] [CrossRef]
- Shi, R.L.; Xiao, G.; Dillon, T.M.; Ricci, M.S.; Bondarenko, P.V. Characterization of therapeutic proteins by cation exchange chromatography-mass spectrometry and top-down analysis. mAbs 2020, 12, 1739825. [Google Scholar] [CrossRef]
- Luo, D.; Mah, N.; Krantz, M.; Wilde, K.; Wishart, D.; Zhang, Y.; Jacobs, F.; Martin, L. Vl-linker-Vh orientation-dependent expression of single chain Fv-containing an engineered disulfide-stabilized bond in the framework regions. J. Biochem. 1995, 118, 825–831. [Google Scholar] [CrossRef]
- Young, N.; MacKenzie, C.; Narang, S.A.; Oomen, R.P.; Baenziger, J.E. Thermal stabilization of a single-chain Fv antibody fragment by introduction of a disulphide bond. FEBS Lett. 1995, 377, 135–139. [Google Scholar] [CrossRef]
- Rajagopal, V.; Pastan, I.; Kreitman, R.J. A form of anti-Tac(Fv) which is both single-chain and disulfide stabilized: Comparison with its single-chain and disulfide-stabilized homologs. Protein Eng. 1997, 10, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Jiang, Y.; Zheng, Y.-L.; Ma, R.; Yu, D.-W. Improved stability and yield of Fv targeted superantigen by introducing both linker and disulfide bond into the targeting moiety. Biochimie 2005, 87, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Sheikholvaezin, A.; Sandström, P.; Eriksson, D.; Norgren, N.; Riklund, K.; Stigbrand, T. Optimizing the Generation of Recombinant Single-Chain Antibodies Against Placental Alkaline Phosphatase. Hybridoma 2006, 25, 181–192. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, L.; Gu, Z.-N.; Chen, H.; Tian, F.; Chen, Y.Q.; Zhang, H.; Chen, W. Stabilization of the Single-Chain Fragment Variable by an Interdomain Disulfide Bond and Its Effect on Antibody Affinity. Int. J. Mol. Sci. 2010, 12, 1. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Chen, I.-C.; Yu, C.-M.; Lee, Y.-C.; Hsu, H.-J.; Ching, A.T.C.; Chang, H.-J.; Yang, A.-S. Engineering Anti-vascular Endothelial Growth Factor Single Chain Disulfide-stabilized Antibody Variable Fragments (sc-dsFv) with Phage-displayed sc-dsFv Libraries. J. Biol. Chem. 2010, 285, 7880–7891. [Google Scholar] [CrossRef]
- Weatherill, E.E.; Cain, K.; Heywood, S.P.; Compson, J.E.; Heads, J.T.; Adams, R.; Humphreys, D.T. Towards a universal disulphide stabilised single chain Fv format: Importance of interchain disulphide bond location and vL-vH orientation. Protein Eng. Des. Sel. 2012, 25, 321–329. [Google Scholar] [CrossRef]
- Cao, M.; Wang, C.; Chung, W.K.; Motabar, D.; Wang, J.; Christian, E.; Lin, S.; Hunter, A.; Wang, X.; Liu, D. Characterization and analysis of scFv-IgG bispecific antibody size variants. mAbs 2018, 10, 1236–1247. [Google Scholar] [CrossRef]
- Trexler-Schmidt, M.; Sargis, S.; Chiu, J.; Sze-Khoo, S.; Mun, M.; Kao, Y.-H.; Laird, M.W. Identification and prevention of antibody disulfide bond reduction during cell culture manufacturing. Biotechnol. Bioeng. 2010, 106, 452–461. [Google Scholar] [CrossRef]
- Koterba, K.L.; Borgschulte, T.; Laird, M.W. Thioredoxin 1 is responsible for antibody disulfide reduction in CHO cell culture. J. Biotechnol. 2012, 157, 261–267. [Google Scholar] [CrossRef]
- Chung, W.K.; Russell, B.; Yang, Y.; Handlogten, M.; Hudak, S.; Cao, M.; Wang, J.; Robbins, D.; Ahuja, S.; Zhu, M. Effects of antibody disulfide bond reduction on purification process performance and final drug substance stability. Biotechnol. Bioeng. 2017, 114, 1264–1274. [Google Scholar] [CrossRef] [PubMed]
- Handlogten, M.W.; Zhu, M.; Ahuja, S. Glutathione and thioredoxin systems contribute to recombinant monoclonal antibody interchain disulfide bond reduction during bioprocessing. Biotechnol. Bioeng. 2017, 114, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Swope, N.; Chung, W.K.; Cao, M.; Motabar, D.; Liu, D.; Ahuja, S.; Handlogten, M.W. Impact of enzymatic reduction on bivalent bispecific antibody fragmentation and loss of product purity upon reoxidation. Biotechnol. Bioeng. 2020, 117, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.L.; Devine, P.W.A.; Phillips, J.J.; Higazi, D.R.; Lloyd, C.; Popovic, B.; Arnold, J.; Buchanan, A.; Lewis, A.; Goodman, J.; et al. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci. Rep. 2016, 6, 38644. [Google Scholar] [CrossRef]
- Geoghegan, J.C.; Fleming, R.; Damschroder, M.; Bishop, S.M.; Sathish, H.A.; Esfandiary, R. Mitigation of reversible self-association and viscosity in a human IgG1 monoclonal antibody by rational, structure-guided Fv engineering. mAbs 2016, 8, 941–950. [Google Scholar] [CrossRef]
- Chow, C.-K.; Allan, B.W.; Chai, Q.; Atwell, S.; Lu, J. Therapeutic Antibody Engineering to Improve Viscosity and Phase Separation Guided by Crystal Structure. Mol. Pharm. 2016, 13, 915–923. [Google Scholar] [CrossRef]
- Du, Q.; Damschroder, M.M.; Pabst, T.M.; Hunter, A.K.; Wang, W.K.; Luo, H. Process optimization and protein engineering mitigated manufacturing challenges of a monoclonal antibody with liquid-liquid phase separation issue by disrupting inter-molecule electrostatic interactions. mAbs 2019, 11, 789–802. [Google Scholar] [CrossRef]
- Raut, A.S.; Kalonia, D.S. Effect of Excipients on Liquid–Liquid Phase Separation and Aggregation in Dual Variable Domain Immunoglobulin Protein Solutions. Mol. Pharm. 2016, 13, 774–783. [Google Scholar] [CrossRef]
- Brereton, H.M.; Taylor, S.D.; Farrall, A.; Hocking, D.; Thiel, M.A.; Tea, M.; Coster, D.J.; Williams, K.A. Influence of format on in vitro penetration of antibody fragments through porcine cornea. Br. J. Ophthalmol 2005, 89, 1205–1209. [Google Scholar] [CrossRef]
- Tesar, D.; Luoma, J.; Wyatt, E.A.; Shi, C.; Shatz, W.; Hass, P.E.; Mathieu, M.; Yi, L.; Corn, J.E.; Maass, K.F.; et al. Protein engineering to increase the potential of a therapeutic antibody Fab for long-acting delivery to the eye. mAbs 2017, 9, 1297–1305. [Google Scholar] [CrossRef]
- Marmor, M.F.; Martin, L.J.; Tharpe, S. Osmotically induced retinal detachment in the rabbit and primate. Electron miscoscopy of the pigment epithelium. Investig. Ophthalmol. Vis. Sci. 1980, 19, 1016–1029. [Google Scholar]
- Rabia, L.A.; Desai, A.A.; Jhajj, H.S.; Tessier, P.M. Understanding and overcoming trade-offs between antibody affinity, specificity, stability and solubility. Biochem. Eng. J. 2018, 137, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Pindrus, M.; Shire, S.J.; Kelley, R.F.; Demeule, B.; Wong, R.; Xu, Y.; Yadav, S. Solubility Challenges in High Concentration Monoclonal Antibody Formulations: Relationship with Amino Acid Sequence and Intermolecular Interactions. Mol. Pharm. 2015, 12, 3896–3907. [Google Scholar] [CrossRef]
- Bethea, D.; Wu, S.-J.; Luo, J.; Hyun, L.; Lacy, E.R.; Teplyakov, A.; Jacobs, S.A.; O’Neil, K.T.; Gilliland, G.L.; Feng, Y. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng. Des. Sel. 2012, 25, 531–538. [Google Scholar] [CrossRef]
- Benschop, R.J.; Chow, C.-K.; Tian, Y.; Nelson, J.; Barmettler, B.; Atwell, S.; Clawson, D.; Chai, Q.; Jones, B.E.; Fitchett, J.; et al. Development of tibulizumab, a tetravalent bispecific antibody targeting BAFF and IL-17A for the treatment of autoimmune disease. mAbs 2019, 11, 1175–1190. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Caffry, I.; Wu, J.; Geng, S.B.; Jain, T.; Sun, T.; Reid, F.; Cao, Y.; Estep, P.; Yu, Y.; et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. mAbs 2014, 6, 483–492. [Google Scholar] [CrossRef]
- Sule, S.V.; Dickinson, C.D.; Lu, J.; Chow, C.-K.; Tessier, P.M. Rapid Analysis of Antibody Self-Association in Complex Mixtures Using Immunogold Conjugates. Mol. Pharm. 2013, 10, 1322–1331. [Google Scholar] [CrossRef]
- Estep, P.; Caffry, I.; Yu, Y.; Sun, T.; Cao, Y.; Lynaugh, H.; Jain, T.; Vásquez, M.; Tessier, P.M.; Xu, Y. An alternative assay to hydrophobic interaction chromatography for high-throughput characterization of monoclonal antibodies. mAbs 2015, 7, 553–561. [Google Scholar] [CrossRef]
- Wu, J.; Schultz, J.S.; Weldon, C.L.; Sule, S.V.; Chai, Q.; Geng, S.B.; Dickinson, C.D.; Tessier, P.M. Discovery of highly soluble antibodies prior to purification using affinity-capture self-interaction nanoparticle spectroscopy. Protein Eng. Des. Sel. 2015, 28, 403–414. [Google Scholar] [CrossRef]
- Kingsbury, J.S.; Saini, A.; Auclair, S.M.; Fu, L.; Lantz, M.M.; Halloran, K.T.; Calero-Rubio, C.; Schwenger, W.; Airiau, C.Y.; Zhang, J.; et al. A single molecular descriptor to predict solution behavior of therapeutic antibodies. Sci. Adv. 2020, 6, eabb0372. [Google Scholar] [CrossRef]
- Li, L.; Kantor, A.; Warne, N. Application of a PEG precipitation method for solubility screening: A tool for developing high protein concentration formulations. Protein Sci. 2013, 22, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.J.; Mccarty, K.; McFadyen, I.J.; Cash, E.; Dalmonte, P.; Hinds, K.D.; Dinerman, A.A.; Alvarez, J.C.; Volkin, D.B. Application of a High-Throughput Screening Procedure with PEG-Induced Precipitation to Compare Relative Protein Solubility During Formulation Development with IgG1 Monoclonal Antibodies. J. Pharm. Sci. 2011, 100, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Yamniuk, A.P.; Ditto, N.; Patel, M.; Dai, J.; Sejwal, P.; Stetsko, P.; Doyle, M.L. Application of a Kosmotrope-Based Solubility Assay to Multiple Protein Therapeutic Classes Indicates Broad Use as a High-Throughput Screen for Protein Therapeutic Aggregation Propensity. J. Pharm. Sci. 2013, 102, 2424–2439. [Google Scholar] [CrossRef]
- Chai, Q.; Shih, J.; Weldon, C.; Phan, S.; Jones, B.E. Development of a high-throughput solubility screening assay for use in antibody discovery. mAbs 2019, 11, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Winzer, M.; Weber, C.; Gieseler, H. Limitations of polyethylene glycol-induced precipitation as predictive tool for protein solubility during formulation development. J. Pharm. Pharmacol. 2017, 70, 648–654. [Google Scholar] [CrossRef]
- Izadi, S.; Patapoff, T.W.; Walters, B.T. Multiscale Coarse-Grained Approach to Investigate Self-Association of Antibodies. Biophys. J. 2020, 118, 2741–2754. [Google Scholar] [CrossRef]
- Sharma, V.K.; Patapoff, T.W.; Kabakoff, B.; Pai, S.; Hilario, E.; Zhang, B.; Li, C.; Borisov, O.; Kelley, R.F.; Chorny, I.; et al. In silico selection of therapeutic antibodies for development: Viscosity, clearance, and chemical stability. Proc. Natl. Acad. Sci. USA 2014, 111, 18601–18606. [Google Scholar] [CrossRef]
- Tomar, D.S.; Li, L.; Broulidakis, M.P.; Luksha, N.G.; Burns, C.T.; Singh, S.K.; Kumar, S. In-silico prediction of concentration-dependent viscosity curves for monoclonal antibody solutions. mAbs 2017, 9, 476–489. [Google Scholar] [CrossRef]
- Calero-Rubio, C.; Saluja, A.; Roberts, C.J. Coarse-Grained Antibody Models for “Weak” Protein–Protein Interactions from Low to High Concentrations. J. Phys. Chem. B 2016, 120, 6592–6605. [Google Scholar] [CrossRef]
- Ferreira, G.M.; Calero-Rubio, C.; Sathish, H.A.; Remmele, R.L.; Roberts, C.J.; Hasige, S. Electrostatically Mediated Protein-Protein Interactions for Monoclonal Antibodies: A Combined Experimental and Coarse-Grained Molecular Modeling Approach. J. Pharm. Sci. 2019, 108, 120–132. [Google Scholar] [CrossRef]
- Wang, G.; Varga, Z.; Hofmann, J.; Zarraga, I.E.; Swan, J.W. Structure and Relaxation in Solutions of Monoclonal Antibodies. J. Phys. Chem. B 2018, 122, 2867–2880. [Google Scholar] [CrossRef] [PubMed]
- Kastelic, M.; Dill, K.A.; Kalyuzhnyi, Y.V.; Vlachy, V. Controlling the viscosities of antibody solutions through control of their binding sites. J. Mol. Liq. 2018, 270, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Terraube, V.; Tam, A.S.P.; Stochaj, W.; Fennell, B.J.; Lin, L.; Stahl, M.; LaVallie, E.R.; Somers, W.; Finlay, W.J.J.; et al. A Combination of Structural and Empirical Analyses Delineates the Key Contacts Mediating Stability and Affinity Increases in an Optimized Biotherapeutic Single-chain Fv (scFv). J. Biol. Chem. 2016, 291, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Tilegenova, C.; Izadi, S.; Yin, J.; Huang, C.S.; Wu, J.; Ellerman, D.; Hymowitz, S.G.; Walters, B.T.; Salisbury, C.; Carter, P.J. Dissecting the molecular basis of high viscosity of monospecific and bispecific IgG antibodies. mAbs 2020, 12, 1692764. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kumar, S.; Buck, P.M.; Burns, C.; Lavoie, J.; Singh, S.K.; Warne, N.W.; Nichols, P.; Luksha, N.; Boardman, D. Concentration Dependent Viscosity of Monoclonal Antibody Solutions: Explaining Experimental Behavior in Terms of Molecular Properties. Pharm. Res. 2014, 31, 3161–3178. [Google Scholar] [CrossRef]
- Yadav, S.; Laue, T.M.; Kalonia, D.S.; Singh, S.N.; Shire, S.J. The Influence of Charge Distribution on Self-Association and Viscosity Behavior of Monoclonal Antibody Solutions. Mol. Pharm. 2012, 9, 791–802. [Google Scholar] [CrossRef]
- Buck, P.M.; Chaudhri, A.; Kumar, S.; Singh, S.K. Highly Viscous Antibody Solutions Are a Consequence of Network Formation Caused by Domain–Domain Electrostatic Complementarities: Insights from Coarse-Grained Simulations. Mol. Pharm. 2015, 12, 127–139. [Google Scholar] [CrossRef]
- Nichols, P.; Li, L.; Kumar, S.; Buck, P.M.; Singh, S.K.; Goswami, S.; Balthazor, B.; Conley, T.R.; Sek, D.; Allen, M.J. Rational design of viscosity reducing mutants of a monoclonal antibody: Hydrophobic versus electrostatic inter-molecular interactions. mAbs 2015, 7, 212–230. [Google Scholar] [CrossRef]
- Kumar, S.; Roffi, K.; Tomar, D.S.; Cirelli, D.; Luksha, N.; Meyer, D.; Mitchell, J.; Allen, M.J.; Li, L. Rational optimization of a monoclonal antibody for simultaneous improvements in its solution properties and biological activity. Protein Eng. Des. Sel. 2018, 31, 313–325. [Google Scholar] [CrossRef]
- Raut, A.S.; Kalonia, D.S. Viscosity Analysis of Dual Variable Domain Immunoglobulin Protein Solutions: Role of Size, Electroviscous Effect and Protein-Protein Interactions. Pharm. Res. 2016, 33, 155–166. [Google Scholar] [CrossRef]
- Woldeyes, M.A.; Josephson, L.L.; Leiske, D.L.; Galush, W.J.; Roberts, C.J.; Furst, E.M. Viscosities and Protein Interactions of Bispecific Antibodies and Their Monospecific Mixtures. Mol. Pharm. 2018, 15, 4745–4755. [Google Scholar] [CrossRef]
- Gallivan, J.P.; Dougherty, D.A. A Computational Study of Cation−π Interactions vs. Salt Bridges in Aqueous Media: Implications for Protein Engineering. J. Am. Chem. Soc. 2000, 122, 870–874. [Google Scholar] [CrossRef]
- Apgar, J.; Tam, A.S.P.; Sorm, R.; Moesta, S.; King, A.C.; Yang, H.; Kelleher, K.; Murphy, D.; D’Antona, A.M.; Yan, G.; et al. Modeling and mitigation of high-concentration antibody viscosity through structure-based computer-aided protein design. PLoS ONE 2020, 15, e0232713. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.J.; Helk, B.; Kumar, S.; Mody, N.; Sathish, H.A.; Samra, H.S.; Buck, P.M.; Li, L.; Trout, B.L. Computational tool for the early screening of monoclonal antibodies for their viscosities. mAbs 2016, 8, 43–48. [Google Scholar] [CrossRef]
- Liu, J.; Nguyen, M.D.; Andya, J.D.; Shire, S.J. Reversible Self-Association Increases the Viscosity of a Concentrated Monoclonal Antibody in Aqueous Solution. J. Pharm. Sci. 2005, 94, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Klibanov, A.M. Hydrophobic salts markedly diminish viscosity of concentrated protein solutions. Biotechnol. Bioeng. 2011, 108, 632–636. [Google Scholar] [CrossRef]
- Kanai, S.; Liu, J.; Patapoff, T.W.; Shire, S.J. Reversible Self-Association of a Concentrated Monoclonal Antibody Solution Mediated by Fab–Fab Interaction That Impacts Solution Viscosity. J. Pharm. Sci. 2008, 97, 4219–4227. [Google Scholar] [CrossRef]
- Inoue, N.; Takai, E.; Arakawa, T.; Shiraki, K. Specific Decrease in Solution Viscosity of Antibodies by Arginine for Therapeutic Formulations. Mol. Pharm. 2014, 11, 1889–1896. [Google Scholar] [CrossRef]
- Ashish; Solanki, A.K.; Boone, C.D.; Krueger, J.K. Global structure of HIV-1 neutralizing antibody IgG1 b12 is asymmetric. Biochem. Biophys. Res. Commun. 2010, 391, 947–951. [Google Scholar] [CrossRef]
- Starr, C.G.; Tessier, P.M. Selecting and engineering monoclonal antibodies with drug-like specificity. Curr. Opin. Biotechnol. 2019, 60, 119–127. [Google Scholar] [CrossRef]
- Jacobs, S.A.; Wu, S.-J.; Feng, Y.; Bethea, D.; O’Neil, K.T. Cross-Interaction Chromatography: A Rapid Method to Identify Highly Soluble Monoclonal Antibody Candidates. Pharm. Res. 2010, 27, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Hötzel, I.; Theil, F.-P.; Bernstein, L.J.; Prabhu, S.; Deng, R.; Quintana, L.; Lutman, J.; Sibia, R.; Chan, P.; Bumbaca, D.; et al. A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012, 4, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Mouquet, H.; Scheid, J.F.; Zoller, M.J.; Krogsgaard, M.; Ott, R.G.; Shukair, S.; Artyomov, M.N.; Pietzsch, J.; Connors, M.; Pereyra, F.; et al. Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nat. Cell Biol. 2010, 467, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Roach, W.; Sun, T.; Jain, T.; Prinz, B.; Yu, T.-Y.; Torrey, J.; Thomas, J.; Bobrowicz, P.; Vásquez, M.; et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: A FACS-based, high-throughput selection and analytical tool. Protein Eng. Des. Sel. 2013, 26, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.L.; Sun, T.; Jain, T.; Caffry, I.; Yu, Y.; Cao, Y.; Lynaugh, H.; Brown, M.; Vásquez, M.; Wittrup, K.D.; et al. High throughput cross-interaction measures for human IgG1 antibodies correlate with clearance rates in mice. mAbs 2015, 7, 770–777. [Google Scholar] [CrossRef]
- Kelly, R.L.; Geoghegan, J.C.; Feldman, J.; Jain, T.; Kauke, M.; Le, D.; Zhao, J.; Wittrup, K.D. Chaperone proteins as single component reagents to assess antibody nonspecificity. mAbs 2017, 9, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.L.; Le, D.; Zhao, J.; Wittrup, K. Reduction of Nonspecificity Motifs in Synthetic Antibody Libraries. J. Mol. Biol. 2018, 430, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.L.; Zhao, J.; Le, D.; Wittrup, K.D. Nonspecificity in a nonimmune human scFv repertoire. mAbs 2017, 9, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Avery, L.B.; Wade, J.; Wang, M.; Tam, A.; King, A.; Piche-Nicholas, N.; Kavosi, M.S.; Penn, S.; Cirelli, D.; Kurz, J.C.; et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. mAbs 2018, 10, 244–255. [Google Scholar] [CrossRef]
- Frese, K.; Eisenmann, M.; Ostendorp, R.; Brocks, B.; Pabst, S. An automated immunoassay for early specificity profiling of antibodies. mAbs 2013, 5, 279–287. [Google Scholar] [CrossRef]
- Li, B.; Tesar, D.; Boswell, C.A.; Cahaya, H.S.; Wong, A.; Zhang, J.; Meng, Y.G.; Eigenbrot, C.; Pantua, H.; Diao, J.; et al. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs 2014, 6, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Dostalek, M.; Prueksaritanont, T.; Kelley, R.F. Pharmacokinetic de-risking tools for selection of monoclonal antibody lead candidates. mAbs 2017, 9, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Kraft, T.E.; Richter, W.F.; Emrich, T.; Knaupp, A.; Schuster, M.; Wolfert, A.; Kettenberger, H. Heparin chromatography as an in vitro predictor for antibody clearance rate through pinocytosis. mAbs 2019, 12, 1683432. [Google Scholar] [CrossRef] [PubMed]
- Schoch, A.; Kettenberger, H.; Mundigl, O.; Winter, G.; Engert, J.; Heinrich, J.; Emrich, T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc. Natl. Acad. Sci. USA 2015, 112, 5997–6002. [Google Scholar] [CrossRef] [PubMed]
- Crowell, S.; Wang, K.; Famili, A.; Shatz, W.; Loyet, K.M.; Chang, V.; Liu, Y.; Prabhu, S.; Kamath, A.V.; Kelley, R.F. Influence of Charge, Hydrophobicity, and Size on Vitreous Pharmacokinetics of Large Molecules. Transl. Vis. Sci. Technol. 2019, 8, 1. [Google Scholar] [CrossRef]
- Schaller, T.H.; Foster, M.W.; Thompson, W.; Spasojevic, I.; Normantaite, D.; Moseley, M.A.; Sanchez-Perez, L.; Sampson, J.H. Pharmacokinetic Analysis of a Novel Human EGFRvIII:CD3 Bispecific Antibody in Plasma and Whole Blood Using a High-Resolution Targeted Mass Spectrometry Approach. J. Proteome Res. 2019, 18, 3032–3041. [Google Scholar] [CrossRef]
- Birtalan, S.; Zhang, Y.; Fellouse, F.A.; Shao, L.; Schaefer, G.; Sidhu, S.S. The Intrinsic Contributions of Tyrosine, Serine, Glycine and Arginine to the Affinity and Specificity of Antibodies. J. Mol. Biol. 2008, 377, 1518–1528. [Google Scholar] [CrossRef]
- Birtalan, S.; Fisher, R.D.; Sidhu, S.S. The functional capacity of the natural amino acids for molecular recognition. Mol. BioSyst. 2010, 6, 1186. [Google Scholar] [CrossRef]
- Tiller, K.E.; Li, L.-J.; Kumar, S.; Julian, M.C.; Garde, S.; Tessier, P.M. Arginine mutations in antibody complementarity-determining regions display context-dependent affinity/specificity trade-offs. J. Biol. Chem. 2017, 292, 16638–16652. [Google Scholar] [CrossRef]
- Wardemann, H.; Yurasov, S.; Schaefer, A.; Young, J.W.; Meffre, E.; Nussenzweig, M.C. Predominant Autoantibody Production by Early Human B Cell Precursors. Science 2003, 301, 1374–1377. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Thangaraju, A.; Leung, D.; Tang, Y.; Witcher, D.R.; Lu, J.; Wroblewski, V.J. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs 2015, 7, 483–493. [Google Scholar] [CrossRef]
- Rabia, L.A.; Zhang, Y.; Ludwig, S.D.; Julian, M.C.; Tessier, P.M. Net charge of antibody complementarity-determining regions is a key predictor of specificity. Protein Eng. Des. Sel. 2018, 31, 409–418. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, L.; Gupta, P.; Desai, A.A.; Smith, M.D.; Rabia, L.A.; Ludwig, S.D.; Tessier, P.M. Physicochemical Rules for Identifying Monoclonal Antibodies with Drug-like Specificity. Mol. Pharm. 2020, 17, 2555–2569. [Google Scholar] [CrossRef]
- Ovacik, M.; Lin, K. Tutorial on Monoclonal Antibody Pharmacokinetics and Its Considerations in Early Development. Clin. Transl. Sci. 2018, 11, 540–552. [Google Scholar] [CrossRef]
- Paci, A.; Desnoyer, A.; Delahousse, J.; Blondel, L.; Maritaz, C.; Chaput, N.; Mir, O.; Broutin, S. Pharmacokinetic/pharmacodynamic relationship of therapeutic monoclonal antibodies used in oncology: Part 1, monoclonal antibodies, antibody-drug conjugates and bispecific T-cell engagers. Eur. J. Cancer 2020, 128, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Datta-Mannan, A.; Croy, J.E.; Schirtzinger, L.; Torgerson, S.; Breyer, M.; Wroblewski, V.J. Aberrant bispecific antibody pharmacokinetics linked to liver sinusoidal endothelium clearance mechanism in cynomolgus monkeys. mAbs 2016, 8, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, P.; Lee, B.; Channappa, D.; Cheung, C.-W.; Crow, D.; Chea, J.; Poku, E.; Li, L.; Andersen, J.T.; Sandlie, I.; et al. A series of anti-CEA/anti-DOTA bispecific antibody formats evaluated for pre-targeting: Comparison of tumor uptake and blood clearance. Protein Eng. Des. Sel. 2012, 26, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.A.; Chang, C.-H.; Cardillo, T.M.; Goldenberg, D.M. Optimization of Multivalent Bispecific Antibodies and Immunocytokines with Improved in Vivo Properties. Bioconjug. Chem. 2012, 24, 63–71. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Brown, R.M.; Fitchett, J.; Heng, A.R.; Balasubramaniam, D.; Pereira, J.; Croy, J.E. Modulation of the Biophysical Properties of Bifunctional Antibodies as a Strategy for Mitigating Poor Pharmacokinetics. Biochemistry 2019, 58, 3116–3132. [Google Scholar] [CrossRef]
- Ghosh, J.G.; Nguyen, A.A.; Bigelow, C.E.; Poor, S.; Qiu, Y.; Rangaswamy, N.; Ornberg, R.; Jackson, B.; Mak, H.; Ezell, T.; et al. Long-acting protein drugs for the treatment of ocular diseases. Nat. Commun. 2017, 8, 14837. [Google Scholar] [CrossRef]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta 2013, 1830, 5526–5534. [Google Scholar] [CrossRef] [PubMed]
- Malm, M.; Bass, T.; Gudmundsdotter, L.; Lord, M.; Frejd, F.; Ståhl, S.; Lofblom, J. Engineering of a bispecific affibody molecule towards HER2 and HER3 by addition of an albumin-binding domain allows for affinity purification and in vivo half-life extension. Biotechnol. J. 2014, 9, 1215–1222. [Google Scholar] [CrossRef]
- Nilvebrant, J.; Åstrand, M.; Georgieva-Kotseva, M.; Björnmalm, M.; Lofblom, J.; Hober, S. Engineering of Bispecific Affinity Proteins with High Affinity for ERBB2 and Adaptable Binding to Albumin. PLoS ONE 2014, 9, e103094. [Google Scholar] [CrossRef] [PubMed]
- Nilvebrant, J.; Alm, T.; Hober, S.; Lofblom, J. Engineering Bispecificity into a Single Albumin-Binding Domain. PLoS ONE 2011, 6, e25791. [Google Scholar] [CrossRef]
- Davé, E.; Adams, R.; Zaccheo, O.; Carrington, B.; Compson, J.E.; Dugdale, S.; Airey, M.; Malcolm, S.; Hailu, H.; Wild, G.; et al. Fab-dsFv: A bispecific antibody format with extended serum half-life through albumin binding. mAbs 2016, 8, 1319–1335. [Google Scholar] [CrossRef]
- Day, S.; Acquah, K.; Mruthyunjaya, P.; Grossman, D.S.; Lee, P.P.; Sloan, F.A. Ocular Complications After Anti–Vascular Endothelial Growth Factor Therapy in Medicare Patients with Age-Related Macular Degeneration. Am. J. Ophthalmol. 2011, 152, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Bustos, R.-H.; Zapata, C.; Garcia, J.; Jauregui, E.; Ashraf, G.M.; Rodríguez, R.H.B.L.F. Immunogenicity in Protein and Peptide Based-Therapeutics: An Overview. Curr. Protein Pept. Sci. 2018, 19, 958–971. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Lagassé, D.; Pedras-Vasconcelos, J.; Golding, B.; Rosenberg, A. Evaluating and Mitigating the Immunogenicity of Therapeutic Proteins. Trends Biotechnol. 2018, 36, 1068–1084. [Google Scholar] [CrossRef]
- Yuseff, M.-I.; Pierobon, P.; Reversat, A.; Lennon-Duménil, A.-M. How B cells capture, process and present antigens: A crucial role for cell polarity. Nat. Rev. Immunol. 2013, 13, 475–486. [Google Scholar] [CrossRef]
- Germain, R.N.; Margulies, D.H. The biochemistry and cell biology of antigen processing and presentation. Annu. Rev. Immunol. 1993, 11, 403–450. [Google Scholar] [CrossRef]
- Paul, S.; Grifoni, A.; Peters, B.; Sette, A. Major Histocompatibility Complex Binding, Eluted Ligands, and Immunogenicity: Benchmark Testing and Predictions. Front. Immunol. 2020, 10, 3151. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.; Sauna, Z.E. Immunogenicity assessment during the development of protein therapeutics. J. Pharm. Pharmacol. 2017, 70, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Gunn, G.R.; Sealey, D.C.F.; Jamali, F.; Meibohm, B.; Ghosh, S.; Shankar, G. From the bench to clinical practice: Understanding the challenges and uncertainties in immunogenicity testing for biopharmaceuticals. Clin. Exp. Immunol. 2016, 184, 137–146. [Google Scholar] [CrossRef] [PubMed]
- FDA. Immunogenicity Testing of Therapeutic Protein Products—Developing and Validating Assays for Anti-Drug Antibody Detection. 2019. Available online: https://www.fda.gov/media/119788/download (accessed on 29 August 2020).
- Shankar, G.; Arkin, S.; Cocea, L.; Devanarayan, V.; Kirshner, S.; Kromminga, A.; Quarmby, V.; Richards, S.; Schneider, C.K.; Subramanyam, M.; et al. Assessment and Reporting of the Clinical Immunogenicity of Therapeutic Proteins and Peptides—Harmonized Terminology and Tactical Recommendations. AAPS J. 2014, 16, 658–673. [Google Scholar] [CrossRef]
- Salazar-Fontana, L.I.; Desai, D.D.; Khan, T.A.; Pillutla, R.C.; Prior, S.; Ramakrishnan, R.; Schneider, J.; Joseph, A. Approaches to Mitigate the Unwanted Immunogenicity of Therapeutic Proteins during Drug Development. AAPS J. 2017, 19, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Tourdot, S.; Hickling, T.P. Nonclinical immunogenicity risk assessment of therapeutic proteins. Bioanalysis 2019, 11, 1631–1643. [Google Scholar] [CrossRef]
- Baumann, A.; Fischmann, S.; Blaich, G.; Friedrich, M. Leverage nonclinical development of bispecifics by translational science. Drug Discov. Today Technol. 2016, 21–22, 95–102. [Google Scholar] [CrossRef]
- Gorovits, B.; Peng, K.; Kromminga, A. Current Considerations on Characterization of Immune Response to Multi-Domain Biotherapeutics. BioDrugs 2019, 34, 39–54. [Google Scholar] [CrossRef]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef]
- Hao, C.-H.; Han, Q.-H.; Shan, Z.-J.; Hu, J.-T.; Zhang, N.; Zhang, X.-P. Effects of different interchain linkers on biological activity of an anti-prostate cancer single-chain bispecific antibody. Theor. Biol. Med. Model. 2015, 12, 14. [Google Scholar] [CrossRef]
- Kibria, G.; Akazawa-Ogawa, Y.; Rahman, N.; Hagihara, Y.; Kuroda, Y. The immunogenicity of an anti-EGFR single domain antibody (VHH) is enhanced by misfolded amorphous aggregation but not by heat-induced aggregation. Eur. J. Pharm. Biopharm. 2020, 152, 164–174. [Google Scholar] [CrossRef]
- Rahman, N.; Islam, M.M.; Unzai, S.; Miura, S.; Kuroda, Y. Nanometer-Sized Aggregates Generated Using Short Solubility Controlling Peptide Tags Do Increase the In Vivo Immunogenicity of a Nonimmunogenic Protein. Mol. Pharm. 2020, 17, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A. Effects of protein aggregates: An immunologic perspective. AAPS J. 2006, 8, E501–E507. [Google Scholar] [CrossRef] [PubMed]
- Broders, O.; Wessels, U.; Zadak, M.; Beckmann, R.; Stubenrauch, K. Novel bioanalytical method for the characterization of the immune response directed against a bispecific F(ab) fragment. Bioanalysis 2020, 12, 509–517. [Google Scholar] [CrossRef]
- Bivi, N.; Moore, T.; Rodgers, G.; Denning, H.; Shockley, T.; Swearingen, C.A.; Gelfanova, V.; Calderon, B.; Peterson, D.A.; Hodsdon, M.E.; et al. Investigation of pre-existing reactivity to biotherapeutics can uncover potential immunogenic epitopes and predict immunogenicity risk. mAbs 2019, 11, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Yang, L.; Trepicchio, W.L.; Wyant, T. Understanding the Supersensitive Anti-Drug Antibody Assay: Unexpected High Anti-Drug Antibody Incidence and Its Clinical Relevance. J. Immunol. Res. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Quarmby, V.; Phung, Q.; Lill, J.R. MAPPs for the identification of immunogenic hotspots of biotherapeutics; an overview of the technology and its application to the biopharmaceutical arena. Expert Rev. Proteom. 2018, 15, 733–748. [Google Scholar] [CrossRef]
- Karle, A.C. Applying MAPPs Assays to Assess Drug Immunogenicity. Front. Immunol. 2020, 11, 698. [Google Scholar] [CrossRef]
- Wu, Y.; Li, C.; Xia, S.; Tian, X.; Kong, Y.; Wang, Z.; Gu, C.; Zhang, R.; Tu, C.; Xie, Y.; et al. Identification of Human Single-Domain Antibodies against SARS-CoV-2. Cell Host Microbe 2020, 27, 891–898.e5. [Google Scholar] [CrossRef]
- Waldmann, H. Human Monoclonal Antibodies: The Benefits of Humanization. Methods Mol. Biol. 2019, 1904, 1–10. [Google Scholar]
- Almagro, J.C.; Fransson, J. Humanization of antibodies. Front. Biosci. 2008, 13, 1619–1633. [Google Scholar] [PubMed]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody humanization methods—A review and update. Biotechnol. Genet. Eng. Rev. 2013, 29, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Wollacott, A.M.; Xue, C.; Qin, Q.; Hua, J.; Bohnuud, T.; Viswanathan, K.; Kolachalama, V.B. Quantifying the nativeness of antibody sequences using long short-term memory networks. Protein Eng. Des. Sel. 2019, 32, 347–354. [Google Scholar] [CrossRef]
- Schmitz, S.; Soto, C.; Crowe, J.E.; Meiler, J.; Crowe, J.E. Human-likeness of antibody biologics determined by back-translation and comparison with large antibody variable gene repertoires. mAbs 2020, 12, 1758291. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Nielsen, M.; Sette, A. T Cell Epitope Predictions. Annu. Rev. Immunol. 2020, 38, 123–145. [Google Scholar] [CrossRef]
- Reynisson, B.; Barra, C.; Kaabinejadian, S.; Hildebrand, W.H.; Peters, B.; Nielsen, M. Improved Prediction of MHC II Antigen Presentation through Integration and Motif Deconvolution of Mass Spectrometry MHC Eluted Ligand Data. J. Proteome Res. 2020, 19, 2304–2315. [Google Scholar] [CrossRef]
- Garde, C.; Ramarathinam, S.H.; Jappe, E.C.; Nielsen, M.; Kringelum, J.V.; Trolle, T.; Purcell, A.W. Improved peptide-MHC class II interaction prediction through integration of eluted ligand and peptide affinity data. Immunogenetics 2019, 71, 445–454. [Google Scholar] [CrossRef]
- Sekiguchi, N.; Kubo, C.; Takahashi, A.; Muraoka, K.; Takeiri, A.; Ito, S.; Yano, M.; Mimoto, F.; Maeda, A.; Iwayanagi, Y.; et al. MHC-associated peptide proteomics enabling highly sensitive detection of immunogenic sequences for the development of therapeutic antibodies with low immunogenicity. mAbs 2018, 10, 1168–1181. [Google Scholar] [CrossRef]
- Barra, C.; Ackaert, C.; Reynisson, B.; Schockaert, J.; Jessen, L.E.; Watson, M.; Jang, A.; Comtois-Marotte, S.; Goulet, J.-P.; Pattijn, S.; et al. Immunopeptidomic Data Integration to Artificial Neural Networks Enhances Protein-Drug Immunogenicity Prediction. Front. Immunol. 2020, 11, 1304. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawant, M.S.; Streu, C.N.; Wu, L.; Tessier, P.M. Toward Drug-Like Multispecific Antibodies by Design. Int. J. Mol. Sci. 2020, 21, 7496. https://doi.org/10.3390/ijms21207496
Sawant MS, Streu CN, Wu L, Tessier PM. Toward Drug-Like Multispecific Antibodies by Design. International Journal of Molecular Sciences. 2020; 21(20):7496. https://doi.org/10.3390/ijms21207496
Chicago/Turabian StyleSawant, Manali S., Craig N. Streu, Lina Wu, and Peter M. Tessier. 2020. "Toward Drug-Like Multispecific Antibodies by Design" International Journal of Molecular Sciences 21, no. 20: 7496. https://doi.org/10.3390/ijms21207496
APA StyleSawant, M. S., Streu, C. N., Wu, L., & Tessier, P. M. (2020). Toward Drug-Like Multispecific Antibodies by Design. International Journal of Molecular Sciences, 21(20), 7496. https://doi.org/10.3390/ijms21207496