Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease




Abstract
1. Introduction
2. The Amyloid Beta Cascade Hypothesis
The Role of Neuroinflammation and Oxidative Stress in Pathophysiology of Alzheimer’s Disease
3. Therapeutic Agents Targeting Amyloid Cascade Events
3.1. Modulation of Secretase Enzymes
3.2. Immunotherapy
3.3. Prevention of Amyloid Aggregation
Peptide Inhibitors of Amyloid Aggregation
3.4. Autacoid Local Injury Antagonist Amides (ALIAmides) as a Novel Therapeutic Strategy in AD
3.5. Other Agents Targeting Aβ Deposits
3.6. Natural Drugs Targeting Amyloids in AD
4. Future Prospects and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gao, H.-M.; Hong, J.-S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Madav, Y.; Wairkar, S.; Prabhakar, B. Recent therapeutic strategies targeting beta amyloid and tauopathies in Alzheimer’s disease. Brain Res. Bull. 2019, 146, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Barker, W.W.; Luis, C.A.; Kashuba, A.; Luis, M.; Harwood, D.G.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 2002, 16, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75. e62. [Google Scholar] [CrossRef]
- Egawa, J.; Pearn, M.L.; Lemkuil, B.P.; Patel, P.M.; Head, B.P. Membrane lipid rafts and neurobiology: Age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 2016, 594, 4565–4579. [Google Scholar] [CrossRef]
- Banning, L.C.; Ramakers, I.H.; Deckers, K.; Verhey, F.R.; Aalten, P. Apolipoprotein E and affective symptoms in mild cognitive impairment and Alzheimer’s disease dementia: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2019, 96, 302–315. [Google Scholar] [CrossRef]
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 1–3. [Google Scholar] [CrossRef]
- Hoie, E. Alzheimer’s Disease: Current Treatments and Potential New Agents. US Pharm 2019, 44, 20–23. [Google Scholar]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef]
- Godyń, J.; Jończyk, J.; Panek, D.; Malawska, B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2016, 68, 127–138. [Google Scholar] [CrossRef]
- Botchway, B.; Iyer, I.C. Alzheimer’s disease–the past, the present and the future. Science 2017, 6, 1–19. [Google Scholar]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.; Lee, B.Y.; Leonenko, Z. Drugs and Drug Delivery Systems Targeting Amyloid-\b {eta} in Alzheimers Disease. AIMS Mol. Sci. 2017. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. New Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Matsuzaki, K. How do membranes initiate Alzheimer’s Disease? Formation of toxic amyloid fibrils by the amyloid β-protein on ganglioside clusters. Acc. Chem. Res. 2014, 47, 2397–2404. [Google Scholar] [CrossRef]
- Cecchi, C.; Stefani, M. The amyloid-cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity. Biophys. Chem. 2013, 182, 30–43. [Google Scholar] [CrossRef]
- Drolle, E.; Gaikwad, R.M.; Leonenko, Z. Nanoscale electrostatic domains in cholesterol-laden lipid membranes create a target for amyloid binding. Biophys. J. 2012, 103, L27–L29. [Google Scholar] [CrossRef]
- Drolle, E.; Hane, F.; Lee, B.; Leonenko, Z. Atomic force microscopy to study molecular mechanisms of amyloid fibril formation and toxicity in Alzheimer’s disease. Drug Metab. Rev. 2014, 46, 207–223. [Google Scholar] [CrossRef]
- Hane, F.; Drolle, E.; Gaikwad, R.; Faught, E.; Leonenko, Z. Amyloid-β aggregation on model lipid membranes: An atomic force microscopy study. J. Alzheimer’s Dis. 2011, 26, 485–494. [Google Scholar] [CrossRef]
- Lal, R.; Lin, H.; Quist, A.P. Amyloid beta ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim. Biophys. Acta (BBA)-Biomembr. 2007, 1768, 1966–1975. [Google Scholar] [CrossRef]
- Arispe, N.; Pollard, H.B.; Rojas, E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes. Proc Natl. Acad. Sci. USA 1993, 90, 10573–10577. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Smith, M.; Parker, I. Single-channel Ca2+ imaging implicates Aβ1–42 amyloid pores in Alzheimer’s disease pathology. J. Cell Biol. 2011, 195, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Plant, L.D.; Boyle, J.P.; Smith, I.F.; Peers, C.; Pearson, H.A. The production of amyloid β peptide is a critical requirement for the viability of central neurons. J. Neurosci. 2003, 23, 5531–5535. [Google Scholar] [CrossRef]
- Giuffrida, M.L.; Caraci, F.; Pignataro, B.; Cataldo, S.; De Bona, P.; Bruno, V.; Molinaro, G.; Pappalardo, G.; Messina, A.; Palmigiano, A. β-amyloid monomers are neuroprotective. J. Neurosci. 2009, 29, 10582–10587. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef]
- Müller, U.C.; Zheng, H. Physiological functions of APP family proteins. Cold Spring Harbor Perspect. Med. 2012, 2, a006288. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Bolduc, D.M.; Montagna, D.R.; Seghers, M.; Selkoe, D.J. P4-071: The Amyloid-B Generating Tri-Peptide Cleavage Mechanism of Gamma-Secretase: Implications for Alzheimer’s Disease. Alzheimer’s Dement. 2016, 12, P1041-P1041. [Google Scholar] [CrossRef]
- Mattson, M.P.; Chan, S.L. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 2003, 34, 385–397. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The role of amyloid precursor protein processing by BACE1, the β-secretase, in Alzheimer disease pathophysiology. J. Biol. Chem. 2008, 283, 29621–29625. [Google Scholar] [CrossRef]
- Sun, J.; Roy, S. The physical approximation of APP and BACE-1: A key event in alzheimer’s disease pathogenesis. Dev. Neurobiol. 2018, 78, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Kurouski, D.; Rasool, S.; Milton, S.; Wu, J.W.; Uversky, V.N.; Lednev, I.K.; Glabe, C.G. Structural differences between amyloid beta oligomers. Biochem. Biophys. Res. Commun. 2016, 477, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.T.; Klementieva, O.; Gouras, G.K. Prion-like seeding and nucleation of intracellular amyloid-β. Neurobiol. Dis. 2018, 113, 1–10. [Google Scholar] [CrossRef]
- Roeters, S.J.; Iyer, A.; Pletikapić, G.; Kogan, V.; Subramaniam, V.; Woutersen, S. Evidence for intramolecular antiparallel beta-sheet structure in alpha-synuclein fibrils from a combination of two-dimensional infrared spectroscopy and atomic force microscopy. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β 1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Sierra-Fonseca, J.A.; Gosselink, K.L. Tauopathy and neurodegeneration: A role for stress. Neurobiol. Stress 2018, 9, 105–112. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Guarnera, E.; Berezovsky, I.N. Insulin-degrading enzyme in the fight against Alzheimer’s disease. Trends Pharmacol. Sci. 2018, 39, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Lanzillotta, C.; Perluigi, M.; Butterfield, D.A. Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 2017, 133, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxidative Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- McQuillan, K.; Lynch, M.A.; Mills, K.H. Activation of mixed glia by Abeta-specific Th1 and Th17 cells and its regulation by Th2 cells. Brain Behav. Immun. 2010, 24, 598–607. [Google Scholar] [CrossRef]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef]
- Suescun, J.; Chandra, S.; Schiess, M.C. The Role of Neuroinflammation in Neurodegenerative Disorders. In Translational Inflammation; Actor, J.K., Smith, K.C., Eds.; Academic Press: Houston, TX, USA, 2019; pp. 241–267. [Google Scholar]
- Medeiros, R.; LaFerla, F.M. Astrocytes: Conductors of the Alzheimer disease neuroinflammatory symphony. Exp. Neurol. 2013, 239, 133–138. [Google Scholar] [CrossRef]
- Das, U.; Wang, L.; Ganguly, A.; Saikia, J.M.; Wagner, S.L.; Koo, E.H.; Roy, S. Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat. Neurosci. 2016, 19, 55–64. [Google Scholar] [CrossRef]
- Asai, M.; Hattori, C.; Iwata, N.; Saido, T.C.; Sasagawa, N.; Szabó, B.; Hashimoto, Y.; Maruyama, K.; Tanuma, S.i.; Kiso, Y. The novel β-secretase inhibitor KMI-429 reduces amyloid β peptide production in amyloid precursor protein transgenic and wild-type mice. J. Neurochem. 2006, 96, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Ghosh, A. Treating transgenic Alzheimer mice with a β-secretase inhibitor, what have we learned? Aging (Albany NY) 2011, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Ozudogru, S.; Lippa, C. Disease modifying drugs targeting β-amyloid. Am. J. Alzheimer’s Dis. Other Dement. 2012, 27, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Daim, M.M.; Shaheen, H.M.; Abushouk, A.I.; Toraih, E.A.; Fawzy, M.S.; Alansari, W.S.; Aleya, L.; Bungau, S. Thymoquinone and diallyl sulfide protect against fipronil-induced oxidative injury in rats. Environ. Sci. Pollut. Res. 2018, 25, 23909–23916. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Abo El-Ela, F.I.; Alshahrani, F.K.; Bin-Jumah, M.; Al-Zharani, M.; Almutairi, B.; Alyousif, M.S.; Bungau, S.; Aleya, L.; Alkahtani, S. Protective effects of thymoquinone against acrylamide-induced liver, kidney and brain oxidative damage in rats. Environ. Sci. Pollut. Res. Int. 2020. [Google Scholar] [CrossRef]
- Chang, W.P.; Huang, X.; Downs, D.; Cirrito, J.R.; Koelsch, G.; Holtzman, D.M.; Ghosh, A.K.; Tang, J. β-Secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 2011, 25, 775–784. [Google Scholar] [CrossRef]
- John, S.; Thangapandian, S.; Sakkiah, S.; Lee, K.W. Potent BACE-1 inhibitor design using pharmacophore modeling, in silico screening and molecular docking studies. BMC Bioinform. 2011, 12, S28. [Google Scholar] [CrossRef]
- Ma, L.; Yang, Z.; Li, C.; Zhu, Z.; Shen, X.; Hu, L. Design, synthesis and SAR study of hydroxychalcone inhibitors of human β-secretase (BACE1). J. Enzym. Inhib. Med. Chem. 2011, 26, 643–648. [Google Scholar] [CrossRef]
- Siman, R.; Salidas, S. Gamma-secretase subunit composition and distribution in the presenilin wild-type and mutant mouse brain. Neuroscience 2004, 129, 615–628. [Google Scholar] [CrossRef]
- Carroll, C.M.; Li, Y.-M. Physiological and pathological roles of the γ-secretase complex. Brain Res. Bull. 2016, 126, 199–206. [Google Scholar] [CrossRef]
- Schroeter, E.H.; Ilagan, M.X.G.; Brunkan, A.L.; Hecimovic, S.; Li, Y.-m.; Xu, M.; Lewis, H.D.; Saxena, M.T.; De Strooper, B.; Coonrod, A. A presenilin dimer at the core of the γ-secretase enzyme: Insights from parallel analysis of Notch 1 and APP proteolysis. Proc. Natl. Acad. Sci. USA 2003, 100, 13075–13080. [Google Scholar] [CrossRef] [PubMed]
- Scannevin, R.H.; Chollate, S.; Brennan, M.S.; Snodgrass-Belt, P.A.; Peng, H.; Xu, L.; Jung, M.-y.; Bussiere, T.; Arastu, M.F.; Talreja, T. BIIB042, a novel γ-secretase modulator, reduces amyloidogenic Aβ isoforms in primates and rodents and plaque pathology in a mouse model of Alzheimer’s disease. Neuropharmacology 2016, 103, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Kounnas, M.Z.; Danks, A.M.; Cheng, S.; Tyree, C.; Ackerman, E.; Zhang, X.; Ahn, K.; Nguyen, P.; Comer, D.; Mao, L. Modulation of γ-secretase reduces β-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron 2010, 67, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Kounnas, M.Z.; Lane-Donovan, C.; Nowakowski, D.W.; Herz, J.; Comer, W.T. NGP 555, a γ-secretase modulator, lowers the amyloid biomarker, Aβ42, in cerebrospinal fluid while preventing Alzheimer’s disease cognitive decline in rodents. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 65–73. [Google Scholar] [CrossRef]
- Stromberg, K.; Eketjall, S.; Georgievska, B.; Tunblad, K.; Eliason, K.; Olsson, F.; Radesater, A.-C.; Klintenberg, R.; Arvidsson, P.I.; von Berg, S. Combining an amyloid-beta (A beta) cleaving enzyme inhibitor with a gamma-secretase modulator results in an additive reduction of A beta production. FEBS J. 2015, 282, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Raven, F.; Ward, J.F.; Zoltowska, K.M.; Wan, Y.; Bylykbashi, E.; Miller, S.J.; Shen, X.; Choi, S.H.; Rynearson, K.D.; Berezovska, O. Soluble Gamma-secretase Modulators Attenuate Alzheimer’s β-amyloid Pathology and Induce Conformational Changes in Presenilin 1. EBioMedicine 2017, 24, 93–101. [Google Scholar] [CrossRef]
- Harrison, T.; Churcher, I.; Beher, D. Gamma-secretase as a target for drug intervention in Alzheimer’s disease. Curr. Opin. Drug Discov. Dev. 2004, 7, 709–719. [Google Scholar]
- Imbimbo, B.P.; Giardina, G.A.M. γ-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: Disappointments and hopes. Curr. Top. Med. Chem. 2011, 11, 1555–1570. [Google Scholar] [CrossRef]
- Morgan, D. Immunotherapy for Alzheimer’s disease. J. Intern. Med. 2011, 269, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Bard, F.; Johnson-Wood, K.; Lee, C.; Hu, K.; Griffith, S.G.; Black, R.S.; Schenk, D.; Seubert, P. Aβ42 immunization in Alzheimer’s disease generates Aβ N-terminal antibodies. Ann. Neurol. 2005, 58, 430–435. [Google Scholar] [CrossRef]
- Lemere, C.A.; Masliah, E. Can Alzheimer disease be prevented by amyloid-β immunotherapy? Nat. Rev. Neurol. 2010, 6, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.; Koller, M.; Black, R.; Jenkins, L.; Griffith, S.; Fox, N.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Kohyama, K.; Matsumoto, Y. Alzheimer’s disease and immunotherapy: What is wrong with clinical trials? ImmunoTargets Ther. 2015, 4, 27. [Google Scholar] [PubMed]
- McLaurin, J.; Cecal, R.; Kierstead, M.; Tian, X.; Phinney, A.L.; Manea, M.; French, J.; Lambermon, M.H.; Darabie, A.A.; Brown, M.E. Therapeutically effective antibodies against amyloid-β peptide target amyloid-β residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat. Med. 2002, 8, 1263–1269. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Ramakrishnan, M.; Holasek, S.S.; Ramirez-Alvarado, M.; Kandimalla, K.K.; Gilles, E.J.; Curran, G.L.; Wengenack, T.M. In vivo targeting of antibody fragments to the nervous system for Alzheimer’s disease immunotherapy and molecular imaging of amyloid plaques. J. Neurochem. 2007, 102, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Moreth, J.; Mavoungou, C.; Schindowski, K. Passive anti-amyloid immunotherapy in Alzheimer’s disease: What are the most promising targets? Immun. Ageing 2013, 10, 18. [Google Scholar] [CrossRef]
- Lee, E.B.; Leng, L.Z.; Zhang, B.; Kwong, L.; Trojanowski, J.Q.; Abel, T.; Lee, V.M. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J. Biol. Chem. 2006, 281, 4292–4299. [Google Scholar] [CrossRef]
- Nell, H.J.; Au, J.L.; Giordano, C.R.; Terlecky, S.R.; Walton, P.A.; Whitehead, S.N.; Cechetto, D.F. Targeted Antioxidant, Catalase–SKL, Reduces Beta-Amyloid Toxicity in the Rat Brain. Brain Pathol. 2017, 27, 86–94. [Google Scholar] [CrossRef]
- Banks, W.A.; Terrell, B.; Farr, S.A.; Robinson, S.M.; Nonaka, N.; Morley, J.E. Passage of amyloid β protein antibody across the blood–brain barrier in a mouse model of Alzheimer’s disease. Peptides 2002, 23, 2223–2226. [Google Scholar] [CrossRef]
- Jordão, J.F.; Ayala-Grosso, C.A.; Markham, K.; Huang, Y.; Chopra, R.; McLaurin, J.; Hynynen, K.; Aubert, I. Antibodies targeted to the brain with image-guided focused ultrasound reduces amyloid-β plaque load in the TgCRND8 mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e10549. [Google Scholar] [CrossRef]
- Kolev, M.V.; Ruseva, M.M.; Harris, C.L.; Morgan, B.P.; Donev, R.M. Implication of complement system and its regulators in Alzheimer’s disease. Curr. Neuropharmacol. 2009, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.; Schmoll, H.; Platt, D.; Popa-Wagner, A.; Riederer, P.; Bauer, J. Complement C1q and C3 mRNA expression in the frontal cortex of Alzheimer’s patients. J. Mol. Med. 1995, 73, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Eckman, E.A.; Reed, D.K.; Eckman, C.B. PROTEIN SYNTHESIS, POST-TRANSLATION MODIFICATION, AND DEGRADATION-Degradation of the Alzheimer’s amyloid b peptide by endothelin-converting enzyme. J. Biol. Chem. 2001, 276, 24540–24548. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.-S.; Wang, D.; Yu, G.-Q.; Zhu, G.; Kharazia, V.N.; Paredes, J.P.; Chang, W.S.; Deitchman, J.K.; Mucke, L.; Messing, R.O. PKCε increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 8215–8220. [Google Scholar] [CrossRef]
- Ledesma, M.D.; Abad-Rodriguez, J.; Galvan, C.; Biondi, E.; Navarro, P.; Delacourte, A.; Dingwall, C.; Dotti, C.G. Raft disorganization leads to reduced plasmin activity in Alzheimer’s disease brains. EMBO Rep. 2003, 4, 1190–1196. [Google Scholar] [CrossRef]
- Ledesma, M.D.; Da Silva, J.S.; Crassaerts, K.; Delacourte, A.; De Strooper, B.; Dotti, C.G. Brain plasmin enhances APP α-cleavage and Aβ degradation and is reduced in Alzheimer’s disease brains. EMBO Rep. 2000, 1, 530–535. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Asahina, M.; Hattori, T. Selective distribution of matrix metalloproteinase-3 (MMP-3) in Alzheimer’s disease brain. Acta Neuropathol. 2000, 99, 91–95. [Google Scholar] [CrossRef]
- Backstrom, J.R.; Lim, G.P.; Cullen, M.J.; Tökés, Z.A. Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-β peptide (1–40). J. Neurosci. 1996, 16, 7910–7919. [Google Scholar] [CrossRef]
- Yan, P.; Hu, X.; Song, H.; Yin, K.; Bateman, R.J.; Cirrito, J.R.; Xiao, Q.; Hsu, F.F.; Turk, J.W.; Xu, J. Matrix metalloproteinase-9 degrades amyloid-β fibrils in vitro and compact plaques in situ. J. Biol. Chem. 2006, 281, 24566–24574. [Google Scholar] [CrossRef]
- Merlo, S.; Sortino, M.A. Estrogen activates matrix metalloproteinases-2 and-9 to increase beta amyloid degradation. Mol. Cell. Neurosci. 2012, 49, 423–429. [Google Scholar] [CrossRef]
- Xiang, Y.; Bu, X.-L.; Liu, Y.-H.; Zhu, C.; Shen, L.-L.; Jiao, S.-S.; Zhu, X.-Y.; Giunta, B.; Tan, J.; Song, W.-H. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V. Clearance systems in the brain—implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, L.O.; Näslund, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
- Ghanta, J.; Shen, C.-L.; Kiessling, L.L.; Murphy, R.M. A strategy for designing inhibitors of β-amyloid toxicity. J. Biol. Chem. 1996, 271, 29525–29528. [Google Scholar] [CrossRef] [PubMed]
- Pallitto, M.M.; Ghanta, J.; Heinzelman, P.; Kiessling, L.L.; Murphy, R.M. Recognition sequence design for peptidyl modulators of β-amyloid aggregation and toxicity. Biochemistry 1999, 38, 3570–3578. [Google Scholar] [CrossRef] [PubMed]
- Austen, B.M.; Paleologou, K.E.; Ali, S.A.; Qureshi, M.M.; Allsop, D.; El-Agnaf, O.M. Designing peptide inhibitors for oligomerization and toxicity of Alzheimer’s β-amyloid peptide. Biochemistry 2008, 47, 1984–1992. [Google Scholar] [CrossRef]
- Taylor, M.; Moore, S.; Mayes, J.; Parkin, E.; Beeg, M.; Canovi, M.; Gobbi, M.; Mann, D.M.; Allsop, D. Development of a proteolytically stable retro-inverso peptide inhibitor of β-amyloid oligomerization as a potential novel treatment for Alzheimer’s disease. Biochemistry 2010, 49, 3261–3272. [Google Scholar] [CrossRef]
- Roy, S.S. Designing Novel Peptidic Inhibitors of Beta Amyloid Oligomerization. Ph.D. Thesis, University of Calgary, Calgary, Canada, 2010. Available online: https://prism.ucalgary.ca/handle/1880/47659?show=full (accessed on 10 August 2020). [CrossRef]
- Gordon, D.; Tappe, R.; Meredith, S. Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits Aβ1–40 fibrillogenesis. J. Pept. Res. 2002, 60, 37–55. [Google Scholar] [CrossRef]
- Porat, Y.; Mazor, Y.; Efrat, S.; Gazit, E. Inhibition of islet amyloid polypeptide fibril formation: A potential role for heteroaromatic interactions. Biochemistry 2004, 43, 14454–14462. [Google Scholar] [CrossRef]
- Hane, F.T.; Lee, B.Y.; Petoyan, A.; Rauk, A.; Leonenko, Z. Testing synthetic amyloid-β aggregation inhibitor using single molecule atomic force spectroscopy. Biosens. Bioelectron. 2014, 54, 492–498. [Google Scholar] [CrossRef]
- Parthsarathy, V.; McClean, P.L.; Hölscher, C.; Taylor, M.; Tinker, C.; Jones, G.; Kolosov, O.; Salvati, E.; Gregori, M.; Masserini, M. A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e54769. [Google Scholar] [CrossRef]
- Robert, R.; Wark, K.L. Engineered antibody approaches for Alzheimer’s disease immunotherapy. Arch. Biochem. Biophys. 2012, 526, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011, 3, ra44–ra84. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. ALIAmides Update: Palmitoylethanolamide and Its Formulations on Management of Peripheral Neuropathic Pain. Int. J. Mol. Sci. 2020, 21, 5330. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Barbierato, M.; Zusso, M.; Bruschetta, G.; Impellizzeri, D.; Cuzzocrea, S.; Giusti, P. N-Palmitoylethanolamine and Neuroinflammation: A Novel Therapeutic Strategy of Resolution. Mol. Neurobiol. 2015, 52, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Cordaro, M.; Campolo, M.; Siracusa, R.; Cornelius, C.; Navarra, M.; Cuzzocrea, S.; Esposito, E. Neuroprotection by association of palmitoylethanolamide with luteolin in experimental Alzheimer’s disease models: The control of neuroinflammation. CNS Neurol. Disord. Drug Targets 2014, 13, 1530–1541. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Cordaro, M.; Bruschetta, G.; Siracusa, R.; Crupi, R.; Esposito, E.; Cuzzocrea, S. N-Palmitoylethanolamine-Oxazoline as a New Therapeutic Strategy to Control Neuroinflammation: Neuroprotective Effects in Experimental Models of Spinal Cord and Brain Injury. J. Neurotrauma 2017, 34, 2609–2623. [Google Scholar] [CrossRef]
- Cipriano, M.; Esposito, G.; Negro, L.; Capoccia, E.; Sarnelli, G.; Scuderi, C.; De Filippis, D.; Steardo, L.; Iuvone, T. Palmitoylethanolamide Regulates Production of Pro-Angiogenic Mediators in a Model of β Amyloid-Induced Astrogliosis In Vitro. CNS Neurol. Disord. Drug Targets 2015, 14, 828–837. [Google Scholar] [CrossRef]
- Orgogozo, J.-M.; Gilman, S.; Dartigues, J.-F.; Laurent, B.; Puel, M.; Kirby, L.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef]
- Black, R.S.; Sperling, R.A.; Safirstein, B.; Motter, R.N.; Pallay, A.; Nichols, A.; Grundman, M. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 198. [Google Scholar] [CrossRef]
- Götz, J.; Ittner, L.; Lim, Y.-A. Common features between diabetes mellitus and Alzheimer’s disease. Cell. Mol. Life Sci. 2009, 66, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; J Rivera, E.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Duffy, K.B.; Ottinger, M.A.; Ray, B.; Bailey, J.A.; Holloway, H.W.; Tweedie, D.; Perry, T.; Mattson, M.P.; Kapogiannis, D. GLP-1 receptor stimulation reduces amyloid-β peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 19, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.; Lahiri, D.K.; Sambamurti, K.; Chen, D.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Glucagon-like peptide-1 decreases endogenous amyloid-β peptide (Aβ) levels and protects hippocampal neurons from death induced by Aβ and iron. J. Neurosci. Res. 2003, 72, 603–612. [Google Scholar] [CrossRef]
- Haag, M.D.; Hofman, A.; Koudstaal, P.J.; Stricker, B.H.; Breteler, M.M. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J. Neurol. Neurosurg. Psychiatry 2009, 80, 13–17. [Google Scholar] [CrossRef]
- Tamboli, I.Y.; Barth, E.; Christian, L.; Siepmann, M.; Kumar, S.; Singh, S.; Tolksdorf, K.; Heneka, M.T.; Lütjohann, D.; Wunderlich, P. Statins promote the degradation of extracellular amyloid β-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem. 2010, 285, 37405–37414. [Google Scholar] [CrossRef]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef]
- Purza, L.; Abdel-Daim, M.; Belba, A.; Iovan, C.; Bumbu, A.; Lazar, L.; Bungau, S.; Tit, D.M. monitoring the effects of various combination of specific drug therapies at different stages of Alzheimer’s dementia. Farmacia 2019, 67, 477–481. [Google Scholar] [CrossRef]
- Group, A.R. Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): Results of a randomized, controlled trial of naproxen and celecoxib. Arch. Neurol. 2008, 65, 896. [Google Scholar]
- Grosu, F.; Ungureanu, A.; Bianchi, E.; Moscu, B.; Coldea, L.; Stupariu, A.L.; Pirici, I.; Roman-Filip, C.C. Multifocal and multicentric low-grade oligoastrocytoma in a young patient. Rom. J. Morphol. Embryol. 2017, 58, 207–210. [Google Scholar]
- Jang, J.; Kim, K.; Yoon, J.; Park, C.B. Piezoelectric materials for ultrasound-driven dissociation of Alzheimer’s β-amyloid aggregate structure. Biomaterials 2020, 120165. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Cohen, A.S. History of the amyloid fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef]
- Feng, B.Y.; Toyama, B.H.; Wille, H.; Colby, D.W.; Collins, S.R.; May, B.C.; Prusiner, S.B.; Weissman, J.; Shoichet, B.K. Small-molecule aggregates inhibit amyloid polymerization. Nat. Chem. Biol. 2008, 4, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.; Edwards, P.; Lai, R.; Roth, M.; Harrington, C. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C.G. Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 2007, 282, 10311–10324. [Google Scholar] [CrossRef]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.; Klang, I.M.; Lithgow, G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 2011, 472, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Herbst, M.; Wiglenda, T.; Friedrich, R.P.; Boeddrich, A.; Schiele, F.; Kleckers, D.; del Amo, J.M.L.; Grüning, B.A.; Wang, Q. Small-molecule conversion of toxic oligomers to nontoxic β-sheet–rich amyloid fibrils. Nat. Chem. Biol. 2012, 8, 93–101. [Google Scholar] [CrossRef]
- Ringman, J.M.; Frautschy, S.A.; Cole, G.M.; Masterman, D.L.; Cummings, J.L. A potential role of the curry spice curcumin in Alzheimer’s disease. Curr. Alzheimer Res. 2005, 2, 131–136. [Google Scholar] [CrossRef]
- Peter, C.; Hongwan, D.; Küpfer, A.; Lauterburg, B. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur. J. Clin. Pharmacol. 2000, 56, 247–250. [Google Scholar] [CrossRef]
- Scheindlin, S. Something old... something blue. Mol. Interv. 2008, 8, 268. [Google Scholar] [CrossRef]
- Bieschke, J. Natural compounds may open new routes to treatment of amyloid diseases. Neurotherapeutics 2013, 10, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Guan, X.; Lin, R.; Liu, X.; Yan, Y.; Lin, R.; Zhang, T.; Chen, X.; Huang, J.; Sun, X. Silibinin inhibits acetylcholinesterase activity and amyloid β peptide aggregation: A dual-target drug for the treatment of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 1792–1807. [Google Scholar] [CrossRef] [PubMed]
- Kanchi, P.K.; Dasmahapatra, A.K. Polyproline chains destabilize the Alzheimer’s amyloid-β protofibrils: A molecular dynamics simulation study. J. Mol. Graph. Model. 2019, 93, 107456. [Google Scholar] [CrossRef]
- Iyaswamy, A.; Krishnamoorthi, S.K.; Song, J.-X.; Yang, C.-B.; Kaliyamoorthy, V.; Zhang, H.; Sreenivasmurthy, S.G.; Malampati, S.; Wang, Z.-Y.; Zhu, Z. NeuroDefend, a novel Chinese medicine, attenuates amyloid-β and tau pathology in experimental Alzheimer’s disease models. J. Food Drug Anal. 2020, 28, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Baptista, F.I.; Henriques, A.G.; Silva, A.M.; Wiltfang, J.; da Cruz e Silva, O.A. Flavonoids as therapeutic compounds targeting key proteins involved in Alzheimer’s disease. ACS Chem. Neurosci. 2014, 5, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Kabir, M.T.; Tewari, D.; Al Mamun, A.; Barreto, G.E.; Bungau, S.G.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Emerging Therapeutic Promise of Ketogenic Diet to Attenuate Neuropathological Alterations in Alzheimer’s Disease. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Agents Targeting Amyloidogenic Events | Action | Ref. |
|---|---|---|
| Novel beta-secretase inhibitor (KMI-429) with hydroxy-methyl-carbonyl (HMC) isostere | Beta-site APP-cleaving enzyme (BACE1) blockers | [52] |
| N-benzoyl-oxy-carbonyl-valine-leucine-leucinal (Z-VLL-CHO, C25H39N3O5) | β-secretase inhibitor, BACE1 blocker | [2] |
| Beta-secretase inhibitor GRL-8234 | β-secretase inhibitor | [54,55,56,57] |
| Iso-liquiritigenin | BACE1 inhibitor | [59] |
| (R)-6-[(1,1′-biphenyl)-4-ylmethoxy]-1,2,3,4-tetrahydro-N,N-dimethyl-2-naphthalene-ethan-amine hydrochloride monohydrate (TAK-070, C27H31NO) | BACE1 inhibitor | [54] |
| BIIB042 | γ-secretase modulator | [63] |
| NGP328 and NGP555 | γ-secretase modulator | [64] |
| SGSM-36 | γ-secretase modulator | [67] |
| LY450139 | γ-secretase inhibitor | [54] |
| Aβ fragments in conjugation with poly-lysine | Active immunotherapeutic agents | [2] |
| NAB61 | Passive immunotherapeutic agent | [78] |
| Anti-beta-amyloid monoclonal antibody (BAM-10) | Passive immunotherapeutic agent | [81] |
| Neprilysin (NEP) | Aβ load-reducing enzyme | [2] |
| Endothelin-converting enzyme (ECE) | Aβ load-reducing enzyme | [84] |
| Protein kinase C epsilon (PKCε) | ECE enhancer, Aβ load reducer | [85] |
| Serine protease plasmin | Amyloid degradation | [87] |
| Matrix metalloproteinases (MMPs) | Amyloid degradation | [88,89,90] |
| Estrogen | MMP-2, -9 enhancer, Aβ clearance | [91] |
| ATP-binding cassette (ABC) transporters | Prevent Aβ accumulation | [2] |
| Low-density lipoprotein receptor-related protein 1 (LRP1) | Prevent Aβ accumulation | [2] |
| Aβ16-20 | Aβ-aggregation inhibitor | [94] |
| Aβ15-25 | Aβ-aggregation inhibitor | [13] |
| OR-2 | Aβ-aggregation inhibitor | [97] |
| RI-OR2 | Aβ-aggregation inhibitor | [98] |
| Aggregated human beta-amyloidAN1792 | Active immunotherapeutic reagent | [71] |
| Bapineuzumab | Monoclonal antibody | [54] |
| Glucagon-like peptide (GLP-1) | Aβ accumulation inhibitor | [115] |
| Statins | Aβ degradation promoter | [54] |
| Non-steroidal anti-inflammatory drugs (NSAIDs) (i.e., ibuprofen) | Curbed Aβ levels and ROS mitigation, γ-secretase modulator | [119,120,121] |
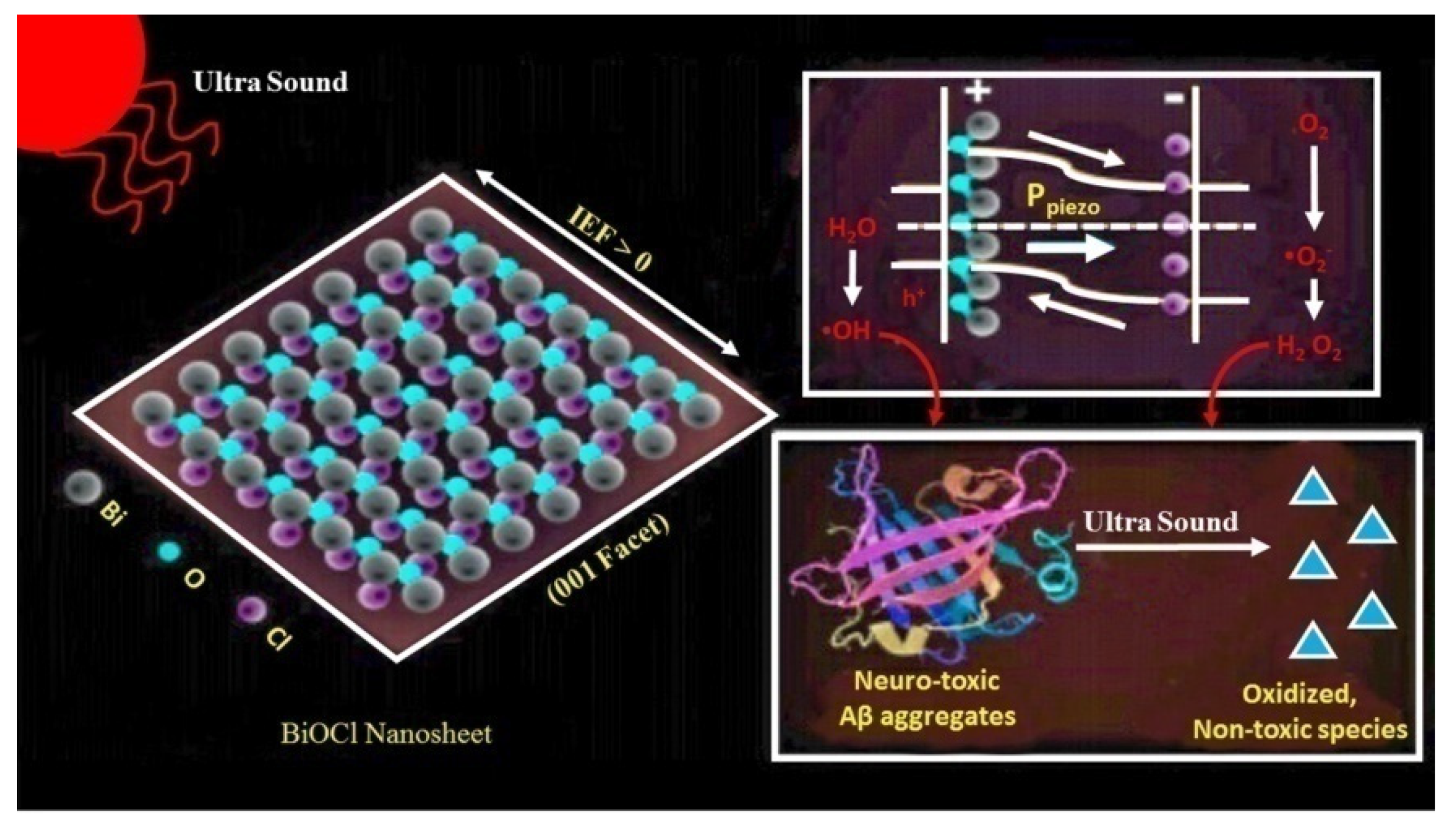
| Bismuth oxychloride (BiOCl) nanosheets | Destabilization of β-sheets | [123] |
| Congo red | Amyloid-inhibiting dye | [124,125] |
| Methylene blue | Amyloid-inhibiting dye | [126,127] |
| Thioflavin T | Amyloid-inhibiting dye | [128] |
| Orcein | Amyloid-inhibiting dye | [129] |
| Curcumin | Amyloid-inhibiting dye | [130] |
| Epi-gallocatechin-3-gallate (EGCG) | Aβ fibrillization inhibitor | [133] |
| Silibinin | Aβ aggregation inhibitor | [134] |
| Proline | β-sheet breaker | [135] |
| NeuroDefend | Aβ load reduction | [136] |
| Naturally obtained dietary flavonoids | CDK-5 and GSK-3β inhibitors, secretase enzyme modulators, Aβ aggregation inhibitors | [137,138] |
| Autacoid local injury antagonist amides (ALIAmides) (palmitoyl ethanol amide, PEA) | Anti-inflammatory, anti-hyperalgesia and lipid metabolism regulator | [107,110] |
| Ultra-micronized form of PEA and luteolin | Neuroinflammation amelioration | [107,108] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7443. https://doi.org/10.3390/ijms21207443
Behl T, Kaur I, Fratila O, Brata R, Bungau S. Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(20):7443. https://doi.org/10.3390/ijms21207443
Chicago/Turabian StyleBehl, Tapan, Ishnoor Kaur, Ovidiu Fratila, Roxana Brata, and Simona Bungau. 2020. "Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 20: 7443. https://doi.org/10.3390/ijms21207443
APA StyleBehl, T., Kaur, I., Fratila, O., Brata, R., & Bungau, S. (2020). Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease. International Journal of Molecular Sciences, 21(20), 7443. https://doi.org/10.3390/ijms21207443