Mitochondrial Quality Control in Age-Related Pulmonary Fibrosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mitochondrial Quality Control Pathways
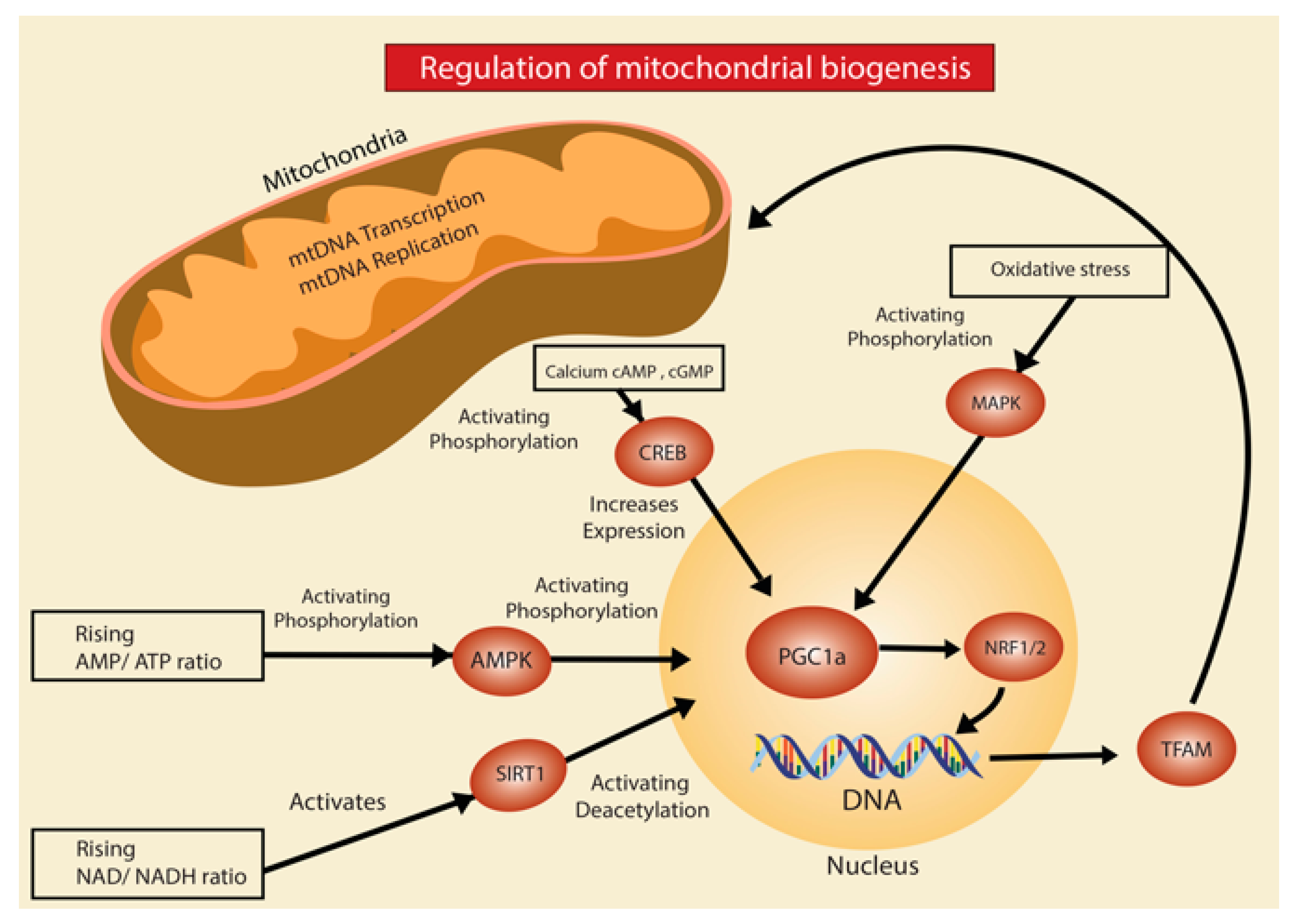
2.1. Mitochondrial Biogenesis
2.2. Mitochondrial Dynamics
2.3. Mitophagy
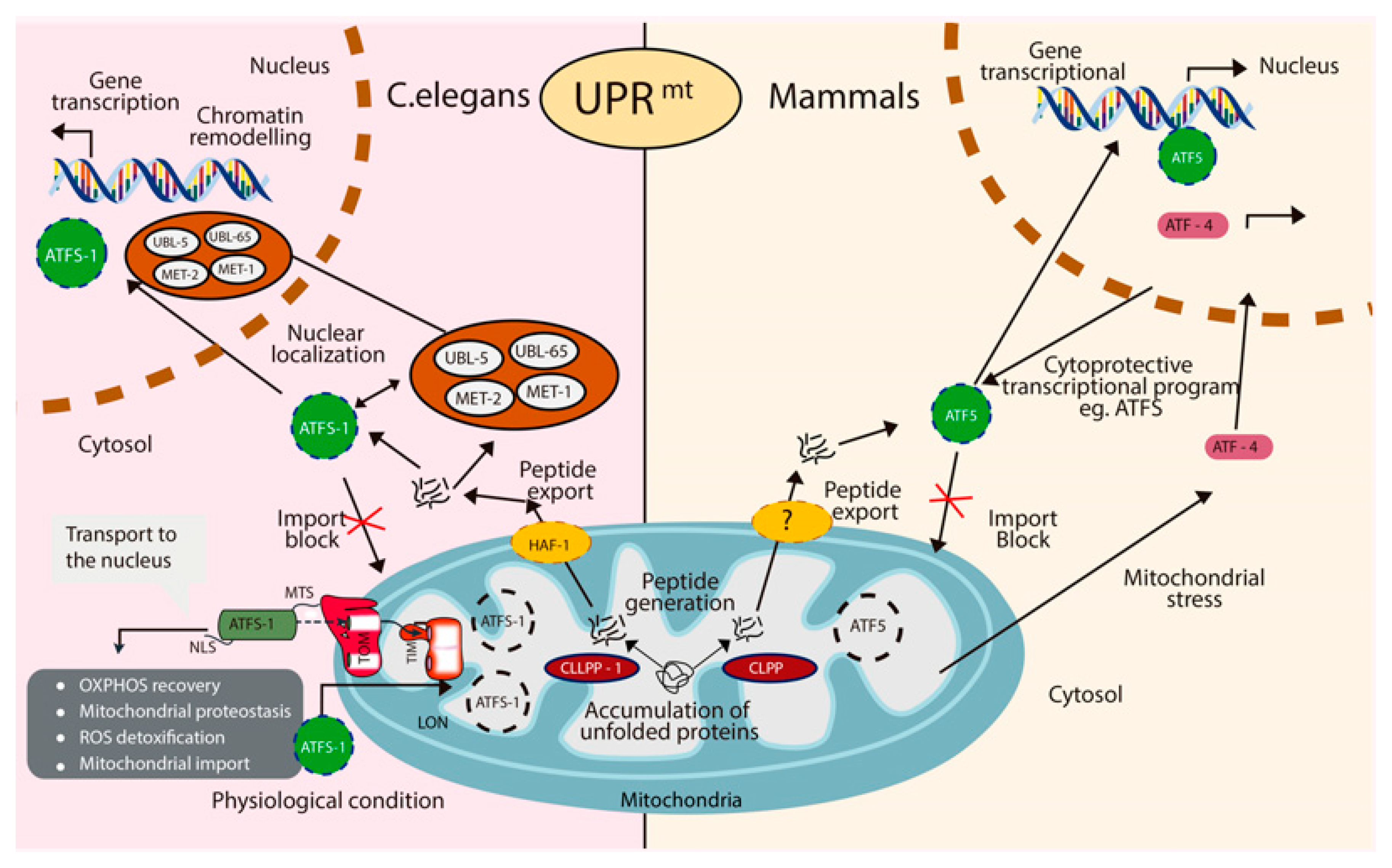
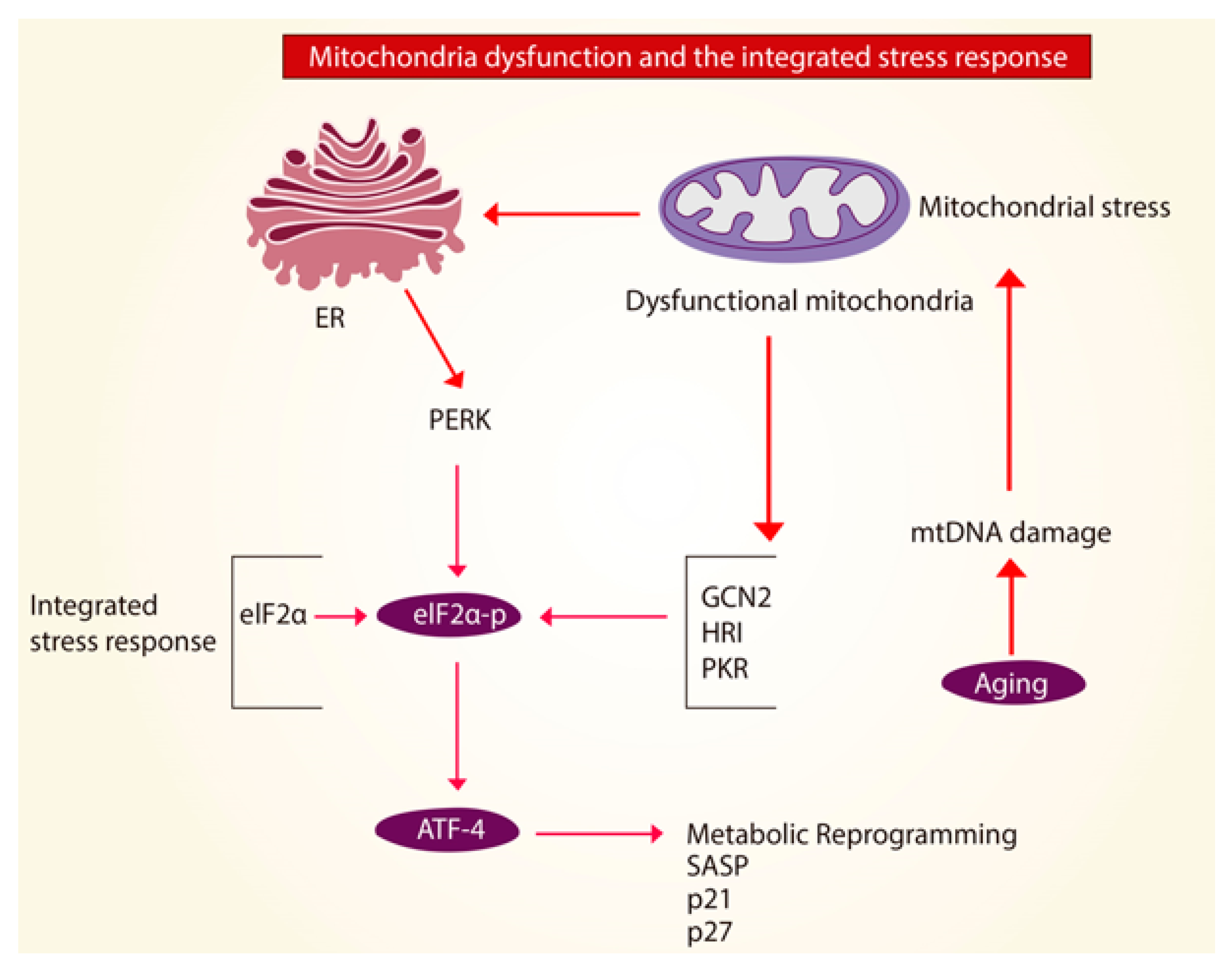
2.4. Mitochondrial Unfolded Protein Response (UPRmt)
2.5. Mitochondrial Quality Control as Therapeutic Target in IPF
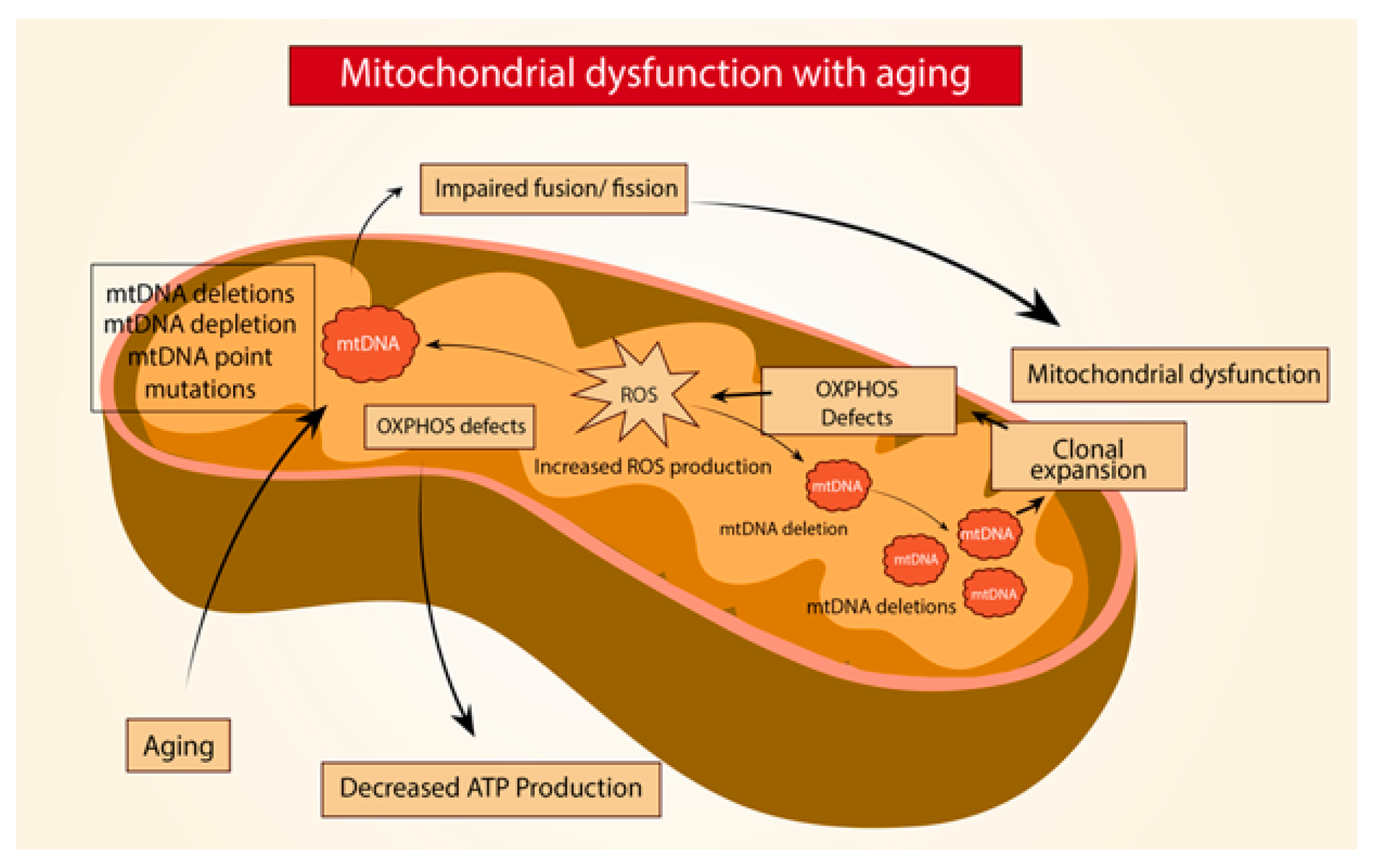
3. Mitochondria in Age-Related Lung Fibrosis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J. Mechanistic links between aging and lung fibrosis. Biogerontology 2013, 14, 609–615. [Google Scholar] [CrossRef]
- Focusing on mitochondrial form and function. Nat. Cell Biol. 2018, 20, 735. [CrossRef] [PubMed]
- Melber, A.; Haynes, C.M. UPRmt regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Shutt, T.E.; McBride, H.M. Staying cool in difficult times: Mitochondrial dynamics, quality control and the stress response. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 417–424. [Google Scholar] [CrossRef]
- Irrcher, I.; Adhihetty, P.J.; Sheehan, T.; Joseph, A.-M.; Hood, D.A. PPARγ coactivator-1α expression during thyroid hormone-and contractile activity-induced mitochondrial adaptations. Am. J. Physiol.-Cell Physiol. 2003, 284, C1669–C1677. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774. [Google Scholar] [CrossRef]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Hannah, J.Y.; Liu, Z.-X.; Dong, J.; Mustard, K.J.; Hawley, S.A. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef]
- Wang, G.; Song, Y.; Feng, W.; Liu, L.; Zhu, Y.; Xie, X.; Pan, Y.; Ke, R.; Li, S.; Li, F. Activation of AMPK attenuates LPS-induced acute lung injury by upregulation of PGC1α and SOD1. Exp. Ther. Med. 2016, 12, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Logsdon, N.J.; Miguel, V.; Benavides, G.A.; Zhang, J.; Carter, A.B.; Darley-Usmar, V.M.; Thannickal, V.J. NADPH oxidase 4 (Nox4) suppresses mitochondrial biogenesis and bioenergetics in lung fibroblasts via a nuclear factor erythroid-derived 2-like 2 (Nrf2)-dependent pathway. J. Biol. Chem. 2017, 292, 3029–3038. [Google Scholar] [CrossRef] [PubMed]
- Susse, S.; Scholz, C.J.; Burkle, A.; Wiesmüller, L. Poly (ADP-ribose) polymerase (PARP-1) and p53 independently function in regulating double-strand break repair in primate cells. Nucleic Acids Res. 2004, 32, 669–680. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Czapski, G.A.; Cieślik, M.; Wencel, P.L.; Wójtowicz, S.; Strosznajder, R.P.; Strosznajder, J.B. Inhibition of poly (ADP-ribose) polymerase-1 alters expression of mitochondria-related genes in PC12 cells: Relevance to mitochondrial homeostasis in neurodegenerative disorders. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 281–288. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.-P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.-P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef]
- Thoreen, C.C. The molecular basis of mTORC1-regulated translation. Biochem. Soc. Trans. 2017, 45, 213–221. [Google Scholar] [CrossRef]
- Aramburu, J.; Ortells, M.C.; Tejedor, S.; Buxade, M.; Lopez-Rodriguez, C. Transcriptional regulation of the stress response by mTOR. Sci. Signal. 2014, 7, re2. [Google Scholar] [CrossRef]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; De Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39. [Google Scholar] [CrossRef]
- Summer, R.; Shaghaghi, H.; Schriner, D.; Roque, W.; Sales, D.; Cuevas-Mora, K.; Desai, V.; Bhushan, A.; Ramirez, M.I.; Romero, F. Activation of the mTORC1/PGC-1 axis promotes mitochondrial biogenesis and induces cellular senescence in the lung epithelium. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 316, L1049–L1060. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 2011, 3, a007559. [Google Scholar] [CrossRef] [PubMed]
- Richter, U.; Ng, K.Y.; Suomi, F.; Marttinen, P.; Turunen, T.; Jackson, C.; Suomalainen, A.; Vihinen, H.; Jokitalo, E.; Nyman, T.A. Mitochondrial stress response triggered by defects in protein synthesis quality control. Life Sci. Alliance 2019, 2, e201800219. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; de Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, L.; Wu, S.; Xing, D. Drp1, Mff, Fis1, and MiD51 are coordinated to mediate mitochondrial fission during UV irradiation–induced apoptosis. FASEB J. 2015, 30, 466–476. [Google Scholar] [CrossRef]
- Cherok, E.; Xu, S.; Li, S.; Das, S.; Meltzer, W.A.; Zalzman, M.; Wang, C.; Karbowski, M. Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5–dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol. Biol. Cell 2017, 28, 396–410. [Google Scholar] [CrossRef]
- Escobar-Henriques, M.; Joaquim, M. Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases. Front. Physiol. 2019, 10, 517. [Google Scholar] [CrossRef]
- Cilenti, L.; Ambivero, C.T.; Ward, N.; Alnemri, E.S.; Germain, D.; Zervos, A.S. Inactivation of Omi/HtrA2 protease leads to the deregulation of mitochondrial Mulan E3 ubiquitin ligase and increased mitophagy. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1295–1307. [Google Scholar] [CrossRef]
- Radke, S.; Chander, H.; Schäfer, P.; Meiss, G.; Krüger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Tsitoura, E.; Vasarmidi, E.; Bibaki, E.; Trachalaki, A.; Koutoulaki, C.; Papastratigakis, G.; Papadogiorgaki, S.; Chalepakis, G.; Tzanakis, N.; Antoniou, K.M. Accumulation of damaged mitochondria in alveolar macrophages with reduced OXPHOS related gene expression in IPF. Respir. Res. 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.; McQuibban, G.A. The mitochondrial rhomboid protease: Its rise from obscurity to the pinnacle of disease-relevant genes. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2916–2925. [Google Scholar] [CrossRef]
- Saita, S.; Tatsuta, T.; Lampe, P.A.; König, T.; Ohba, Y.; Langer, T. PARL partitions the lipid transfer protein STARD7 between the cytosol and mitochondria. EMBO J. 2018, 37, e97909. [Google Scholar] [CrossRef]
- Dhingra, A.; Alexander, D.; Reyes-Reveles, J.; Sharp, R.; Boesze-Battaglia, K. Microtubule-associated protein 1 light chain 3 (LC3) isoforms in RPE and Retina. In Retinal Degenerative Diseases; Springer: Cham, Switzerland, 2018; pp. 609–616. [Google Scholar]
- Roca-Agujetas, V.; de Dios, C.; Lestón, L.; Marí, M.; Morales, A.; Colell, A. Recent Insights into the Mitochondrial Role in Autophagy and Its Regulation by Oxidative Stress. Oxidative Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Henriques, M.; Langer, T. Dynamic survey of mitochondria by ubiquitin. EMBO Rep. 2014, 15, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.H.; Nair, V.R.; Scharn, C.R.; Xavier, R.J.; Torrealba, J.R.; Shiloh, M.U.; Levine, B. The ubiquitin ligase Smurf1 functions in selective autophagy of Mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe 2017, 21, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jia, Y.; Zhang, Y.; Ma, F.; Zhu, Y.; Hong, X.; Zhou, Q.; He, R.; Zhang, H.; Jin, J. Ubiquitination of UVRAG by SMURF1 promotes autophagosome maturation and inhibits hepatocellular carcinoma growth. Autophagy 2019, 15, 1130–1149. [Google Scholar] [CrossRef] [PubMed]
- Fei, C.; He, X.; Xie, S.; Miao, H.; Zhou, Z.; Li, L. Smurf1-mediated axin ubiquitination requires Smurf1 C2 domain and is cell cycle-dependent. J. Biol. Chem. 2014, 289, 14170–14177. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Zank, D.; Buendia-Roldán, I.; Fiedler, K.; Mays, B.G.; Alvarez, D.; Sembrat, J.; Kimball, B.; Bullock, J.K.; Martin, J.L. PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses. PLoS ONE 2019, 14, e0218003. [Google Scholar] [CrossRef]
- Birch, J.; Barnes, P.J.; Passos, J.F. Mitochondria, telomeres and cell senescence: Implications for lung ageing and disease. Pharmacol. Ther. 2018, 183, 34–49. [Google Scholar] [CrossRef]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; St. Croix, C.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF 3 represses PINK 1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17, e12720. [Google Scholar] [CrossRef]
- Aggarwal, S.; Mannam, P.; Zhang, J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 311, L433–L452. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ze’ev, A.R. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Investig. 2018, 128, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Romero, F.; Summer, R. Protein folding and the challenges of maintaining endoplasmic reticulum proteostasis in idiopathic pulmonary fibrosis. Ann. Am. Thorac. Soc. 2017, 14, S410–S413. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Araya, J.; Minagawa, S.; Hara, H.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Tsubouchi, K.; Kurita, Y. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J. Immunol. 2016, 197, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399. [Google Scholar] [CrossRef]
- Semren, N.; Welk, V.; Korfei, M.; Keller, I.E.; Fernandez, I.E.; Adler, H.; Günther, A.; Eickelberg, O.; Meiners, S. Regulation of 26S proteasome activity in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1089–1101. [Google Scholar] [CrossRef]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria retrograde signaling and the UPRmt: Where are we in mammals? Int. J. Mol. Sci. 2015, 16, 18224–18251. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.K.; Li, C.; Ahmad, A.; Abrams, O.; Gorbatyuk, M.S.; Harrod, K.S.; Wek, R.C.; Afaq, F.; Athar, M. ATF4 regulates arsenic trioxide-mediated NADPH oxidase, ER-mitochondrial crosstalk and apoptosis. Arch. Biochem. Biophys. 2016, 609, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Münch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 1–9. [Google Scholar] [CrossRef]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef]
- Ahn, B.-H.; Kim, H.-S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.-X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef]
- Li, Y.; Ma, Y.; Song, L.; Yu, L.; Zhang, L.; Zhang, Y.; Xing, Y.; Yin, Y.; Ma, H. SIRT3 deficiency exacerbates p53/Parkin-mediated mitophagy inhibition and promotes mitochondrial dysfunction: Implication for aged hearts. Int. J. Mol. Med. 2018, 41, 3517–3526. [Google Scholar] [CrossRef]
- Jablonski, R.P.; Kim, S.-J.; Cheresh, P.; Williams, D.B.; Morales-Nebreda, L.; Cheng, Y.; Yeldandi, A.; Bhorade, S.; Pardo, A.; Selman, M. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J. 2017, 31, 2520–2532. [Google Scholar] [CrossRef]
- van’t Wout, E.F.; Hiemstra, P.S.; Marciniak, S.J. The integrated stress response in lung disease. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1005–1009. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.L.; LeBon, L.; Basso, A.M.; Kohlhaas, K.L.; Nikkel, A.L.; Robb, H.M.; Donnelly-Roberts, D.L.; Prakash, J.; Swensen, A.M.; Rubinstein, N.D. eIF2B activator prevents neurological defects caused by a chronic integrated stress response. Elife 2019, 8, e42940. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-S.; Liu, C.-C.; Lin, J.-H.; Hsu, T.-W.; Hsu, J.-W.; Su, K.; Hung, S.-C. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci. Rep. 2017, 7, 14272. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Ebert, S.M.; Dyle, M.C. Role of ATF4 in skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.M.; Radde, B.N.; Son, J.; Mehta, F.F.; Chung, S.-H.; Klinge, C.M. Estradiol and tamoxifen regulate NRF-1 and mitochondrial function in mouse mammary gland and uterus. J. Mol. Endocrinol. 2013, 51, 233. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Montopoli, M.; Perli, E.; Orlandi, M.; Fantin, M.; Ross-Cisneros, F.N.; Caparrotta, L.; Martinuzzi, A.; Ragazzi, E.; Ghelli, A. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain 2010, 134, 220–234. [Google Scholar] [CrossRef]
- Sbert-Roig, M.; Bauzá-Thorbrügge, M.; Galmés-Pascual, B.M.; Capllonch-Amer, G.; García-Palmer, F.J.; Lladó, I.; Proenza, A.M.; Gianotti, M. GPER mediates the effects of 17β-estradiol in cardiac mitochondrial biogenesis and function. Mol. Cell. Endocrinol. 2016, 420, 116–124. [Google Scholar] [CrossRef]
- Smith, L.C.; Moreno, S.; Robertson, L.; Robinson, S.; Gant, K.; Bryant, A.J.; Sabo-Attwood, T. Transforming growth factor beta1 targets estrogen receptor signaling in bronchial epithelial cells. Respir. Res. 2018, 19, 160. [Google Scholar] [CrossRef]
- Ito, I.; Hanyu, A.; Wayama, M.; Goto, N.; Katsuno, Y.; Kawasaki, S.; Nakajima, Y.; Kajiro, M.; Komatsu, Y.; Fujimura, A. Estrogen inhibits transforming growth factor β signaling by promoting Smad2/3 degradation. J. Biol. Chem. 2010, 285, 14747–14755. [Google Scholar] [CrossRef]
- Reiners, J., Jr.; Caruso, J.; Mathieu, P.; Chelladurai, B.; Yin, X.-M.; Kessel, D. Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves Bid cleavage. Cell Death Differ. 2002, 9, 934. [Google Scholar] [CrossRef]
- Yel, L.; Brown, L.E.; Su, K.; Gollapudi, S.; Gupta, S. Thimerosal induces neuronal cell apoptosis by causing cytochrome c and apoptosis-inducing factor release from mitochondria. Int. J. Mol. Med. 2005, 16, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Bouchier-Hayes, L.; Oberst, A.; McStay, G.P.; Connell, S.; Tait, S.W.; Dillon, C.P.; Flanagan, J.M.; Beere, H.M.; Green, D.R. Characterization of cytoplasmic caspase-2 activation by induced proximity. Mol. Cell 2009, 35, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.S.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M.; Group, P.S. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Hertz, N.T.; Berthet, A.; Sos, M.L.; Thorn, K.S.; Burlingame, A.L.; Nakamura, K.; Shokat, K.M. A neo-substrate that amplifies catalytic activity of Parkinson’s-disease-related kinase PINK1. Cell 2013, 154, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Akamata, K.; Wei, J.; Bhattacharyya, M.; Cheresh, P.; Bonner, M.Y.; Arbiser, J.L.; Raparia, K.; Gupta, M.P.; Kamp, D.W.; Varga, J. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget 2016, 7, 69321. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.R.; Martinez, A.; Lane, J.D.; Mayor, U.; Clague, M.J.; Urbé, S. USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep. 2015, 16, 618–627. [Google Scholar] [CrossRef]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160. [Google Scholar] [CrossRef]
- Haas, R.H. Mitochondrial Dysfunction in Aging and Diseases of Aging; Multidisciplinary Digital Publishing Institute: Basel, Switzerland, 2019. [Google Scholar]
- Bota, D.A.; Davies, K.J. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 2016, 100, 188–198. [Google Scholar] [CrossRef]
- Ngo, J.K.; Pomatto, L.C.; Davies, K.J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Yin, P.-H.; Chi, C.-W.; Wei, Y.-H. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J. Biomed. Sci. 2002, 9, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Vasileiou, P.V.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.-G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [PubMed]
- Deepa, S.S.; Bhaskaran, S.; Ranjit, R.; Qaisar, R.; Nair, B.C.; Liu, Y.; Walsh, M.E.; Fok, W.C.; Van Remmen, H. Down-regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic. Biol. Med. 2016, 91, 281–292. [Google Scholar] [CrossRef]
- Wu, G.; Xiong, Q.; Wei, X.; Wang, Y.; Hu, X.; He, G.; Liu, L.; Lai, Q.; Dai, Z.; Anushesh, D. Mitochondrial unfolded protein response gene CLPP changes mitochondrial dynamics and affects mitochondrial function. PeerJ 2019, 7, e7209. [Google Scholar] [CrossRef]
- Wang, T.; Babayev, E.; Jiang, Z.; Li, G.; Zhang, M.; Esencan, E.; Horvath, T.; Seli, E. Mitochondrial unfolded protein response gene Clpp is required to maintain ovarian follicular reserve during aging, for oocyte competence, and development of pre-implantation embryos. Aging Cell 2018, 17, e12784. [Google Scholar] [CrossRef]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivelä, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 2017, 26, 419–428. [Google Scholar] [CrossRef]
- Sears, T.K.; Angelastro, J.M. The transcription factor ATF5: Role in cellular differentiation, stress responses, and cancer. Oncotarget 2017, 8, 84595. [Google Scholar] [CrossRef]
- Sheng, Z.; Ma, L.; Sun, J.E.; Zhu, L.J.; Green, M.R. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood 2011, 118, 2840–2848. [Google Scholar] [CrossRef]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481. [Google Scholar] [CrossRef] [PubMed]
- Wortel, I.M.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving stress: Modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Fusakio, M.E.; Willy, J.A.; Wang, Y.; Mirek, E.T.; Al Baghdadi, R.J.; Adams, C.M.; Anthony, T.G.; Wek, R.C. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell 2016, 27, 1536–1551. [Google Scholar] [CrossRef] [PubMed]
- Mahavadi, P.; Korfei, M.; Henneke, I.; Liebisch, G.; Schmitz, G.; Gochuico, B.R.; Markart, P.; Bellusci, S.; Seeger, W.; Ruppert, C. Epithelial stress and apoptosis underlie Hermansky-Pudlak syndrome–associated interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2010, 182, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Mahavadi, P.; Henneke, I.; Ruppert, C.; Knudsen, L.; Venkatesan, S.; Liebisch, G.; Chambers, R.C.; Ochs, M.; Schmitz, G.; Vancheri, C. Altered surfactant homeostasis and alveolar epithelial cell stress in amiodarone-induced lung fibrosis. Toxicol. Sci. 2014, 142, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, Y.; Zhang, Z.; He, L.; Zhu, J.; Zhang, M.; He, X.; Cheng, Z.; Ao, Q.; Cao, Y. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol. Ther. 2016, 24, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Mazzacurati, L.; Zhu, X.; Tong, T.; Song, Y.; Shujuan, S.; Petrik, K.L.; Rajasekaran, B.; Wu, M.; Zhan, Q. Gadd45a contributes to p53 stabilization in response to DNA damage. Oncogene 2003, 22, 8536. [Google Scholar] [CrossRef]
- Mishra, A.; Doyle, N.A.; Martin, W.J. Bleomycin-mediated pulmonary toxicity: Evidence for a p53-mediated response. Am. J. Respir. Cell Mol. Biol. 2000, 22, 543–549. [Google Scholar] [CrossRef]
- Akram, K.M.; Lomas, N.J.; Forsyth, N.R.; Spiteri, M.A. Alveolar epithelial cells in idiopathic pulmonary fibrosis display upregulation of TRAIL, DR4 and DR5 expression with simultaneous preferential over-expression of pro-apoptotic marker p53. Int. J. Clin. Exp. Pathol. 2014, 7, 552. [Google Scholar]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roque, W.; Cuevas-Mora, K.; Romero, F. Mitochondrial Quality Control in Age-Related Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 643. https://doi.org/10.3390/ijms21020643
Roque W, Cuevas-Mora K, Romero F. Mitochondrial Quality Control in Age-Related Pulmonary Fibrosis. International Journal of Molecular Sciences. 2020; 21(2):643. https://doi.org/10.3390/ijms21020643
Chicago/Turabian StyleRoque, Willy, Karina Cuevas-Mora, and Freddy Romero. 2020. "Mitochondrial Quality Control in Age-Related Pulmonary Fibrosis" International Journal of Molecular Sciences 21, no. 2: 643. https://doi.org/10.3390/ijms21020643
APA StyleRoque, W., Cuevas-Mora, K., & Romero, F. (2020). Mitochondrial Quality Control in Age-Related Pulmonary Fibrosis. International Journal of Molecular Sciences, 21(2), 643. https://doi.org/10.3390/ijms21020643