SLMP53-1 Inhibits Tumor Cell Growth through Regulation of Glucose Metabolism and Angiogenesis in a P53-Dependent Manner

 , , , , , ,
, , , , , ,  and
and 
Abstract
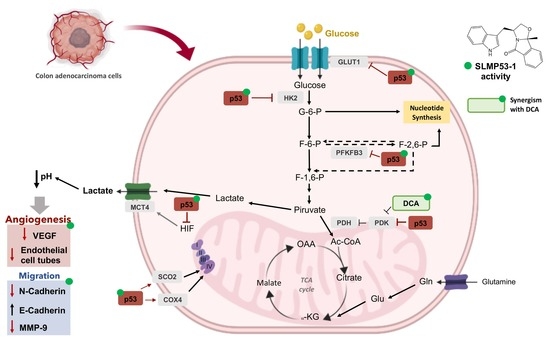
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
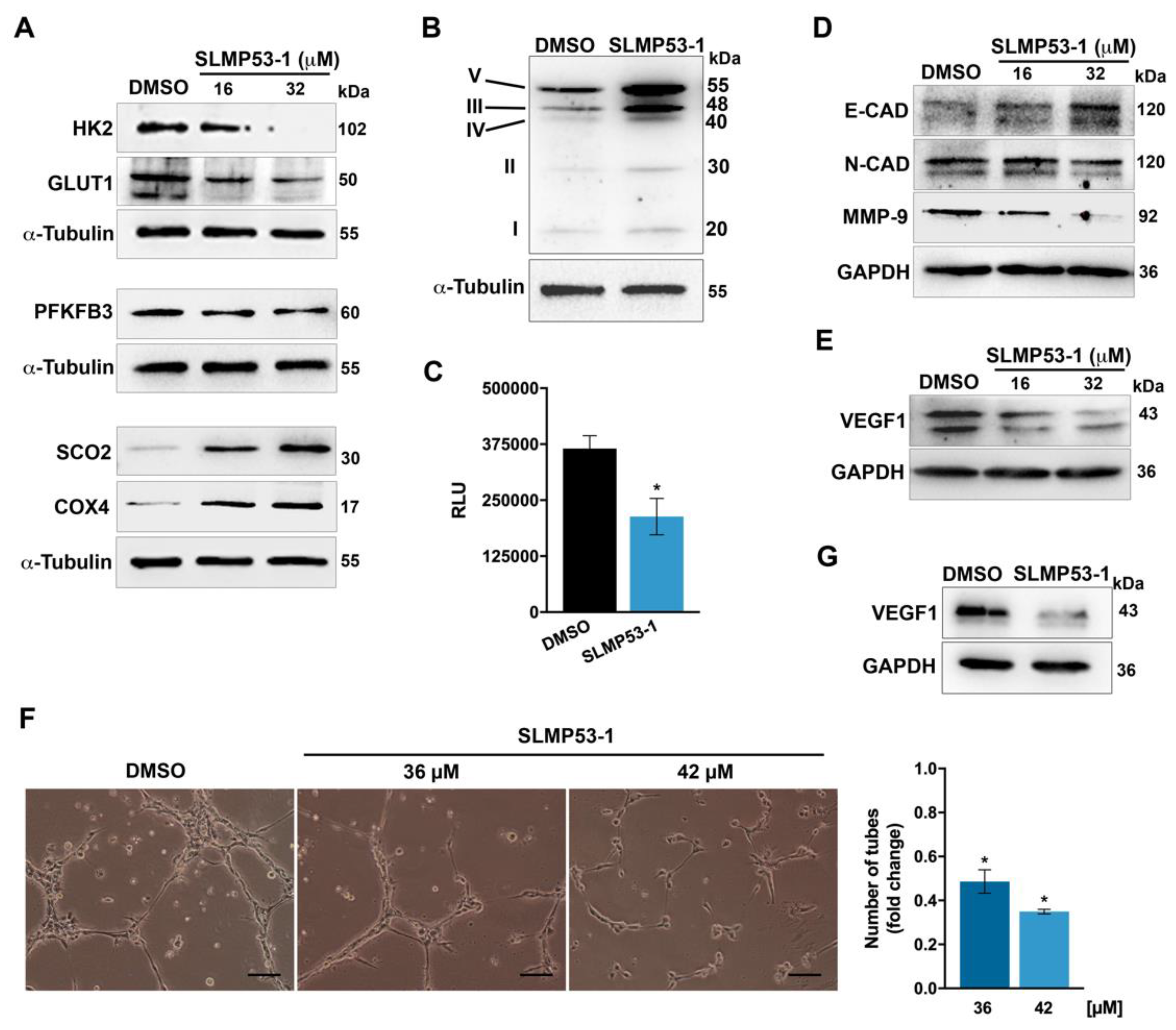
2.1. SLMP53-1 Regulates the Warburg Effect and Angiogenesis in Cancer Cells, with Interference in ECM Remodeling and EMT Events
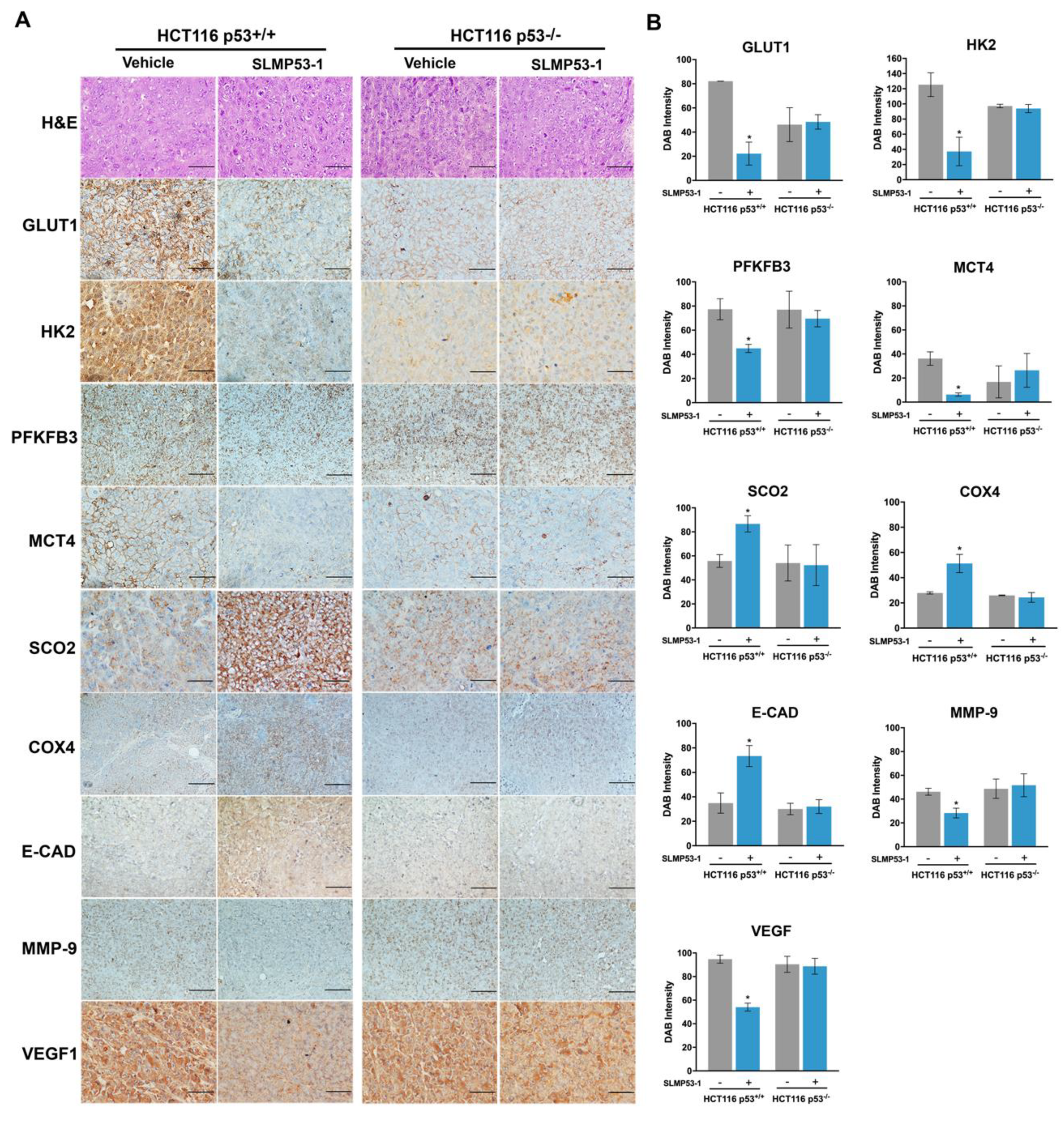
2.2. SLMP53-1 Regulates the Warburg Effect and Angiogenesis, Interfering with ECM Remodeling and EMT Events, in a p53-Dependent Manner, in Tumor Tissues of Xenograft Mouse Models
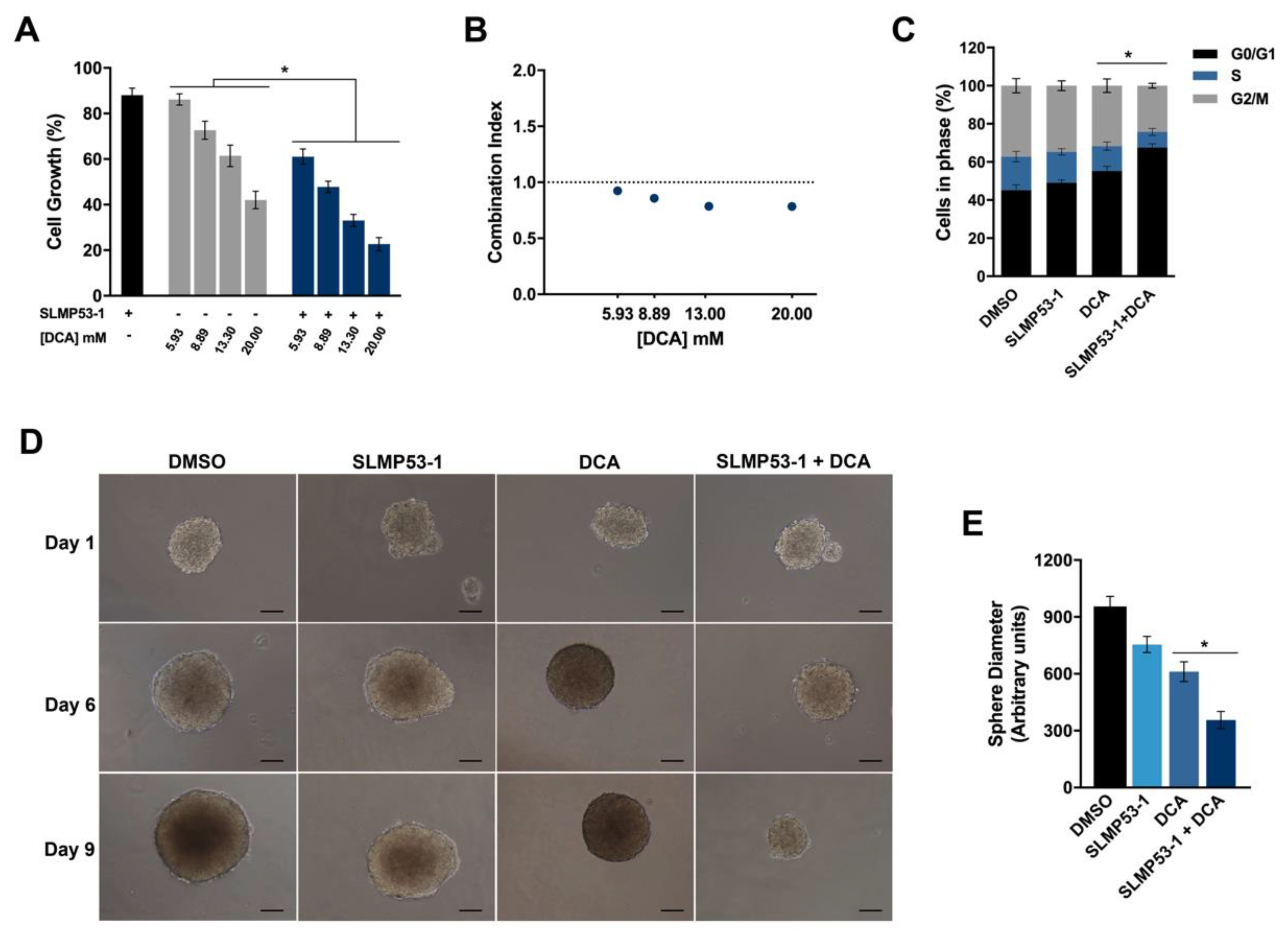
2.3. SLMP53-1 Synergizes with Dichloroacetic Acid (DCA), Enhancing Its Antitumor Efficacy in Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Compounds and Reagents
4.2. Human Cell Lines and Growth Conditions
4.3. Immunohistochemistry (IHC)
4.4. Western Blot
4.5. Measurement of Extracellular Lactate
4.6. Angiogenesis Assay
4.7. Generation of Colon Cancer Spheroids
4.8. Combination Therapy Assays
4.9. Cell Cycle Analysis
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Smallbone, K.; Gatenby, R.A.; Gillies, R.J.; Maini, P.K.; Gavaghan, D.J. Metabolic changes during carcinogenesis: Potential impact on invasiveness. J. Biol. 2007, 244, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Bianchini, F.; Calorini, L. Extracellular acidity, a “reappreciated” trait of tumor environment driving malignancy: Perspectives in diagnosis and therapy. Cancer Metastasis Rev. 2014, 33, 823–832. [Google Scholar] [CrossRef]
- Suzuki, A.; Maeda, T.; Baba, Y.; Shimamura, K.; Kato, Y. Acidic extracellular pH promotes epithelial mesenchymal transition in Lewis lung carcinoma model. Cancer Cell Int. 2014, 14, 129. [Google Scholar] [CrossRef]
- Shi, Q.; Le, X.; Wang, B.; Abbruzzese, J.L.; Xiong, Q.; He, Y.; Xie, K. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 2001, 20, 3751–3756. [Google Scholar] [CrossRef]
- Beckert, S.; Farrahi, F.; Aslam, R.S.; Scheuenstuhl, H.; Konigsrainer, A.; Hussain, M.Z.; Hunt, T.K. Lactate stimulates endothelial cell migration. Wound Repair Regen. 2006, 14, 321–324. [Google Scholar] [CrossRef]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Semenza, G.L.; Zhang, H.F. Hypoxia-inducible factor 1 and breast cancer metastasis. J. Zhejiang Univ. Sci. B 2015, 16, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Ruckenstuhl, C.; Buttner, S.; Carmona-Gutierrez, D.; Eisenberg, T.; Kroemer, G.; Sigrist, S.J.; Frohlich, K.U.; Madeo, F. The Warburg effect suppresses oxidative stress induced apoptosis in a yeast model for cancer. PLoS ONE 2009, 4, e4592. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Soares, J.; Saraiva, L. p53 and glucose metabolism: An orchestra to be directed in cancer therapy. Pharm. Res. 2018, 131, 75–86. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef]
- Franklin, D.A.; He, Y.; Leslie, P.L.; Tikunov, A.P.; Fenger, N.; Macdonald, J.M.; Zhang, Y. p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci. Rep. 2016, 6, 38067. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of AIF expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef]
- Simabuco, F.M.; Morale, M.G.; Pavan, I.C.B.; Morelli, A.P.; Silva, F.R.; Tamura, R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget 2018, 9, 23780–23823. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Raimundo, L.; Pereira, N.A.; Monteiro, A.; Gomes, S.; Bessa, C.; Pereira, C.; Queiroz, G.; Bisio, A.; Fernandes, J.; et al. Reactivation of wild-type and mutant p53 by tryptophanolderived oxazoloisoindolinone SLMP53-1, a novel anticancer small-molecule. Oncotarget 2016, 7, 4326–4343. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lago, C.U.; Sung, H.J.; Ma, W.; Wang, P.Y.; Hwang, P.M. p53, aerobic metabolism, and cancer. Antioxid Redox Signal. 2011, 15, 1739–1748. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Gomes, S.; Loureiro, J.B.; Soares, J.; Barcherini, V.; Monti, P.; Fronza, G.; Oliveira, C.; Domingues, L.; et al. SLMP53-1 interacts with wild-type and mutant p53 DNA-binding domain and reactivates multiple hotspot mutations. Biochim. Biophys. Acta Gen. Subj. 2019. [Google Scholar] [CrossRef]
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell Biol. 2003, 23, 8576–8585. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Madan, E.; Gogna, R.; Kuppusamy, P.; Bhatt, M.; Mahdi, A.A.; Pati, U. SCO2 induces p53-mediated apoptosis by Thr845 phosphorylation of ASK-1 and dissociation of the ASK-1-Trx complex. Mol. Cell Biol. 2013, 33, 1285–1302. [Google Scholar] [CrossRef]
- Goodwin, M.L.; Gladden, L.B.; Nijsten, M.W.; Jones, K.B. Lactate and cancer: Revisiting the warburg effect in an era of lactate shuttling. Front Nutr. 2014, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Goubran, H.A.; Kotb, R.R.; Stakiw, J.; Emara, M.E.; Burnouf, T. Regulation of tumor growth and metastasis: The role of tumor microenvironment. Cancer Growth Metastasis 2014, 7, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef]
- Obacz, J.; Pastorekova, S.; Vojtesek, B.; Hrstka, R. Cross-talk between HIF and p53 as mediators of molecular responses to physiological and genotoxic stresses. Mol. Cancer 2013, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.C.; Baltazar, F. Role of monocarboxylate transporters in human cancers: State of the art. J. Bioenerg. Biomembr. 2012, 44, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Baek, G.; Tse, Y.F.; Hu, Z.; Cox, D.; Buboltz, N.; McCue, P.; Yeo, C.J.; White, M.A.; DeBerardinis, R.J.; Knudsen, E.S.; et al. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014, 9, 2233–2249. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep. 2018, 25, 3047–3058.e3044. [Google Scholar] [CrossRef]
- Polanski, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef]
- Pal, S.; Datta, K.; Mukhopadhyay, D. Central role of p53 on regulation of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in mammary carcinoma. Cancer Res. 2001, 61, 6952–6957. [Google Scholar]
- Qin, G.; Kishore, R.; Dolan, C.M.; Silver, M.; Wecker, A.; Luedemann, C.N.; Thorne, T.; Hanley, A.; Curry, C.; Heyd, L.; et al. Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc. Natl. Acad. Sci. USA 2006, 103, 11015–11020. [Google Scholar] [CrossRef]
- Wu, M.; Zhou, J.; Cheng, M.; Boriboun, C.; Biyashev, D.; Wang, H.; Mackie, A.; Thorne, T.; Chou, J.; Wu, Y.; et al. E2F1 suppresses cardiac neovascularization by down-regulating VEGF and PlGF expression. Cardiovasc. Res. 2014, 104, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. The pharmacology of dichloroacetate. Metabolism 1989, 38, 1124–1144. [Google Scholar] [CrossRef]
- Knoechel, T.R.; Tucker, A.D.; Robinson, C.M.; Phillips, C.; Taylor, W.; Bungay, P.J.; Kasten, S.A.; Roche, T.E.; Brown, D.G. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry 2006, 45, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Liang, Y.; Hou, L.; Li, L.; Li, L.; Zhu, L.; Wang, Y.; Huang, X.; Hou, Y.; Zhu, D.; Zou, H.; et al. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149-3p/PDK2-mediated glucose metabolic pathway. Oncogene 2019. [Google Scholar] [CrossRef]
- Agnoletto, C.; Melloni, E.; Casciano, F.; Rigolin, G.M.; Rimondi, E.; Celeghini, C.; Brunelli, L.; Cuneo, A.; Secchiero, P.; Zauli, G. Sodium dichloroacetate exhibits anti-leukemic activity in B-chronic lymphocytic leukemia (B-CLL) and synergizes with the p53 activator Nutlin-3. Oncotarget 2014, 5, 4347–4360. [Google Scholar] [CrossRef][Green Version]
- Bessa, C.; Soares, J.; Raimundo, L.; Loureiro, J.B.; Gomes, C.; Reis, F.; Soares, M.L.; Santos, D.; Dureja, C.; Chaudhuri, S.R.; et al. Discovery of a small-molecule protein kinase Cdelta-selective activator with promising application in colon cancer therapy. Cell Death Dis. 2018, 9, 23. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos, H.; Calheiros, J.; Almeida, J.; Barcherini, V.; Santos, S.; Carvalho, A.T.P.; Santos, M.M.M.; Saraiva, L. SLMP53-1 Inhibits Tumor Cell Growth through Regulation of Glucose Metabolism and Angiogenesis in a P53-Dependent Manner. Int. J. Mol. Sci. 2020, 21, 596. https://doi.org/10.3390/ijms21020596
Ramos H, Calheiros J, Almeida J, Barcherini V, Santos S, Carvalho ATP, Santos MMM, Saraiva L. SLMP53-1 Inhibits Tumor Cell Growth through Regulation of Glucose Metabolism and Angiogenesis in a P53-Dependent Manner. International Journal of Molecular Sciences. 2020; 21(2):596. https://doi.org/10.3390/ijms21020596
Chicago/Turabian StyleRamos, Helena, Juliana Calheiros, Joana Almeida, Valentina Barcherini, Sónia Santos, Alexandra T. P. Carvalho, Maria M.M. Santos, and Lucília Saraiva. 2020. "SLMP53-1 Inhibits Tumor Cell Growth through Regulation of Glucose Metabolism and Angiogenesis in a P53-Dependent Manner" International Journal of Molecular Sciences 21, no. 2: 596. https://doi.org/10.3390/ijms21020596
APA StyleRamos, H., Calheiros, J., Almeida, J., Barcherini, V., Santos, S., Carvalho, A. T. P., Santos, M. M. M., & Saraiva, L. (2020). SLMP53-1 Inhibits Tumor Cell Growth through Regulation of Glucose Metabolism and Angiogenesis in a P53-Dependent Manner. International Journal of Molecular Sciences, 21(2), 596. https://doi.org/10.3390/ijms21020596