Assembly and Maintenance of Sarcomere Thin Filaments and Associated Diseases

Abstract

1. Introduction
2. Thin Filament Assembly in the Sarcomere
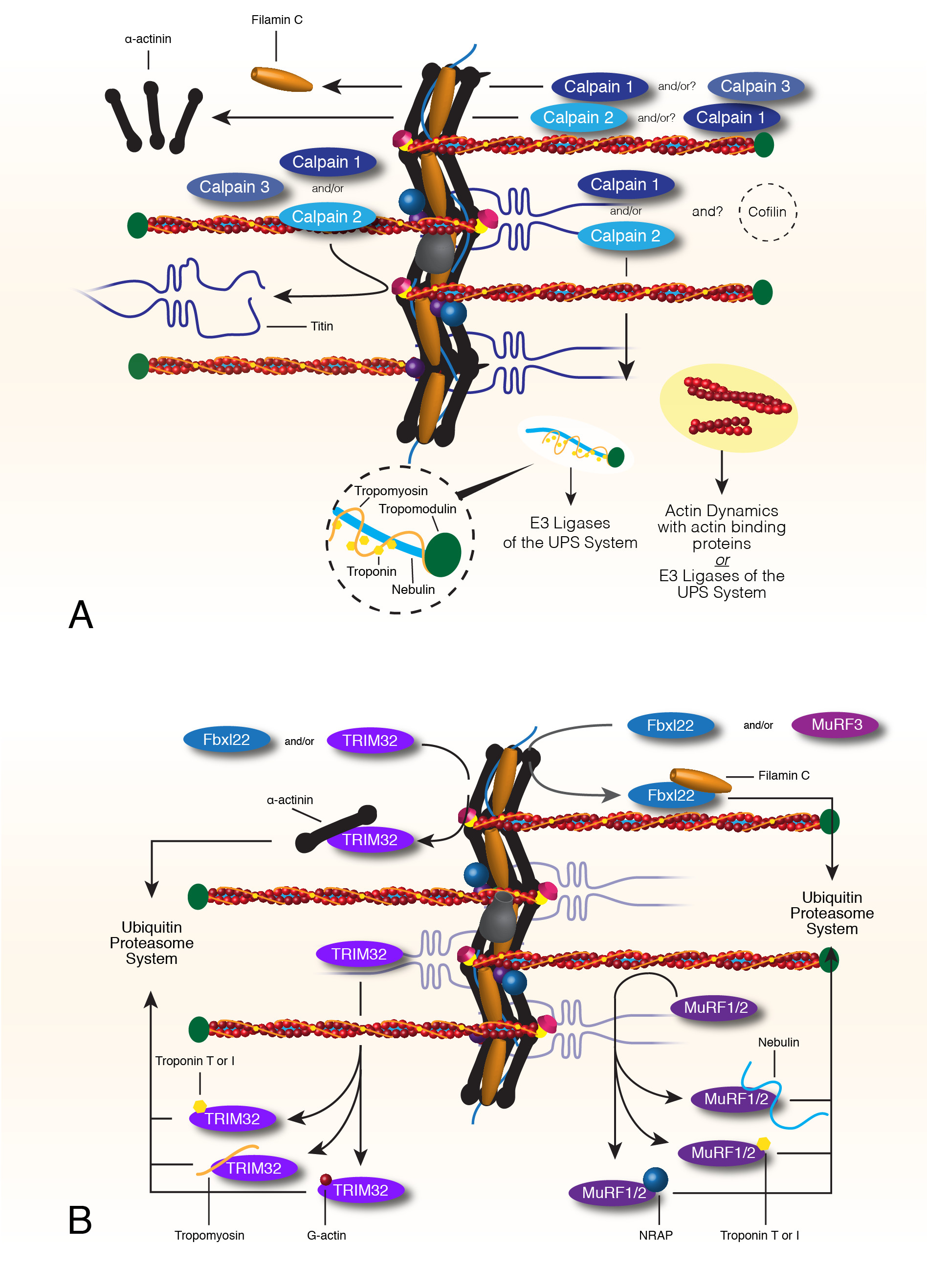
3. Sarcomere Thin Filament Maintenance
3.1. Thin Filament Maintenance and Turnover
3.2. Calpain Mediated Breakdown of Z-Disc and Thin Filaments
3.3. Turnover of Thin Filament Components via E3 Ligases and the Ubiquitin Proteasome System
3.4. Autophagy-Dependent Mechanisms for Thin Filament Maintenance
3.5. Dynamic Actin Turnover
4. Diseases of Components of Striated Muscle Thin Filaments
4.1. Mutations That Interfere with Proper Thin Filament Assembly
4.2. Mutations that Interfere with Thin Filament Maintenance and Turnover
5. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Katzemich, A.; Liao, K.A.; Czerniecki, S.; Schöck, F. Alp/Enigma Family Proteins Cooperate in Z-Disc Formation and Myofibril Assembly. PLoS Genet. 2013, 9, e1003342. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Ruiz-Lozano, P.; Martone, M.E.; Chen, J. Cypher, a striated muscle-restricted PDZ and LIM domain-containing protein, binds to α-actinin-2 and protein kinase C. J. Biol. Chem. 1999, 274, 19807–19813. [Google Scholar] [CrossRef]
- Passier, R.; Richardson, J.A.; Olson, E.N. Oracle, a novel PDZ-LIM domain protein expressed in heart and skeletal muscle. Mech. Dev. 2000, 92, 277–284. [Google Scholar] [CrossRef]
- Jani, K.; Schöck, F. Zasp is required for the assembly of functional integrin adhesion sites. J. Cell Biol. 2007, 179, 1583–1597. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.L.; Herrera, A.H.; Horowits, R. Targeting and functional role of N-RAP, a nebulin-related LIM protein, during myofibril assembly in cultured chick cardiomyocytes. J. Cell Sci. 2001, 114, 4229–4238. [Google Scholar]
- Carroll, S.; Lu, S.; Herrera, A.H.; Horowits, R. N-RAP scaffolds I-Z-I assembly during myofibrillogenesis in cultured chick cardiomyocytes. J. Cell Sci. 2004, 117, 105–114. [Google Scholar] [CrossRef]
- Manisastry, S.M.; Zaal, K.J.M.; Horowits, R. Myofibril assembly visualized by imaging N-RAP, alpha-actinin, and actin in living cardiomyocytes. Exp. Cell Res. 2009, 315, 2126–2139. [Google Scholar] [CrossRef]
- Van Der Ven, P.F.M.; Obermann, W.M.J.; Lemke, B.; Gautel, M.; Weber, K.; Fürst, D.O. Characterization of muscle filamin isoforms suggests a possible role of γ-Filamin/ABP-L in sarcomeric Z-disc formation. Cell Motil. Cytoskelet. 2000, 45, 149–162. [Google Scholar] [CrossRef]
- González-Morales, N.; Holenka, T.K.; Schöck, F. Filamin actin-binding and titin-binding fulfill distinct functions in Z-disc cohesion. PLoS Genet. 2017, 13, e1006880. [Google Scholar] [CrossRef]
- Linnemann, A.; Vakeel, P.; Bezerra, E.; Orfanos, Z.; Djinović-Carugo, K.; Van Der Ven, P.F.M.; Kirfel, G.; Fürst, D.O. Myopodin is an F-actin bundling protein with multiple independent actin-binding regions. J. Muscle Res. Cell Motil. 2013, 34, 61–69. [Google Scholar] [CrossRef]
- Wang, J.; Shaner, N.; Mittal, B.; Zhou, Q.; Chen, J.; Sanger, J.M.; Sanger, J.W. Dynamics of Z-band based proteins in developing skeletal muscle cells. Cell Motil. Cytoskelet. 2005, 61, 34–48. [Google Scholar] [CrossRef]
- Faulkner, G.; Pallavicini, A.; Comelli, A.; Salamon, M.; Bortoletto, G.; Ievolella, C.; Trevisan, S.; Kojić, S.; Vecchia, F.D.; Laveder, P.; et al. FATZ, a filamin-, actinin-, and telethonin-binding protein of the Z-disc of skeletal muscle. J. Biol. Chem. 2000, 275, 41234–41242. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Richardson, J.A.; Olson, E.N. Calsarcins, a novel family of sarcomeric calcineurin-binding proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 14632–14637. [Google Scholar] [CrossRef]
- Chu, M.; Gregorio, C.C.; Pappas, C.T. Nebulin, a multi-functional giant. J. Exp. Biol. 2016, 219, 146–152. [Google Scholar] [CrossRef]
- Moncman, C.L.; Wang, K. Assembly of nebulin into the sarcomeres of avian skeletal muscle. Cell Motil. Cytoskelet. 1996, 34, 167–184. [Google Scholar] [CrossRef]
- Labeit, S.; Kolmerer, B. The complete primary structure of human nebulinand its correlation to muscle structure. J. Mol. Biol. 1995, 248, 308–315. [Google Scholar] [CrossRef]
- Kazmierski, S.T.; Antin, P.B.; Witt, C.C.; Huebner, N.; McElhinny, A.S.; Labeit, S.; Gregorio, C.C. The complete mouse nebulin gene sequence and the identification of cardiac nebulin. J. Mol. Biol. 2003, 328, 835–846. [Google Scholar] [CrossRef]
- Millevoi, S.; Trombitas, K.; Kolmerer, B.; Kostin, S.; Schaper, J.; Pelin, K.; Granzier, H.; Labeit, S. Characterization of nebulette and nebulin and emerging concepts of their roles for vertebrate Z-discs. J. Mol. Biol. 1998, 282, 111–123. [Google Scholar] [CrossRef]
- Moncman, C.L.; Wang, K. Targeted disruption of nebulette protein expression alters cardiac myofibril assembly and function. Exp. Cell Res. 2002, 273, 204–218. [Google Scholar] [CrossRef]
- Esham, M.; Bryan, K.; Milnes, J.; Holmes, W.B.; Moncman, C.L. Expression of nebulette during early cardiac development. Cell Motil. Cytoskelet. 2007, 64, 258–273. [Google Scholar] [CrossRef]
- Moncman, C.L.; Wang, K. Nebulette: A 107 kD nebulin-like protein in cardiac muscle. Cell Motil. Cytoskelet. 1995, 32, 205–225. [Google Scholar] [CrossRef]
- Bang, M.L.; Chen, J. Roles of nebulin family members in the heart. Circ. J. 2015, 79, 2081–2087. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, C.C.; Connelly, P.S.; Daniels, M.P.; Horowits, R. Krp1 (Sarcosin) promotes lateral fusion of myofibril assembly intermediates in cultured mouse cardiomyocytes. Exp. Cell Res. 2008, 314, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Jirka, C.; Pak, J.H.; Grosgogeat, C.A.; Marchetii, M.M.; Gupta, V.A. Dysregulation of NRAP degradation by KLHL41 contributes to pathophysiology in nemaline myopathy. Hum. Mol. Genet. 2019, 28, 2549–2560. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Martinez, A.; Cenik, B.K.; Bezprozvannaya, S.; Chen, B.; Bassel-Duby, R.; Liu, N.; Olson, E.N. KLHL41 stabilizes skeletal muscle sarcomeres by nonproteolytic ubiquitination. Elife 2017, 6, e26439. [Google Scholar] [CrossRef]
- Garg, A.; O’Rourke, J.; Long, C.; Doering, J.; Ravenscroft, G.; Bezprozvannaya, S.; Nelson, B.R.; Beetz, N.; Li, L.; Chen, S.; et al. KLHL40 deficiency destabilizes thin filament proteins and promotes Nemaline myopathy. J. Clin. Investig. 2014, 124, 3529–3539. [Google Scholar] [CrossRef]
- Ottenheijm, C.A.C.; Hooijman, P.; DeChene, E.T.; Stienen, G.J.; Beggs, A.H.; Granzier, H. Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2). J. Struct. Biol. 2010, 170, 334–343. [Google Scholar] [CrossRef]
- Witt, C.C.; Burkart, C.; Labeit, D.; McNabb, M.; Wu, Y.; Granzier, H.; Labeit, S. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO J. 2006, 25, 3843–3855. [Google Scholar] [CrossRef]
- Tskhovrebova, L.; Trinick, J. Titin and Nebulin in thick and thin filament length regulation. Subcell. Biochem. 2017, 285–318. [Google Scholar] [CrossRef]
- Bang, M.L.; Li, X.; Littlefield, R.; Bremner, S.; Thor, A.; Knowlton, K.U.; Lieber, R.L.; Chen, J. Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J. Cell Biol. 2006, 173, 905–916. [Google Scholar] [CrossRef]
- Pappas, C.T.; Krieg, P.A.; Gregorio, C.C. Nebulin regulates actin filament lengths by a stabilization mechanism. J. Cell Biol. 2010, 189, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Labeit, S.; Gibson, T.; Lakey, A.; Leonard, K.; Zeviani, M.; Knight, P.; Wardale, J.; Trinick, J. Evidence that nebulin is a protein-ruler in muscle thin filaments. FEBS Lett. 1991, 282, 313–316. [Google Scholar] [CrossRef]
- Castillo, A.; Nowak, R.; Littlefield, K.P.; Fowler, V.M.; Littlefield, R.S. A nebulin ruler does not dictate thin filament lengths. Biophys. J. 2009, 96, 1856–1865. [Google Scholar] [CrossRef]
- Gokhin, D.S.; Fowler, V.M. A two-segment model for thin filament architecture in skeletal muscle. Nat. Rev. Mol. Cell Biol. 2013, 14, 113. [Google Scholar] [CrossRef]
- Salmikangas, P.; van der Ven, P.F.M.; Lalowski, M.; Taivainen, A.; Zhao, F.; Suila, H.; Schröder, R.; Lappalainen, P.; Fürst, D.O.; Carpén, O. Myotilin, the limb-girdle muscular dystrophy 1A (LGMD1A) protein, cross-links actin filaments and controls sarcomere assembly. Hum. Mol. Genet. 2003, 12, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Sanger, J.W.; Wang, J.; Holloway, B.; Du, A.; Sanger, J.M. Myofibrillogenesis in skeletal muscle cells in zebrafish. Cell Motil. Cytoskelet. 2009, 66, 556–566. [Google Scholar] [CrossRef]
- Salmikangas, P.; Mykkänen, O.M.; Grönholm, M.; Heiska, L.; Kere, J.; Carpén, O. Myotilin, a novel sarcomeric protein with two Ig-like domains, is encoded by a candidate gene for limb-girdle muscular dystrophy. Hum. Mol. Genet. 1999, 8, 1329–1336. [Google Scholar] [CrossRef]
- Moza, M.; Mologni, L.; Trokovic, R.; Faulkner, G.; Partanen, J.; Carpen, O. Targeted Deletion of the Muscular Dystrophy Gene myotilin Does Not Perturb Muscle Structure or Function in Mice. Mol. Cell. Biol. 2007, 27, 244–252. [Google Scholar] [CrossRef]
- Schafer, D.A.; Waddle, J.A.; Cooper, J.A. Localization of CapZ during myofibrillogenesis in cultured chicken muscle. Cell Motil. Cytoskelet. 1993, 25, 317–335. [Google Scholar] [CrossRef]
- Hishiya, A.; Kitazawa, T.; Takayama, S. BAG3 and Hsc70 interact with actin capping protein CapZ to maintain myofibrillar integrity under mechanical stress. Circ. Res. 2010, 107, 1220–1231. [Google Scholar] [CrossRef]
- Yamashita, A.; Maeda, K.; Maéda, Y. Crystal structure of CapZ: Structural basis for actin filament barbed end capping. EMBO J. 2003, 22, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.A.; Hug, C.; Cooper, J.A. Inhibition of CapZ during myofibrillogenesis alters assembly of actin filaments. J. Cell Biol. 1995, 128, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Warren, C.M.; Li, J.; McKinsey, T.A.; Russell, B. Myofibril growth during cardiac hypertrophy is regulated through dual phosphorylation and acetylation of the actin capping protein CapZ. Cell. Signal. 2016, 28, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Swanson, E.R.; Li, J.; Mkrtschjan, M.A.; Russell, B. Cyclic mechanical strain of myocytes modifies CapZβ1 post translationally via PKCε. J. Muscle Res. Cell Motil. 2015, 36, 329–337. [Google Scholar] [CrossRef]
- Sanger, J.W.; Wang, J.; Fan, Y.; White, J.; Mi-Mi, L.; Dube, D.K.; Sanger, J.M.; Pruyne, D. Assembly and Maintenance of Myofibrils in Striated Muscle. In The Actin Cytoskeleton; Handbook of Experimental Pharmacology; Jockusch, B., Ed.; Springer: Cham, Switzerland, 2016; Volume 235. [Google Scholar]
- David Pruyne, L.M.M. Loss of Sarcomere-associated Formins Disrupts Z-line Organization, but does not Prevent Thin Filament Assembly in Caenorhabditis elegans Muscle. J. Cytol. Histol. 2015, 6, 318. [Google Scholar] [CrossRef]
- Aspenström, P. Formin-binding proteins: Modulators of formin-dependent actin polymerization. Biochim. Biophys. Acta -Mol. Cell Res. 2010, 1803, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Ajima, R.; Bisson, J.A.; Helt, J.C.; Nakaya, M.A.; Habas, R.; Tessarollo, L.; He, X.; Morrisey, E.E.; Yamaguchi, T.P.; Cohen, E.D. DAAM1 and DAAM2 are co-required for myocardial maturation and sarcomere assembly. Dev. Biol. 2015, 408, 126–139. [Google Scholar] [CrossRef]
- Molnár, I.; Migh, E.; Szikora, S.; Kalmár, T.; Végh, A.G.; Molnár, I.; Migh, E.; Szikora, S.; Kalmár, T.; Végh, A.G.; et al. DAAM Is Required for Thin Filament Formation and Sarcomerogenesis during Muscle Development in Drosophila. PLoS Genet. 2014, 10, e1004166. [Google Scholar] [CrossRef]
- Xu, Y.; Moseley, J.B.; Sagot, I.; Poy, F.; Pellman, D.; Goode, B.L.; Eck, M.J. Crystal structures of a formin homology-2 domain reveal a tethered dimer architecture. Cell 2004, 116, 711–723. [Google Scholar] [CrossRef]
- Paul, A.S.; Pollard, T.D. Review of the mechanism of processive actin filament elongation by formins. Cell Motil. Cytoskelet. 2009, 66, 606–617. [Google Scholar] [CrossRef]
- Paul, A.; Pollard, T. The Role of the FH1 Domain and Profilin in Formin-Mediated Actin-Filament Elongation and Nucleation. Curr. Biol. 2008, 18, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Vavylonis, D.; Kovar, D.R.; O’Shaughnessy, B.; Pollard, T.D. Model of formin-associated actin filament elongation. Mol. Cell 2006, 21, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Takeya, R.; Suetsugu, S.; Kan-o, M.; Narusawa, M.; Shiose, A.; Tominaga, R.; Sumimoto, H. Mammalian formin Fhod3 regulates actin assembly and sarcomere organization in striated muscles. J. Biol. Chem. 2009, 284, 29873–29881. [Google Scholar] [CrossRef]
- Goode, B.L.; Eck, M.J. Mechanism and Function of Formins in the Control of Actin Assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, N.; Kan-O, M.; Ushijima, T.; Kage, Y.; Tominaga, R.; Sumimoto, H.; Takeya, R. Transgenic expression of the formin protein fhod3 selectively in the embryonic heart: Role of actin-binding activity of fhod3 and its sarcomeric localization during myofibrillogenesis. PLoS ONE 2016, 11, e0148472. [Google Scholar] [CrossRef] [PubMed]
- Iskratsch, T.; Lange, S.; Dwyer, J.; Kho, A.L.; Dos Remedios, C.; Ehler, E. Formin follows function: A muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. J. Cell Biol. 2010, 191, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Iskratsch, T.; Reijntjes, S.; Dwyer, J.; Toselli, P.; Dégano, I.R.; Dominguez, I.; Ehler, E. Two distinct phosphorylation events govern the function of muscle FHOD3. Cell. Mol. Life Sci. 2013, 70, 893–908. [Google Scholar] [CrossRef]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef]
- Fenix, A.M.; Neininger, A.C.; Taneja, N.; Hyde, K.; Visetsouk, M.R.; Garde, R.J.; Liu, B.; Nixon, B.R.; Manalo, A.E.; Becker, J.R.; et al. Muscle-specific stress fibers give rise to sarcomeres in cardiomyocytes. Elife 2018, 7, e42144. [Google Scholar] [CrossRef]
- Berger, J.; Berger, S.; Li, M.; Jacoby, A.S.; Arner, A.; Bavi, N.; Stewart, A.G.; Currie, P.D. In Vivo Function of the Chaperonin TRiC in α-Actin Folding during Sarcomere Assembly. Cell Rep. 2018, 22, 313–322. [Google Scholar] [CrossRef]
- Lutsch, G.; Vetter, R.; Offhauss, U.; Wieske, M.; Gröne, H.J.; Klemenz, R.; Schimke, I.; Stahl, J.; Benndorf, R. Abundance and location of the small heat shock proteins HSP25 and αB- crystallin in rat and human heart. Circulation 1997, 96, 3466–3476. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.D.; Christine, K.S.; Showell, C.; Conlon, F.L. Small heat shock protein Hsp27 is required for proper heart tube formation. Genesis 2007, 45, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Mu, Y.; Bogomolovas, J.; Fang, X.; Veevers, J.; Nowak, R.B.; Pappas, C.T.; Gregorio, C.C.; Evans, S.M.; Fowler, V.M.; et al. HSPB7 is indispensable for heart development by modulating actin filament assembly. Proc. Natl. Acad. Sci. USA 2017, 114, 11956–11961. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, M.I.; Kiff, R.M.; Goulding, D.A.; Stemple, D.L. Troponin T is essential for sarcomere assembly in zebrafish skeletal muscle. J. Cell Sci. 2011, 124, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, R.; Xu, X. Myofibrillogenesis in the developing zebrafish heart: A functional study of tnnt2. Dev. Biol. 2009, 331, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Marco-Ferreres, R.; Arredondo, J.J.; Fraile, B.; Cervera, M. Overexpression of troponin T in Drosophila muscles causes a decrease in the levels of thin-filament proteins. Biochem. J. 2005, 386, 145–152. [Google Scholar] [CrossRef]
- Sehnert, A.J.; Huq, A.; Weinstein, B.M.; Walker, C.; Fishman, M.; Stainier, D.Y.R. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat. Genet. 2002, 31, 106. [Google Scholar] [CrossRef]
- Hinkle, A.; Goranson, A.; Butters, C.A.; Tobacman, L.S. Roles for the troponin tail domain in thin filament assembly and regulation: A deletional study of cardiac troponin T. J. Biol. Chem. 1999, 274, 7157–7164. [Google Scholar] [CrossRef]
- Chereau, D.; Boczkowska, M.; Skwarek-Maruszewska, A.; Fujiwara, I.; Hayes, D.B.; Rebowski, G.; Lappalainen, P.; Pollard, T.D.; Dominguez, R. Leiomodin is an actin filament nucleator in muscle cells. Science 2008, 320, 239–243. [Google Scholar] [CrossRef]
- Szatmári, D.; Bugyi, B.; Ujfalusi, Z.; Grama, L.; Dudás, R.; Nyitrai, M. Cardiac leiomodin2 binds to the sides of actin filaments and regulates the ATPase activity of myosin. PLoS ONE 2017, 12, e0186288. [Google Scholar] [CrossRef]
- Pappas, C.T.; Farman, G.P.; Mayfield, R.M.; Konhilas, J.P.; Gregorio, C.C. Cardiac-specific knockout of Lmod2 results in a severe reduction in myofilament force production and rapid cardiac failure. J. Mol. Cell. Cardiol. 2018, 122, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Gokhin, D.S.; Tierney, M.T.; Sui, Z.; Sacco, A.; Fowler, V.M. Calpain-mediated proteolysis of tropomodulin isoforms leads to thin filament elongation in dystrophic skeletal muscle. Mol. Biol. Cell 2014, 25, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Bliss, K.T.; Tsukada, T.; Novak, S.M.; Dorovkov, M.V.; Shah, S.P.; Nworu, C.; Kostyukova, A.S.; Gregorio, C.C. Phosphorylation of tropomodulin1 contributes to the regulation of actin filament architecture in cardiac muscle. FASEB J. 2014, 28, 3987–3995. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, C.; Prill, K.; Pilgrim, D. Chaperones and the proteasome system: Regulating the construction and demolition of striated muscle. Int. J. Mol. Sci. 2018, 19, 32. [Google Scholar] [CrossRef]
- Ono, Y.; Sorimachi, H. Calpains—An elaborate proteolytic system. Biochim. Biophys. Acta-Proteins Proteom. 2012, 1824, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Portbury, A.L.; Willis, M.S.; Patterson, C. Tearin’ up my heart: Proteolysis in the cardiac sarcomere. J. Biol. Chem. 2011, 286, 9929–9934. [Google Scholar] [CrossRef]
- Reimann, L.; Wiese, H.; Leber, Y.; Schwable, A.N.; Fricke, A.L.; Rohland, A.; Knapp, B.; Peikert, C.D.; Drepper, F.; Van Der Ven, P.F.M.; et al. Myofibrillar Z-discs are a protein phosphorylation hot spot with protein kinase C (PKCα) modulating protein dynamics. Mol. Cell. Proteom. 2017, 16, 346–367. [Google Scholar] [CrossRef]
- Letavernier, E.; Zafrani, L.; Perez, J.; Letavernier, B.; Haymann, J.P.; Baud, L. The role of calpains in myocardial remodelling and heart failure. Cardiovasc. Res. 2012, 96, 38–45. [Google Scholar] [CrossRef]
- Koohmaraie, M. Ovine skeletal muscle multicatalytic proteinase complex (proteasome): Purification, characterization, and comparison of its effects on myofibrils with μ-calpains. J. Anim. Sci. 1992, 70, 3697–3708. [Google Scholar] [CrossRef]
- Dayton, W.R.; Goll, D.; Stromer, M.; Reville, W.; Zeece, M.; Robson, R. Some properties of a Ca2+-activated protease that may be involved in myofibrillar protein turnover. Cold Spring Harb. Conf. Cell Prolif. 1975, 2, 551–577. [Google Scholar]
- Di Lisa, F.; De Tullio, R.; Salamino, F.; Barbato, R.; Melloni, E.; Siliprandi, N.; Schiaffino, S.; Pontremoli, S. Specific degradation of troponin T and I by μ-calpain and its modulation by substrate phosphorylation. Biochem. J. 1995, 308, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Huff-Lonergan, E.; Mitsuhashi, T.; Beekman, D.D.; Parrish, F.C.; Olson, D.G.; Robson, R.M. Proteolysis of Specific Muscle Structural Proteins by μ-Calpain at Low pH and Temperature is Similar to Degradation in Postmortem Bovine Muscle. J. Anim. Sci. 1996, 74, 993–1008. [Google Scholar] [CrossRef] [PubMed]
- Goll, D.E.; Thompson, V.F.; Taylor, R.G.; Ouali, A. The calpain system and skeletal muscle growth. Can. J. Anim. Sci. 1998, 78, 503–512. [Google Scholar] [CrossRef]
- Bullard, B.; Sainsbury, G.; Miller, N. Digestion of proteins associated with the Z-disc by calpain. J. Muscle Res. Cell Motil. 1990, 11, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.R.; Kudryashova, E.; Potts, A.; Dalkilic, I.; Brosius, M.A.; Thompson, T.G.; Beckmann, J.S.; Kunkel, L.M.; Spencer, M.J. Calpain 3 cleaves filamin C and regulates its ability to interact with γ- and δ-sarcoglycans. Muscle Nerve 2003, 28, 472–483. [Google Scholar] [CrossRef]
- Goll, D.E.; Thompson, V.F.; Taylor, R.G.; Christiansen, J.A. Role of the calpain system in muscle growth. Biochimie 1992, 74, 225–237. [Google Scholar] [CrossRef]
- Taveau, M.; Bourg, N.; Sillon, G.; Roudaut, C.; Bartoli, M.; Richard, I. Calpain 3 Is Activated through Autolysis within the Active Site and Lyses Sarcomeric and Sarcolemmal Components. Mol. Cell. Biol. 2003, 23, 9127–9135. [Google Scholar] [CrossRef]
- Hayashi, C.; Ono, Y.; Doi, N.; Kitamura, F.; Tagami, M.; Mineki, R.; Arai, T.; Taguchi, H.; Yanagida, M.; Hirner, S.; et al. Multiple molecular interactions implicate the connectin/titin N2A region as a modulating scaffold for p94/calpain 3 activity in skeletal muscle. J. Biol. Chem. 2008, 283, 14801–14814. [Google Scholar] [CrossRef]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef]
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.H.; Rockman, H.A.; Patterson, C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140. [Google Scholar] [CrossRef]
- Chen, S.N.; Czernuszewicz, G.; Tan, Y.; Lombardi, R.; Jin, J.; Willerson, J.T.; Marian, A.J. Human molecular genetic and functional studies identify TRIM63, encoding muscle RING finger protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ. Res. 2012, 111, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J. Cell Biol. 2012, 198, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, E.; Kudryashov, D.; Kramerova, I.; Spencer, M.J. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J. Mol. Biol. 2005, 354, 413424. [Google Scholar] [CrossRef] [PubMed]
- Panicucci, C.; Traverso, M.; Baratto, S.; Romeo, C.; Iacomino, M.; Gemelli, C.; Tagliafico, A.; Broda, P.; Zara, F.; Bruno, C.; et al. Novel TRIM32 mutation in sarcotubular myopathy. Acta Myol. 2019, 38, 8–12. [Google Scholar] [PubMed]
- Spencer, J.A.; Eliazer, S.; Ilaria, R.L.; Richardson, J.A.; Olson, E.N. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. J. Cell Biol. 2000, 150, 771–784. [Google Scholar] [CrossRef]
- Fielitz, J.; Van Rooij, E.; Spencer, J.A.; Shelton, J.M.; Latif, S.; Van Der Nagel, R.; Bezprozvannaya, S.; De Windt, L.; Richardson, J.A.; Bassel-Duby, R.; et al. Loss of muscle-specific RING-finger 3 predisposes the heart to cardiac rupture after myocardial infarction. Proc. Natl. Acad. Sci. USA 2007, 104, 4377–4382. [Google Scholar] [CrossRef]
- Macqueen, D.J.; Fuentes, E.N.; Valdés, J.A.; Molina, A.; Martin, S.A.M. The vertebrate muscle-specific RING finger protein family includes MuRF4—A novel, conserved E3-ubiquitin ligase. FEBS Lett. 2014, 588, 4390–4397. [Google Scholar] [CrossRef]
- Zhou, W.; Wei, W.; Sun, Y. Genetically engineered mouse models for functional studies of SKP1-CUL1-F-box-protein (SCF) E3 ubiquitin ligases. Cell Res. 2013, 23, 599. [Google Scholar] [CrossRef]
- Spaich, S.; Will, R.D.; Just, S.; Spaich, S.; Kuhn, C.; Frank, D.; Berger, I.M.; Wiemann, S.; Korn, B.; Koegl, M.; et al. F-box and leucine-rich repeat protein 22 is a cardiac-enriched f-box protein that regulates sarcomeric protein turnover and is essential for maintenance of contractile function in vivo. Circ. Res. 2012, 111, 1504–1516. [Google Scholar] [CrossRef]
- Al-Yacoub, N.; Shaheen, R.; Awad, S.M.; Kunhi, M.; Dzimiri, N.; Nguyen, H.C.; Xiong, Y.; Al-Buraiki, J.; Al-Habeeb, W.; Alkuraya, F.S.; et al. FBXO32, encoding a member of the SCF complex, is mutated in dilated cardiomyopathy. Genome Biol. 2016, 17, 2. [Google Scholar] [CrossRef]
- Li, H.H.; Kedar, V.; Zhang, C.; McDonough, H.; Arya, R.; Wang, D.Z.; Patterson, C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J. Clin. Investig. 2004, 114, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Reed, J.C. Molecular chaperone targeting and regulation by BAG family proteins. Nat. Cell Biol. 2001, 3, E237. [Google Scholar] [CrossRef] [PubMed]
- Haus, U.; Trommler, P.; Fisher, P.R.; Hartmann, H.; Lottspeich, F.; Noegel, A.A.; Schleicher, M. The heat shock cognate protein from Dictyostelium affects actin polymerization through interaction with the actin-binding protein cap32/34. EMBO J. 1993, 12, 3763–3771. [Google Scholar] [CrossRef] [PubMed]
- Tardieux, I.; Baines, I.; Mossakowska, M.; Ward, G.E. Actin-binding proteins of invasive malaria parasites and the regulation of actin polymerization by a complex of 32/34-kDa proteins associated with heat shock protein 70kDa. Mol. Biochem. Parasitol. 1998, 93, 295–308. [Google Scholar] [CrossRef]
- Benaroudj, N.; Tarcsa, E.; Cascio, P.; Goldberg, A.L. The unfolding of substrates and ubiquitin-independent protein degradation by proteasomes. Biochimie 2001, 83, 311–318. [Google Scholar] [CrossRef]
- Bedford, L.; Paine, S.; Sheppard, P.W.; Mayer, R.J.; Roelofs, J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010, 20, 391–401. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931. [Google Scholar] [CrossRef]
- Shintani, T.; Mizushima, N.; Ogawa, Y.; Matsuura, A.; Noda, T.; Ohsumi, Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 1999, 18, 5234–5241. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Gottlieb, R.A. Recycle or die: The role of autophagy in cardioprotection. J. Mol. Cell. Cardiol. 2008, 44, 654–661. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619. [Google Scholar] [CrossRef]
- Kostin, S.; Pool, L.; Elsässer, A.; Hein, S.; Drexler, H.C.A.; Arnon, E.; Hayakawa, Y.; Zimmermann, R.; Bauer, E.; Klövekorn, W.P.; et al. Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 2003, 92, 715–724. [Google Scholar] [CrossRef]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Fürst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-Assisted Selective Autophagy Is Essential for Muscle Maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef]
- Klimek, C.; Kathage, B.; Wördehoff, J.; Höhfeld, J. BAG3-mediated proteostasis at a glance. J. Cell Sci. 2017, 130, 2781–2788. [Google Scholar] [CrossRef]
- Ulbricht, A.; Höhfeld, J. Tension-induced autophagy: May the chaperone be with you. Autophagy 2013, 9, 920–922. [Google Scholar] [CrossRef]
- Ulbricht, A.; Eppler, F.J.; Tapia, V.E.; Van Der Ven, P.F.M.; Hampe, N.; Hersch, N.; Vakeel, P.; Stadel, D.; Haas, A.; Saftig, P.; et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr. Biol. 2013, 23, 430–435. [Google Scholar] [CrossRef]
- Carra, S.; Seguin, S.J.; Landry, J. HspB8 and Bag3: A new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 2008, 4, 237–239. [Google Scholar] [CrossRef]
- Joshi, V.; Amanullah, A.; Upadhyay, A.; Mishra, R.; Kumar, A.; Mishra, A. A decade of boon or burden: What has the chip ever done for cellular protein quality control mechanism implicated in neurodegeneration and aging? Front. Mol. Neurosci. 2016, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Lázaro-Diéguez, F.; Aguado, C.; Mato, E.; Sánchez-Ruíz, Y.; Esteban, I.; Alberch, J.; Knecht, E.; Egea, G. Dynamics of an F-actin aggresome generated by the actin-stabilizing toxin jasplakinolide. J. Cell Sci. 2008, 121, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Russell, B. Phosphatidylinositol 4,5-bisphosphate regulates CapZβ1 and actin dynamics in response to mechanical strain. Am. J. Physiol.-Heart Circ. Physiol. 2013, 305, H1614–H1623. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Minami, N.; Abe, H.; Obinata, T. Characterization of a novel cofilin isoform that is predominantly expressed in mammalian skeletal muscle. J. Biol. Chem. 1994, 269, 15280–15286. [Google Scholar]
- Gurniak, C.B.; Chevessier, F.; Jokwitz, M.; Jönsson, F.; Perlas, E.; Richter, H.; Matern, G.; Boyl, P.P.; Chaponnier, C.; Fürst, D.; et al. Severe protein aggregate myopathy in a knockout mouse model points to an essential role of cofilin2 in sarcomeric actin exchange and muscle maintenance. Eur. J. Cell Biol. 2014, 93, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.B.; Joshi, M.; Savic, T.; Chen, Z.; Beggs, A.H. Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout demonstrates requirement of cofilin-2 for muscle maintenance. Hum. Mol. Genet. 2012, 21, 2341–2356. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Wan, P.; Chu, D.; Nie, J.; Cao, Y.; Luo, W.; Lu, S.; Chen, J.; Yang, Z. A cardiomyocyte-specific Wdr1 knockout demonstrates essential functional roles for actin disassembly during myocardial growth and maintenance in mice. Am. J. Pathol. 2014, 184, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Kremneva, E.; Makkonen, M.H.; Skwarek-Maruszewska, A.; Gateva, G.; Michelot, A.; Dominguez, R.; Lappalainen, P. Cofilin-2 controls actin filament length in muscle sarcomeres. Dev. Cell 2014, 31, 215–226. [Google Scholar] [CrossRef]
- McGough, A.; Pope, B.; Chiu, W.; Weeds, A. Cofilin changes the twist of F-actin: Implications for actin filament dynamics and cellular function. J. Cell Biol. 1997, 138, 771–781. [Google Scholar] [CrossRef]
- Bamburg, J.R. Proteins of the ADF/Cofilin Family: Essential Regulators of Actin Dynamics. Annu. Rev. Cell Dev. Biol. 1999, 15, 185–230. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Sato, N.; Nakagaki, T.; Abe, H.; Ono, S.; Obinata, T. Two mouse cofilin isoforms, muscle-type (MCF) and non-muscle type (NMCF), interact with F-actin with different efficiencies. J. Biochem. 2005, 138, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Maciver, S.K.; Zot, H.G.; Pollard, T.D. Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J. Cell Biol. 1991, 115, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.N.; Huot, J.; Landry, J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997, 110, 357–368. [Google Scholar]
- Lavoie, J.N.; Gingras-Breton, G.; Tanguay, R.M.; Landry, J. Induction of Chinese hamster HSP27 gene expression in mouse cells confers resistance to heat shock. HSP27 stabilization of the microfilament organization. J. Biol. Chem. 1993, 268, 3420–3429. [Google Scholar]
- Yoshida, K.I.; Aki, T.; Harada, K.; Shama, K.M.A.; Kamoda, Y.; Suzuki, A.; Ohno, S. Translocation of HSP27 and MKBP in ischemic heart. Cell Struct. Funct. 1999, 24, 181–185. [Google Scholar] [CrossRef]
- Sakamoto, K.; Urushidani, T.; Nagao, T. Translocation of HSP27 to sarcomere induced by ischemic preconditioning in isolated rat hearts. Biochem. Biophys. Res. Commun. 2000, 269, 137–142. [Google Scholar] [CrossRef]
- Morita, T.; Hayashi, K. G-actin sequestering protein thymosin-β4 regulates the activity of myocardin-related transcription factor. Biochem. Biophys. Res. Commun. 2013, 437, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Ono, S. The role of cyclase-associated protein in regulating actin filament dynamics—more than a monomer-sequestration factor. J. Cell Sci. 2013, 126, 3249–3258. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Section 18.2, The Dynamics of Actin Assembly. In Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK21594/ (accessed on 1 September 2019).
- Schaus, T.E.; Taylor, E.W.; Borisy, G.G. Self-organization of actin filament orientation in the dendritic- nucleation/array-treadmilling model. Proc. Natl. Acad. Sci. USA 2007, 104, 7086–7091. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.K.; McKane, M.; Houtman, J.C.D.; Rubenstein, P.A. Control of the ability of profilin to bind and facilitate nucleotide exchange from G-actin. J. Biol. Chem. 2008. [Google Scholar] [CrossRef]
- Carlier, M.F. Actin: Protein structure and filament dynamics. J. Biol. Chem. 1991, 266, 1–4. [Google Scholar]
- Mockrin, S.C.; Korn, E.D. Acanthamoeba Profilin Interacts with G-Actin to Increase the Rate of Exchange of Actin-Bound Adenosine 5’-Triphosphate. Biochemistry 1980, 19, 5359–5362. [Google Scholar] [CrossRef]
- Smart, N.; Riegler, J.; Turtle, C.W.; Lygate, C.A.; McAndrew, D.J.; Gehmlich, K.; Dubé, K.N.; Price, A.N.; Muthurangu, V.; Taylor, A.M.; et al. Aberrant developmental titin splicing and dysregulated sarcomere length in Thymosin β4 knockout mice. J. Mol. Cell. Cardiol. 2017, 102, 94–107. [Google Scholar] [CrossRef]
- Skruber, K.; Read, T.A.; Vitriol, E.A. Reconsidering an active role for G-actin in cytoskeletal regulation. J. Cell Sci. 2018, 131, jcs203760. [Google Scholar] [CrossRef]
- Ushijima, T.; Fujimoto, N.; Matsuyama, S.; Kan-O, M.; Kiyonari, H.; Shioi, G.; Kage, Y.; Yamasaki, S.; Takeya, R.; Sumimoto, H. The actin-organizing formin protein Fhod3 is required for postnatal development and functional maintenance of the adult heart in mice. J. Biol. Chem. 2018, 293, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Takeya, R.; Ishikawa, T.; Yamano, T.; Matsuo, A.; Tatsumi, T.; Nomura, T.; Sumimoto, H.; Kimura, A. Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ. J. 2013, 2990–2996. [Google Scholar] [CrossRef] [PubMed]
- Kan-o, M.; Takeya, R.; Taniguchi, K.; Tanoue, Y.; Tominaga, R.; Sumimoto, H. Expression and subcellular localization of mammalian formin Fhod3 in the embryonic and adult heart. PLoS ONE 2012, 7, e34765. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Kage, Y.; Fujimoto, N.; Ushijima, T.; Tsuruda, T.; Kitamura, K.; Shiose, A.; Asada, Y.; Sumimoto, H.; Takeya, R. Interaction between cardiac myosin-binding protein C and formin Fhod3. Proc. Natl. Acad. Sci. USA 2018, 115, E4386–E4395. [Google Scholar] [CrossRef]
- Skwarek-Maruszewska, A.; Boczkowska, M.; Zajac, A.L.; Kremneva, E.; Svitkina, T.; Dominguez, R.; Lappalainen, P. Different localizations and cellular behaviors of leiomodin and tropomodulin in mature cardiomyocyte sarcomeres. Mol. Biol. Cell 2010, 21, 3352–3361. [Google Scholar] [CrossRef]
- Yotti, R.; Seidman, C.E.; Seidman, J.G. Advances in the Genetic Basis and Pathogenesis of Sarcomere Cardiomyopathies. Annu. Rev. Genom. Hum. Genet. 2019, 20, 129–153. [Google Scholar] [CrossRef]
- Tsukada, T.; Pappas, C.T.; Moroz, N.; Antin, P.B.; Kostyukova, A.S.; Gregorio, C.C. Leiomodin-2 is an antagonist of tropomodulin-1 at the pointed end of the thin filaments in cardiac muscle. J. Cell Sci. 2010, 123, 3136–3145. [Google Scholar] [CrossRef]
- Zheng, J.; Chen, S.; Chen, Y.; Zhu, M.; Hong, D. A novel mutation in the PDZ-like motif of ZASP causes distal ZASP-related myofibrillar myopathy. Neuropathology 2017, 37, 45–51. [Google Scholar] [CrossRef]
- Selcen, D.; Engel, A.G. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann. Neurol. 2005, 57, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Theis, J.L.; Martijn Bos, J.; Bartleson, V.B.; Will, M.L.; Binder, J.; Vatta, M.; Towbin, J.A.; Gersh, B.J.; Ommen, S.R.; Ackerman, M.J. Echocardiographic-determined septal morphology in Z-disc hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 351, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Truszkowska, G.T.; Bilińska, Z.T.; Muchowicz, A.; Pollak, A.; Biernacka, A.; Kozar-Kamińska, K.; Stawiński, P.; Gasperowicz, P.; Kosińska, J.; Zieliński, T.; et al. Homozygous truncating mutation in NRAP gene identified by whole exome sequencing in a patient with dilated cardiomyopathy. Sci. Rep. 2017, 7, 3362. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Crawford, G.L.; Dore, J.; Anderson, S.A.; DesPres, D.; Horowits, R. Cardiac-specific NRAP overexpression causes right ventricular dysfunction in mice. Exp. Cell Res. 2011, 317, 1226–1237. [Google Scholar] [CrossRef] [PubMed]
- Papizan, J.B.; Garry, G.A.; Brezprozvannaya, S.; McAnally, J.R.; Bassel-Duby, R.; Liu, N.; Olson, E.N. Deficiency in Kelch protein Klhl31 causes congenital myopathy in mice. J. Clin. Investig. 2017, 127, 3730–3740. [Google Scholar] [CrossRef] [PubMed]
- Hishiya, A.; Salman, M.N.; Carra, S.; Kampinga, H.H.; Takayama, S. BAG3 directly interacts with mutated alphaB-crystallin to suppress its aggregation and toxicity. PLoS ONE 2011, 6, e16828. [Google Scholar] [CrossRef]
- Homma, S.; Iwasaki, M.; Shelton, G.D.; Engvall, E.; Reed, J.C.; Takayama, S. BAG3 deficiency results in fulminant myopathy and early lethality. Am. J. Pathol. 2006, 169, 761–773. [Google Scholar] [CrossRef]
- Arimura, T.; Ishikawa, T.; Nunoda, S.; Kawai, S.; Kimura, A. Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum. Mutat. 2011, 32, 1481–1491. [Google Scholar] [CrossRef]
- Myers, V.D.; McClung, J.M.; Wang, J.F.; Tahrir, F.G.; Gupta, M.K.; Gordon, J.; Kontos, C.H.; Khalili, K.; Cheung, J.Y.; Feldman, A.M. The Multifunctional Protein BAG3: A Novel Therapeutic Target in Cardiovascular Disease. JACC Basic Transl. Sci. 2018, 3, 122–131. [Google Scholar] [CrossRef]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Züchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Common Name | Alternative Name(s) | Common Name | Alternative Name(s) |
|---|---|---|---|
| ZASP | Cypher, Oracle | HSPB7 | Cardiovascular HSP |
| actinin | α-actinin | Muscle LIM Protein (MLP) | Cardiac LIM protein (CLP) |
| NRAP | N-RAP | T-CAP | TCAP, telethonin |
| Filamin C | γ-filamin, ABP-L, sarcomere filamin | Calpain 1 | μ-calpain, mu-calpain, calpain I |
| myopodin | SYNPO2 | Calpain 2 | m-calpain, calpain II |
| Calsarcin | FATZ, myozenin | Calpain 3 | p94 |
| Calcineurin | Protein phosphatase 2B (PP2B) | MuRF1 | TRIM63 |
| KLHL41 | Krp1, sarcosin | MuRF2 | TRIM55 |
| Titin | Connectin | MuRF3 | TRIM54 |
| CapZ | β-actinin, CapZβ, CapZβ1 | Atrogin-1 | MAFbx1, Fbxo32 |
| Myotilin | TTID | Hsc70 | HSPA8 |
| TRiC | CCT, chaperonin | HspB8 | Hsp22 |
| Prefoldin | GimC | Cofilin2 | m-cofilin, ADF |
| Hsp25/27 | Hspβ1, Hsp25, Hsp27 | Wdr-1 | AIP-1 |
| αβ-crystallin | CRYAB, Hsp20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prill, K.; Dawson, J.F. Assembly and Maintenance of Sarcomere Thin Filaments and Associated Diseases. Int. J. Mol. Sci. 2020, 21, 542. https://doi.org/10.3390/ijms21020542
Prill K, Dawson JF. Assembly and Maintenance of Sarcomere Thin Filaments and Associated Diseases. International Journal of Molecular Sciences. 2020; 21(2):542. https://doi.org/10.3390/ijms21020542
Chicago/Turabian StylePrill, Kendal, and John F. Dawson. 2020. "Assembly and Maintenance of Sarcomere Thin Filaments and Associated Diseases" International Journal of Molecular Sciences 21, no. 2: 542. https://doi.org/10.3390/ijms21020542
APA StylePrill, K., & Dawson, J. F. (2020). Assembly and Maintenance of Sarcomere Thin Filaments and Associated Diseases. International Journal of Molecular Sciences, 21(2), 542. https://doi.org/10.3390/ijms21020542