Human Induced Pluripotent Stem Cells Derived from a Cardiac Somatic Source: Insights for an In-Vitro Cardiomyocyte Platform




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
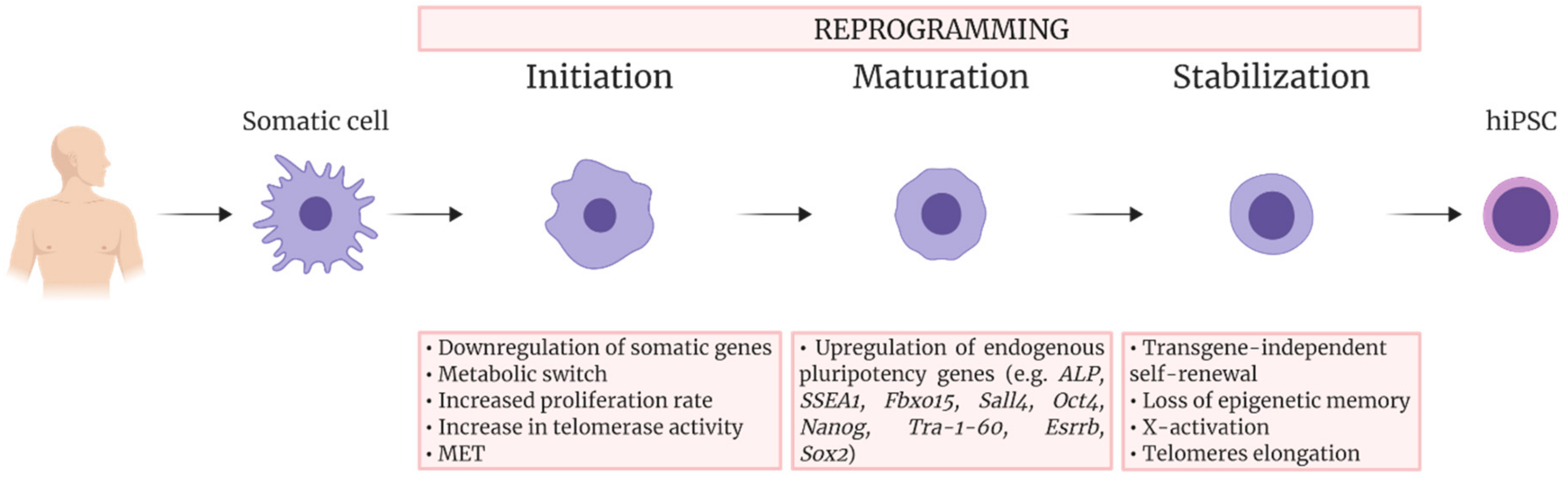
2. Reprogramming
3. Cardiac Differentiation
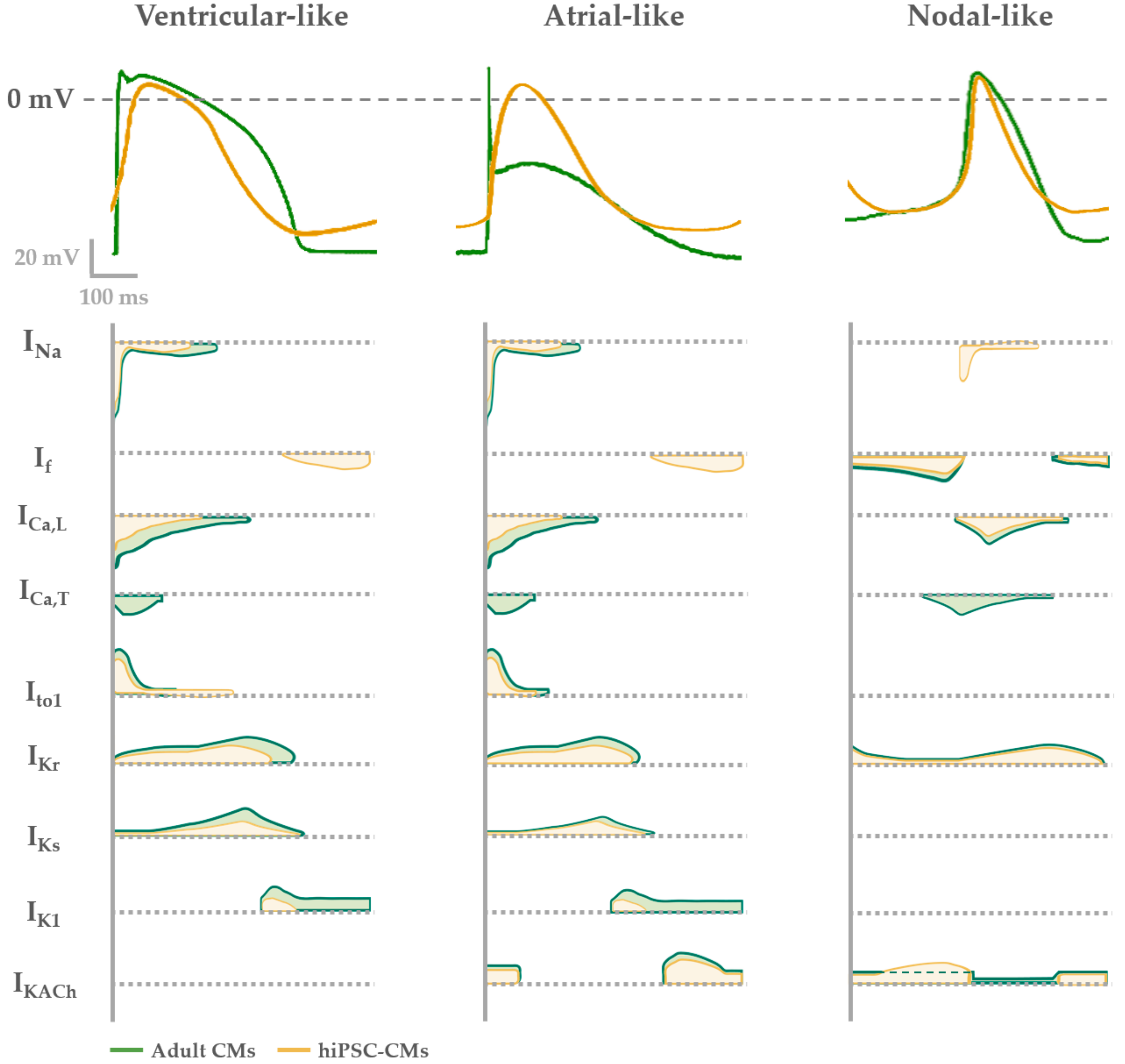
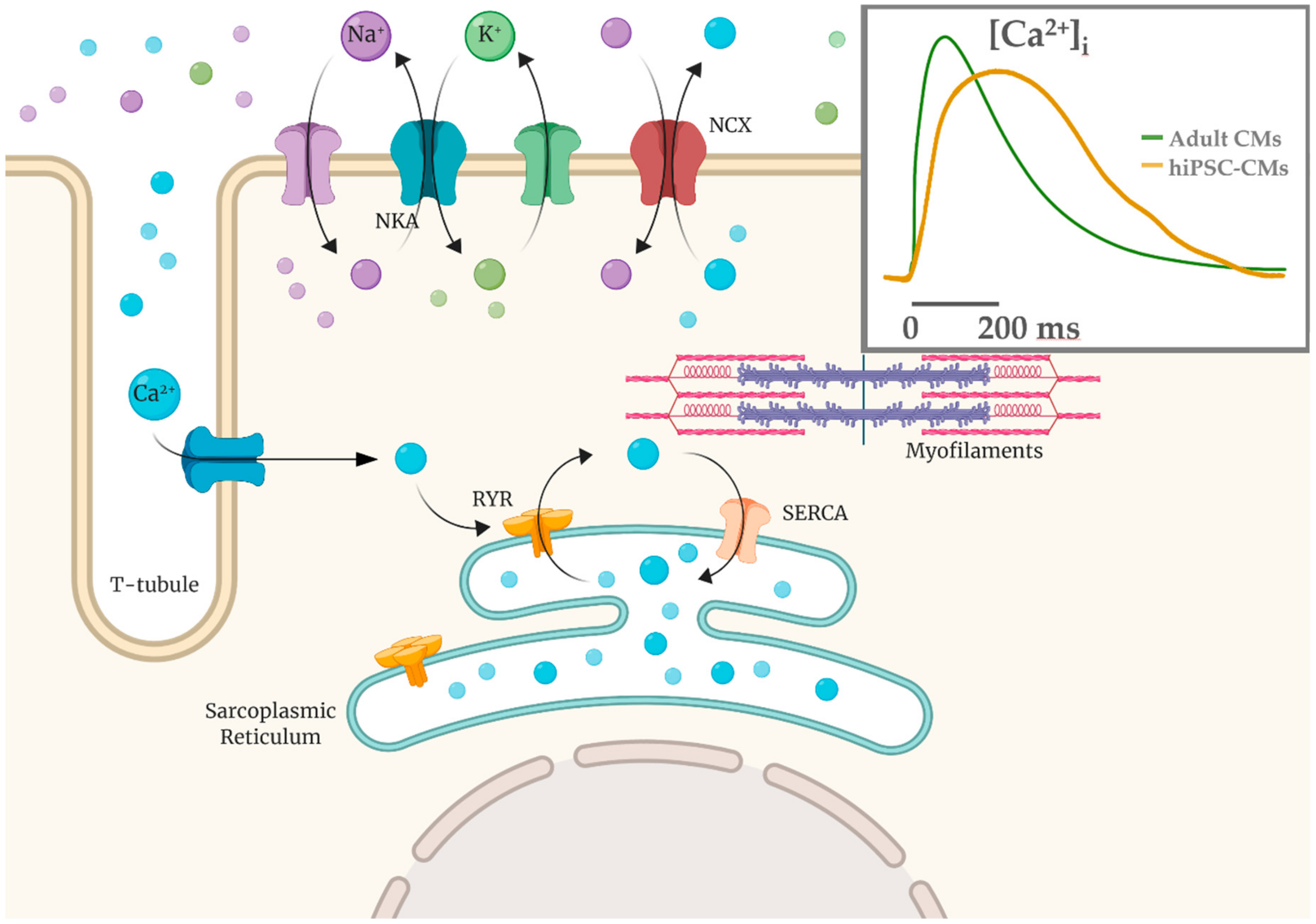
4. Functional Properties of hiPSC-CMs: Overview and Limitations
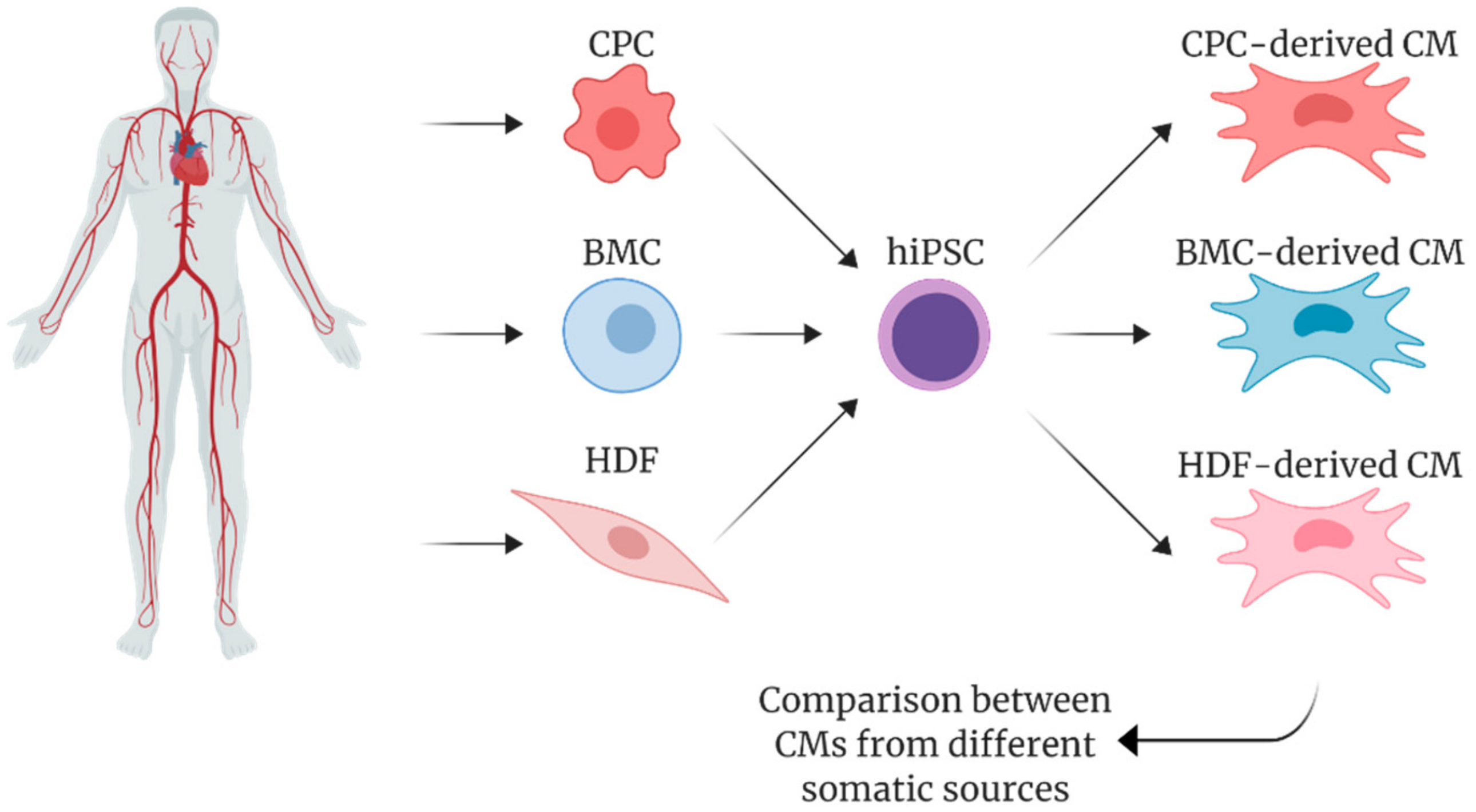
5. Pluripotency and Cardiac Differentiation of hiPSCs Derived from Cardiac vs. Non-Cardiac Sources
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| hiPSC | Human Induced Pluripotent Stem Cell |
| CM | Cardiomyocyte |
| ESC | Embryonic Stem Cell |
| MET | Mesenchymal to Epithelial Transition |
| CVD | Cardiovascular Disease |
| CPC | Cardiac Progenitor Cell |
| BMC | Bone Marrow-derived stem Cell |
| HDF | Human Dermal Fibroblast |
| Na+ | Sodium |
| K+ | Potassium |
| Ca2+ | Calcium |
| Ediast | Diastolic Membrane Potential |
| AP | Action Potential |
| APA | Action Potential Amplitude |
| APD | Action Potential Duration |
| MDP | Maximum Diastolic Potential |
| MEA | Multielectrode Array |
| ECG | Electrocardiogram |
| TdP | Torsade de Pointes |
| LQTS | Long QT Syndrome |
| CPVT | Catecholaminergic Polymorphic Ventricular Tachycardia |
| DCM | Dilated Cardiomyopathies |
| HCM | Hypertrophic Cardiomyopathies |
| QSP | Quantitative System Pharmacology |
| CiPA | Comprehensive In Vitro Proarrythmia Assay |
References
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Minami, I.; Shiozaki, M.; Yu, L.; Yajima, S.; Miyagawa, S.; Shiba, Y.; Morone, N.; Fukushima, S.; Yoshioka, M.; et al. Human Pluripotent Stem Cell-Derived Cardiac Tissue-like Constructs for Repairing the Infarcted Myocardium. Stem Cell Rep. 2017, 9, 1546–1559. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, S.; Miki, K.; Takaki, T.; Okubo, C.; Hatani, T.; Chonabayashi, K.; Nishikawa, M.; Takei, I.; Oishi, A.; Narita, M.; et al. Enhanced engraftment, proliferation, and therapeutic potential in heart using optimized human iPSC-derived cardiomyocytes. Sci. Rep. 2016, 6, 19111. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, M.; Sala, L.; Dreizehnter, L.; Crotti, L.; Sinnecker, D.; Mura, M.; Pane, L.S.; Altomare, C.; Torre, E.; Mostacciuolo, G.; et al. Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc. Res. 2017, 113, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Laksman, Z.; Wauchop, M.; Lin, E.; Protze, S.; Lee, J.; Yang, W.; Izaddoustdar, F.; Shafaattalab, S.; Gepstein, L.; Tibbits, G.F.; et al. Modeling Atrial Fibrillation using Human Embryonic Stem Cell-Derived Atrial Tissue. Sci. Rep. 2017, 7, 5268. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, P.A.; Darche, F.F.; Ullrich, N.D.; Geschwill, P.; Greber, B.; Rivinius, R.; Seyler, C.; Müller-Decker, K.; Draguhn, A.; Utikal, J.; et al. Subtype-specific differentiation of cardiac pacemaker cell clusters from human induced pluripotent stem cells. Stem Cell Res. Ther. 2017, 8, 229. [Google Scholar] [CrossRef]
- Noguchi, H.; Miyagi-Shiohira, C.; Nakashima, Y. Induced Tissue-Specific Stem Cells and Epigenetic Memory in Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2018, 19, 930. [Google Scholar] [CrossRef]
- Robertson, C.; Tran, D.D.; George, S.C. Concise review: Maturation phases of human pluripotent stem cell-derived cardiomyocytes. Stem Cells 2013, 31, 829–837. [Google Scholar] [CrossRef]
- Aigha, I.; Raynaud, C. Maturation of pluripotent stem cell derived cardiomyocytes: The new challenge. Glob. Cardiol. Sci. Pract. 2016, 2016, e201606. [Google Scholar] [CrossRef]
- Bedada, F.B.; Wheelwright, M.; Metzger, J.M. Maturation status of sarcomere structure and function in human iPSC-derived cardiac myocytes. Biochim. Biophys. Acta 2016, 1863, 1829–1838. [Google Scholar] [CrossRef]
- Hu, D.; Linders, A.; Yamak, A.; Correia, C.; Kijlstra, J.D.; Garakani, A.; Xiao, L.; Milan, D.J.; van der Meer, P.; Serra, M.; et al. Metabolic Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes by Inhibition of HIF1α and LDHA. Circ. Res. 2018, 123, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Dougherty, J.A.; Manring, H.R.; Elmadbouh, I.; Mergaye, M.; Czirok, A.; Greta Isai, D.; Belevych, A.E.; Yu, L.; Janssen, P.M.L.; et al. Assessment of temporal functional changes and miRNA profiling of human iPSC-derived cardiomyocytes. Sci. Rep. 2019, 9, 13188. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Athanasopoulos, T.; Munye, M.M.; Yáñez-Muñoz, R.J. Nonintegrating Gene Therapy Vectors. Hematol. Oncol. Clin. North. Am. 2017, 31, 753–770. [Google Scholar] [CrossRef]
- Somers, A.; Jean, J.C.; Sommer, C.A.; Omari, A.; Ford, C.C.; Mills, J.A.; Ying, L.; Sommer, A.G.; Jean, J.M.; Smith, B.W.; et al. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells 2010, 28, 1728–1740. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Churko, J.M.; Burridge, P.W.; Wu, J.C. Generation of human iPSCs from human peripheral blood mononuclear cells using non-integrative Sendai virus in chemically defined conditions. Methods Mol. Biol. 2013, 1036, 81–88. [Google Scholar]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef]
- Burridge, P.W.; Thompson, S.; Millrod, M.A.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS ONE 2011, 6, e18293. [Google Scholar] [CrossRef]
- Diecke, S.; Lu, J.; Lee, J.; Termglinchan, V.; Kooreman, N.G.; Burridge, P.W.; Ebert, A.D.; Churko, J.M.; Sharma, A.; Kay, M.A.; et al. Novel codon-optimized mini-intronic plasmid for efficient, inexpensive, and xeno-free induction of pluripotency. Sci. Rep. 2015, 5, 8081. [Google Scholar] [CrossRef]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef]
- Loh, Y.H.; Agarwal, S.; Park, I.H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar] [CrossRef]
- Cai, J.; Li, W.; Su, H.; Qin, D.; Yang, J.; Zhu, F.; Xu, J.; He, W.; Guo, X.; Labuda, K.; et al. Generation of human induced pluripotent stem cells from umbilical cord matrix and amniotic membrane mesenchymal cells. J. Biol. Chem. 2010, 285, 11227–11234. [Google Scholar] [CrossRef]
- Xue, Y.; Cai, X.; Wang, L.; Liao, B.; Zhang, H.; Shan, Y.; Chen, Q.; Zhou, T.; Li, X.; Hou, J.; et al. Generating a Non-Integrating Human Induced Pluripotent Stem Cell Bank from Urine-Derived Cells. PLoS ONE 2013, 8, e70573. [Google Scholar] [CrossRef]
- Novak, A.; Lorber, A.; Itskovitz-Eldor, J.; Binah, O. Modeling Catecholaminergic Polymorphic Ventricular Tachycardia using Induced Pluripotent Stem Cell-derived Cardiomyocytes. Rambam Maimonides Med. J. 2012, 3, e0015. [Google Scholar] [CrossRef]
- Altomare, C.; Pianezzi, E.; Cervio, E.; Bolis, S.; Biemmi, V.; Benzoni, P.; Camici, G.G.; Moccetti, T.; Barile, L.; Vassalli, G. Human-induced pluripotent stem cell-derived cardiomyocytes from cardiac progenitor cells: Effects of selective ion channel blockade. Europace 2016, 18, iv67–iv76. [Google Scholar]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional Genomics Reveals a BMP-Driven Mesenchymal-to-Epithelial Transition in the Initiation of Somatic Cell Reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef]
- David, L.; Polo, J.M. Phases of reprogramming. Stem Cell Res. 2014, 12, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Downing, T.L.; Soto, J.; Morez, C.; Houssin, T.; Fritz, A.; Yuan, F.; Chu, J.; Patel, S.; Schaffer, D.V.; Li, S. Biophysical regulation of epigenetic state and cell reprogramming. Nat. Mater. 2013, 12, 1154. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A Molecular Roadmap of Reprogramming Somatic Cells into iPS Cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Nakamura, M.; Narita, M.; Takahashi, K.; Yamanaka, S. Maturation, not initiation, is the major roadblock during reprogramming toward pluripotency from human fibroblasts. Proc. Natl. Acad. Sci. USA 2013, 110, 12172–12179. [Google Scholar] [CrossRef]
- Golipour, A.; David, L.; Liu, Y.; Jayakumaran, G.; Hirsch, C.L.; Trcka, D.; Wrana, J.L. A Late Transition in Somatic Cell Reprogramming Requires Regulators Distinct from the Pluripotency Network. Cell Stem Cell 2012, 11, 769–782. [Google Scholar] [CrossRef]
- Pan, G.; Li, J.; Zhou, Y.; Zheng, H.; Pei, D. A negative feedback loop of transcription factors that controls stem cell pluripotency and self-renewal. FASEB J. 2006, 20, 1730–1732. [Google Scholar] [CrossRef]
- Jaenisch, R.; Young, R. Stem Cells, the Molecular Circuitry of Pluripotency and Nuclear Reprogramming. Cell 2008, 132, 567–582. [Google Scholar] [CrossRef]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc Regulates Transcriptional Pause Release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef]
- Maekawa, M.; Yamaguchi, K.; Nakamura, T.; Shibukawa, R.; Kodanaka, I.; Ichisaka, T.; Kawamura, Y.; Mochizuki, H.; Goshima, N.; Yamanaka, S. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 2011, 474, 225–229. [Google Scholar] [CrossRef]
- Bilic, J.; Izpisua Belmonte, J.C. Concise review: Induced pluripotent stem cells versus embryonic stem cells: Close enough or yet too far apart? Stem Cells 2012, 30, 33–41. [Google Scholar] [CrossRef]
- Saric, T.; Hescheler, J. Stem cells and nuclear reprogramming. Minim. Invasive Ther. Allied Technol. 2008, 17, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Südhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Suzuki, M.; Kasahara, H.; Shimizu, N.; Shichita, T.; Sekiya, T.; Kimura, A.; Sasaki, K.; Yasukawa, H.; Yoshimura, A. ETS transcription factor ETV2 directly converts human fibroblasts into functional endothelial cells. Proc. Natl. Acad. Sci. USA 2015, 112, 160–165. [Google Scholar] [CrossRef]
- Sekiya, S.; Suzuki, A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 2011, 475, 390–393. [Google Scholar] [CrossRef]
- Yin, S.; Cen, L.; Wang, C.; Zhao, G.; Sun, J.; Liu, W.; Cao, Y.; Cui, L. Chondrogenic transdifferentiation of human dermal fibroblasts stimulated with cartilage-derived morphogenetic protein 1. Tissue Eng. Part A 2010, 16, 1633–1643. [Google Scholar] [CrossRef]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Südhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–223. [Google Scholar] [CrossRef]
- Boularaoui, S.M.; Abdel-Raouf, K.M.A.; Alwahab, N.S.A.; Kondash, M.E.; Truskey, G.A.; Teo, J.C.M.; Christoforou, N. Efficient transdifferentiation of human dermal fibroblasts into skeletal muscle: Efficient human skeletal muscle transdifferentiation. J. Tissue Eng. Regen. Med. 2018, 12, e918–e936. [Google Scholar] [CrossRef]
- Shimizu, K.; Ohsumi, S.; Kishida, T.; Mazda, O.; Honda, H. Fabrication of contractile skeletal muscle tissues using directly converted myoblasts from human fibroblasts. J. Biosci. Bioeng. 2019. [Google Scholar] [CrossRef]
- World Health Organization. Noncommunicable Diseases Country Profiles 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Hoppe, U.C.; Beuckelmann, D.J. Characterization of the hyperpolarization-activated inward current in isolated human atrial myocytes. Cardiovasc. Res. 1998, 38, 788–801. [Google Scholar] [CrossRef]
- Cerbai, E.; Sartiani, L.; DePaoli, P.; Pino, R.; Maccherini, M.; Bizzarri, F.; DiCiolla, F.; Davoli, G.; Sani, G.; Mugelli, A. The properties of the pacemaker current I(F)in human ventricular myocytes are modulated by cardiac disease. J. Mol. Cell. Cardiol. 2001, 33, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Dell’Era, P.; Benzoni, P.; Crescini, E.; Valle, M.; Xia, E.; Consiglio, A.; Memo, M. Cardiac disease modeling using induced pluripotent stem cell-derived human cardiomyocytes. World J. Stem Cells 2015, 7, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.A.; Spinale, F.G. Large animal models of heart failure: A critical link in the translation of basic science to clinical practice. Circ. Heart Fail. 2009, 2, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.R.; Vlaminckx, E.; Hermans, A.N.; Rohrbacher, J.; Van Ammel, K.; Towart, R.; Pugsley, M.; Gallacher, D.J. Predicting drug-induced changes in QT interval and arrhythmias: QT-shortening drugs point to gaps in the ICHS7B Guidelines. Br. J. Pharmacol. 2008, 154, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Loskill, P.; Shao, K.; Huebsch, N.; Hong, S.; Marcus, S.G.; Marks, N.; Mandegar, M.; Conklin, B.R.; Lee, L.P.; et al. Human iPSC-based Cardiac Microphysiological System for Drug Screening Applications. Sci. Rep. 2015, 5, 1–7. [Google Scholar] [CrossRef]
- Itskovitz-Eldor, J.; Schuldiner, M.; Karsenti, D.; Eden, A.; Yanuka, O.; Amit, M.; Soreq, H.; Benvenisty, N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 2000, 6, 88–95. [Google Scholar] [CrossRef]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef]
- Synnergren, J.; Åkesson, K.; Dahlenborg, K.; Vidarsson, H.; Améen, C.; Steel, D.; Lindahl, A.; Olsson, B.; Sartipy, P. Molecular Signature of Cardiomyocyte Clusters Derived from Human Embryonic Stem Cells. Stem Cells 2008, 26, 1831–1840. [Google Scholar] [CrossRef]
- Cao, F.; Wagner, R.A.; Wilson, K.D.; Xie, X.; Fu, J.-D.; Drukker, M.; Lee, A.; Li, R.A.; Gambhir, S.S.; Weissman, I.L.; et al. Transcriptional and Functional Profiling of Human Embryonic Stem Cell-Derived Cardiomyocytes. PLoS ONE 2008, 3, e3474. [Google Scholar] [CrossRef]
- Xu, X.Q.; Soo, S.Y.; Sun, W.; Zweigerdt, R. Global Expression Profile of Highly Enriched Cardiomyocytes Derived from Human Embryonic Stem Cells. Stem Cells 2009, 27, 2163–2174. [Google Scholar] [CrossRef] [PubMed]
- Mummery, C.; Ward-van Oostwaard, D.; Doevendans, P.; Spijker, R.; van den Brink, S.; Hassink, R.; van der Heyden, M.; Opthof, T.; Pera, M.; de la Riviere, A.B.; et al. Differentiation of human embryonic stem cells to cardiomyocytes: Role of coculture with visceral endoderm-like cells. Circulation 2003, 107, 2733–2740. [Google Scholar] [CrossRef] [PubMed]
- Passier, R.; Oostwaard, D.W.; Snapper, J.; Kloots, J.; Hassink, R.J.; Kuijk, E.; Roelen, B.; de la Riviere, A.B.; Mummery, C. Increased Cardiomyocyte Differentiation from Human Embryonic Stem Cells in Serum-Free Cultures. Stem Cells 2005, 23, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Willems, E.; Bushway, P.J.; Mercola, M. Natural and Synthetic Regulators of Embryonic Stem Cell Cardiogenesis. Pediatr. Cardiol. 2009, 30, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Freund, C.; Davis, R.P.; Gkatzis, K.; Ward-van Oostwaard, D.; Mummery, C.L. The first reported generation of human induced pluripotent stem cells (iPS cells) and iPS cell-derived cardiomyocytes in the Netherlands. Neth. Heart J. 2010, 18, 51–54. [Google Scholar] [PubMed]
- Kang, Y.; Nagy, J.M.; Polak, J.M.; Mantalaris, A. Proteomic Characterization of the Conditioned Media Produced by the Visceral Endoderm-Like Cell Lines HepG2 and END2: Toward a Defined Medium for the Osteogenic/Chondrogenic Differentiation of Embryonic Stem Cells. Stem Cells Dev. 2008, 18, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Arrell, D.K.; Niederländer, N.J.; Faustino, R.S.; Behfar, A.; Terzic, A. Cardioinductive Network Guiding Stem Cell Differentiation Revealed by Proteomic Cartography of Tumor Necrosis Factor α-Primed Endodermal Secretome. Stem Cells 2008, 26, 387–400. [Google Scholar] [CrossRef]
- Freund, C.; Oostwaard, D.W.; Monshouwer-Kloots, J.; van den Brink, S.; van Rooijen, M.; Xu, X.; Zweigerdt, R.; Mummery, C.; Passier, R. Insulin Redirects Differentiation from Cardiogenic Mesoderm and Endoderm to Neuroectoderm in Differentiating Human Embryonic Stem Cells. Stem Cells 2008, 26, 724–733. [Google Scholar] [CrossRef]
- Xu, X.Q.; Graichen, R.; Soo, S.Y.; Balakrishnan, T.; Bte Rahmat, S.N.; Sieh, S.; Tham, S.C.; Freund, C.; Moore, J.; Mummery, C.; et al. Chemically defined medium supporting cardiomyocyte differentiation of human embryonic stem cells. Differentiation 2008, 76, 958–970. [Google Scholar] [CrossRef]
- Winnier, G.; Blessing, M.; Labosky, P.A.; Hogan, B.L. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995, 9, 2105–2116. [Google Scholar] [CrossRef]
- Marvin, M.J.; Di Rocco, G.; Gardiner, A.; Bush, S.M.; Lassar, A.B. Inhibition of Wnt activity induces heart formation from posterior mesoderm. Genes Dev. 2001, 15, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Naujok, O.; Diekmann, U.; Lenzen, S. The Generation of Definitive Endoderm from Human Embryonic Stem Cells is Initially Independent from Activin A but Requires Canonical Wnt-Signaling. Stem Cell Rev. Rep. 2014, 10, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Police, S.; Hassanipour, M.; Gold, J.D. Cardiac Bodies: A Novel Culture Method for Enrichment of Cardiomyocytes Derived from Human Embryonic Stem Cells. Stem Cells Dev. 2006, 15, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Chen, K.Y.; Naumova, A.V.; Muskheli, V.; Fugate, J.A.; Dupras, S.K.; Reinecke, H.; Xu, C.; Hassanipour, M.; Police, S.; et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 2007, 25, 1015–1024. [Google Scholar] [CrossRef]
- Naito, A.T.; Shiojima, I.; Akazawa, H.; Hidaka, K.; Morisaki, T.; Kikuchi, A.; Komuro, I. Developmental stage-specific biphasic roles of Wnt/β-catenin signaling in cardiomyogenesis and hematopoiesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19812–19817. [Google Scholar] [CrossRef]
- Ueno, S.; Weidinger, G.; Osugi, T.; Kohn, A.D.; Golob, J.L.; Pabon, L.; Reinecke, H.; Moon, R.T.; Murry, C.E. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9685–9690. [Google Scholar] [CrossRef]
- Tran, T.H.; Wang, X.; Browne, C.; Zhang, Y.; Schinke, M.; Izumo, S.; Burcin, M. Wnt3a-Induced Mesoderm Formation and Cardiomyogenesis in Human Embryonic Stem Cells. Stem Cells 2009, 27, 1869–1878. [Google Scholar] [CrossRef]
- Yang, L.; Soonpaa, M.H.; Adler, E.D.; Roepke, T.K.; Kattman, S.J.; Kennedy, M.; Henckaerts, E.; Bonham, K.; Abbott, G.W.; Linden, R.M.; et al. Human cardiovascular progenitor cells develop from a KDR + embryonic-stem-cell-derived population. Nature 2008, 453, 524–528. [Google Scholar] [CrossRef]
- Schlange, T.; Andrée, B.; Arnold, H.H.; Brand, T. BMP2 is required for early heart development during a distinct time period. Mech. Dev. 2000, 91, 259–270. [Google Scholar] [CrossRef]
- Ross, S.; Holliday, M.; Lim, S.; Semsarian, C. Characterization of the first induced pluripotent stem cell line generated from a patient with autosomal dominant catecholaminergic polymorphic ventricular tachycardia due to a heterozygous mutation in cardiac calsequestrin-2. Stem Cell Res. 2019, 37, 101450. [Google Scholar] [CrossRef]
- Matsa, E.; Burridge, P.W.; Wu, J.C. Human stem cells for modeling heart disease and for drug discovery. Sci. Transl. Med. 2014, 6, 239ps6. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human induced pluripotent stem cell-derived cardiomyocytes: Insights into molecular, cellular, and functional phenotypes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Chao, B.S.; Wu, J.C. Strategies for Improving the Maturity of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2018, 123, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef]
- Ma, J.; Guo, L.; Fiene, S.J.; Anson, B.D.; Thomson, J.A.; Kamp, T.J.; Kolaja, K.L.; Swanson, B.J.; January, C.T. High purity human-induced pluripotent stem cell-derived cardiomyocytes: Electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2006–H2017. [Google Scholar] [CrossRef]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 2011, 471, 230–234. [Google Scholar] [CrossRef]
- Davis, R.P.; Casini, S.; van den Berg, C.W.; Hoekstra, M.; Remme, C.A.; Dambrot, C.; Salvatori, D.; Oostwaard, D.W.; Wilde, A.A.M.; Bezzina, C.R.; et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 2012, 125, 3079–3091. [Google Scholar] [CrossRef]
- Ivashchenko, C.Y.; Pipes, G.C.; Lozinskaya, I.M.; Lin, Z.; Xiaoping, X.; Needle, S.; Grygielko, E.T.; Hu, E.; Toomey, J.R.; Lepore, J.J.; et al. Human-induced pluripotent stem cell-derived cardiomyocytes exhibit temporal changes in phenotype. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H913–H922. [Google Scholar] [CrossRef]
- Goversen, B.; van der Heyden, M.A.G.; van Veen, T.A.B.; de Boer, T.P. The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: Special focus on IK1. Pharmacol. Ther. 2018, 183, 127–136. [Google Scholar] [CrossRef]
- Otsuji, T.G.; Minami, I.; Kurose, Y.; Yamauchi, K.; Tada, M.; Nakatsuji, N. Progressive maturation in contracting cardiomyocytes derived from human embryonic stem cells: Qualitative effects on electrophysiological responses to drugs. Stem Cell Res. 2010, 4, 201–213. [Google Scholar] [CrossRef]
- Ben-Ari, M.; Naor, S.; Zeevi-Levin, N.; Schick, R.; Ben Jehuda, R.; Reiter, I.; Raveh, A.; Grijnevitch, I.; Barak, O.; Rosen, M.R.; et al. Developmental changes in electrophysiological characteristics of human-induced pluripotent stem cell-derived cardiomyocytes. Heart Rhythm. 2016, 13, 2379–2387. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Mengarelli, I.; Lodder, E.M.; Kosmidis, G.; Bellin, M.; Zhang, M.; Dittmann, S.; Guan, K.; Wilde, A.A.M.; Schulze-Bahr, E.; et al. Switch from Fetal to Adult SCN5A Isoform in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Unmasks the Cellular Phenotype of a Conduction Disease-Causing Mutation. J. Am. Heart Assoc. 2017, 6, e005135. [Google Scholar] [CrossRef] [PubMed]
- Schwach, V.; Verkerk, A.O.; Mol, M.; Monshouwer-Kloots, J.J.; Devalla, H.D.; Orlova, V.V.; Anastassiadis, K.; Mummery, C.L.; Davis, R.P.; Passier, R. A COUP-TFII Human Embryonic Stem Cell Reporter Line to Identify and Select Atrial Cardiomyocytes. Stem Cell Rep. 2017, 9, 1765–1779. [Google Scholar] [CrossRef] [PubMed]
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Chuva de Sousa Lopes, S.M.; Mummery, C.L.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410. [Google Scholar] [CrossRef]
- Argenziano, M.; Lambers, E.; Hong, L.; Sridhar, A.; Zhang, M.; Chalazan, B.; Menon, A.; Savio-Galimberti, E.; Wu, J.C.; Rehman, J.; et al. Electrophysiologic Characterization of Calcium Handling in Human Induced Pluripotent Stem Cell-Derived Atrial Cardiomyocytes. Stem Cell Rep. 2018, 10, 1867–1878. [Google Scholar] [CrossRef]
- Elliott, D.A.; Braam, S.R.; Koutsis, K.; Ng, E.S.; Jenny, R.; Lagerqvist, E.L.; Biben, C.; Hatzistavrou, T.; Hirst, C.E.; Yu, Q.C.; et al. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat. Methods 2011, 8, 1037–1040. [Google Scholar] [CrossRef]
- Doss, M.X.; Di Diego, J.M.; Goodrow, R.J.; Wu, Y.; Cordeiro, J.M.; Nesterenko, V.V.; Barajas-Martínez, H.; Hu, D.; Urrutia, J.; Desai, M.; et al. Maximum Diastolic Potential of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Depends Critically on IKr. PLoS ONE 2012, 7, e40288. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Markandeya, Y.S.; Kamp, T.J.; Makielski, J.C.; January, C.T.; Eckhardt, L.L. IK1-enhanced human-induced pluripotent stem cell-derived cardiomyocytes: An improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1611–H1621. [Google Scholar] [CrossRef]
- Meijer van Putten, R.M.E.; Mengarelli, I.; Guan, K.; Zegers, J.G.; van Ginneken, A.C.G.; Verkerk, A.O.; Wilders, R. Ion channelopathies in human induced pluripotent stem cell derived cardiomyocytes: A dynamic clamp study with virtual IK1. Front. Physiol. 2015, 6, 7. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Veerman, C.C.; Zegers, J.G.; Mengarelli, I.; Bezzina, C.R.; Wilders, R. Patch-Clamp Recording from Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Improving Action Potential Characteristics through Dynamic Clamp. Int. J. Mol. Sci. 2017, 18, 1873. [Google Scholar] [CrossRef]
- Zeng, H.; Wang, J.; Clouse, H.; Lagrutta, A.; Sannajust, F. Human-induced pluripotent stem cell-derived cardiomyocytes have limited IKs for repolarization reserve as revealed by specific KCNQ1/KCNE1 blocker. JRSM Cardiovasc. Dis. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Braam, S.R.; Tertoolen, L.; Casini, S.; Matsa, E.; Lu, H.R.; Teisman, A.; Passier, R.; Denning, C.; Gallacher, D.J.; Towart, R.; et al. Repolarization reserve determines drug responses in human pluripotent stem cell derived cardiomyocytes. Stem Cell Res. 2013, 10, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Jost, N.; Virág, L.; Comtois, P.; Ordög, B.; Szuts, V.; Seprényi, G.; Bitay, M.; Kohajda, Z.; Koncz, I.; Nagy, N.; et al. Ionic mechanisms limiting cardiac repolarization reserve in humans compared to dogs. J. Physiol. 2013, 591, 4189–4206. [Google Scholar] [CrossRef] [PubMed]
- Mauritz, C.; Schwanke, K.; Reppel, M.; Neef, S.; Katsirntaki, K.; Maier, L.S.; Nguemo, F.; Menke, S.; Haustein, M.; Hescheler, J.; et al. Generation of functional murine cardiac myocytes from induced pluripotent stem cells. Circulation 2008, 118, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Rapoport, S.; Huber, I.; Mizrahi, I.; Zwi-Dantsis, L.; Arbel, G.; Schiller, J.; Gepstein, L. Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS ONE 2011, 6, e18037. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Ng, K.M.; Lai, W.-H.; Chan, Y.C.; Lau, Y.-M.; Lian, Q.; Tse, H.F.; Siu, C.-W. Calcium homeostasis in human induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Rev. Rep. 2011, 7, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Gherghiceanu, M.; Barad, L.; Novak, A.; Reiter, I.; Itskovitz-Eldor, J.; Binah, O.; Popescu, L.M. Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: Comparative ultrastructure. J. Cell. Mol. Med. 2011, 15, 2539–2551. [Google Scholar] [CrossRef]
- Lundy, S.D.; Zhu, W.Z.; Regnier, M.; Laflamme, M.A. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013, 22, 1991–2002. [Google Scholar] [CrossRef]
- Cadet, J.S.; Kamp, T.J. A Recipe for T-Tubules in Human iPS Cell-Derived Cardiomyocytes. Circ. Res. 2017, 121, 1294–1295. [Google Scholar] [CrossRef]
- Pioner, J.M.; Guan, X.; Klaiman, J.M.; Racca, A.W.; Pabon, L.; Muskheli, V.; Macadangdang, J.; Ferrantini, C.; Hoopmann, M.R.; Moritz, R.L.; et al. Absence of full-length dystrophin impairs normal maturation and contraction of cardiomyocytes derived from human induced pluripotent stem cells. Cardiovasc. Res. 2019, cvz109. [Google Scholar] [CrossRef]
- Li, Z.; Garnett, C.; Strauss, D.G. Quantitative Systems Pharmacology Models for a New International Cardiac Safety Regulatory Paradigm: An Overview of the Comprehensive In Vitro Proarrhythmia Assay In Silico Modeling Approach. Cpt Pharmacomet. Syst. Pharmacol. 2019, 8, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.K.; Yue, Y.; Bronson, J.; Palmer, C.; Numann, R. Human stem cell-derived cardiomyocytes detect drug-mediated changes in action potentials and ion currents. J. Pharmacol. Toxicol. Methods 2014, 70, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.R.; Whittaker, R.; Price, J.H.; Vega, R.; Pfeiffer, E.R.; Cerignoli, F.; Towart, R.; Gallacher, D.J. High Throughput Measurement of Ca ++ Dynamics in Human Stem Cell-Derived Cardiomyocytes by Kinetic Image Cytometery: A Cardiac Risk Assessment Characterization Using a Large Panel of Cardioactive and Inactive Compounds. Toxicol. Sci. 2015, 148, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Stohlman, J.; Dang, Q.; Strauss, D.G.; Blinova, K. Assessment of Proarrhythmic Potential of Drugs in Optogenetically Paced Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Toxicol. Sci. 2019, 170, 167–179. [Google Scholar] [CrossRef]
- Colatsky, T.; Fermini, B.; Gintant, G.; Pierson, J.B.; Sager, P.; Sekino, Y.; Strauss, D.G.; Stockbridge, N. The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative—Update on progress. J. Pharmacol. Toxicol. Methods 2016, 81, 15–20. [Google Scholar] [CrossRef]
- Weiss, J.N.; Garfinkel, A.; Karagueuzian, H.S.; Chen, P.S.; Qu, Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010, 7, 1891–1899. [Google Scholar] [CrossRef]
- Kussauer, S.; David, R.; Lemcke, H. hiPSCs Derived Cardiac Cells for Drug and Toxicity Screening and Disease Modeling: What Micro- Electrode-Array Analyses Can Tell Us. Cells 2019, 8, 1331. [Google Scholar] [CrossRef]
- Kanda, Y.; Yamazaki, D.; Osada, T.; Yoshinaga, T.; Sawada, K. Development of torsadogenic risk assessment using human induced pluripotent stem cell-derived cardiomyocytes: Japan iPS Cardiac Safety Assessment (JiCSA) update. J. Pharmacol. Sci. 2018, 138, 233–239. [Google Scholar] [CrossRef]
- O’Hara, T.; Virág, L.; Varró, A.; Rudy, Y. Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Comput. Biol. 2011, 7, e1002061. [Google Scholar] [CrossRef]
- Paci, M.; Hyttinen, J.; Aalto-Setälä, K.; Severi, S. Computational models of ventricular- and atrial-like human induced pluripotent stem cell derived cardiomyocytes. Ann. Biomed. Eng. 2013, 41, 2334–2348. [Google Scholar] [CrossRef]
- Passini, E.; Britton, O.J.; Lu, H.R.; Rohrbacher, J.; Hermans, A.N.; Gallacher, D.J.; Greig, R.J.H.; Bueno-Orovio, A.; Rodriguez, B. Human in Silico Drug Trials Demonstrate Higher Accuracy than Animal Models in Predicting Clinical Pro-Arrhythmic Cardiotoxicity. Front. Physiol. 2017, 8, 668. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Pabon, L.; Murry, C.E. Engineering adolescence: Maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 2014, 114, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res. 2014, 103, 530–541. [Google Scholar] [CrossRef]
- Pianezzi, E.; Altomare, C.; Bolis, S.; Balbi, C.; Torre, T.; Rinaldi, A.; Camici, G.G.; Barile, L.; Vassalli, G. Role of somatic cell sources in the maturation degree of human induced pluripotent stem cell-derived cardiomyocytes. Biochim. Biophys. Acta Mol. Cell Res. 2019, 28, 118538. [Google Scholar] [CrossRef]
- Sanchez-Freire, V.; Lee, A.S.; Hu, S.; Abilez, O.J.; Liang, P.; Lan, F.; Huber, B.C.; Ong, S.G.; Hong, W.X.; Huang, M.; et al. Effect of human donor cell source on differentiation and function of cardiac induced pluripotent stem cells. J. Am. Coll. Cardiol. 2014, 64, 436–448. [Google Scholar] [CrossRef]
- Meraviglia, V.; Wen, J.; Piacentini, L.; Campostrini, G.; Wang, C.; Florio, M.C.; Azzimato, V.; Fassina, L.; Langes, M.; Wong, J.; et al. Higher cardiogenic potential of iPSCs derived from cardiac versus skin stromal cells. Front. Biosci. 2016, 21, 719–743. [Google Scholar]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Huo, H.; Loh, Y.-H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef]
- Sheng, X.; Reppel, M.; Nguemo, F.; Mohammad, F.I.; Kuzmenkin, A.; Hescheler, J.; Pfannkuche, K. Human pluripotent stem cell-derived cardiomyocytes: Response to TTX and lidocain reveals strong cell to cell variability. PLoS ONE 2012, 7, e45963. [Google Scholar] [CrossRef]
- Giacomelli, E.; Mummery, C.L.; Bellin, M. Human heart disease: Lessons from human pluripotent stem cell-derived cardiomyocytes. Cell. Mol. Life Sci. 2017, 74, 3711–3739. [Google Scholar] [CrossRef]
- Fatima, A.; Kaifeng, S.; Dittmann, S.; Xu, G.; Gupta, M.K.; Linke, M.; Zechner, U.; Nguemo, F.; Milting, H.; Farr, M.; et al. The disease-specific phenotype in cardiomyocytes derived from induced pluripotent stem cells of two long QT syndrome type 3 patients. PLoS ONE 2013, 8, e83005. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; Gnecchi, M.; Schwartz, P.J. Long QT Syndrome Modelling with Cardiomyocytes Derived from Human-induced Pluripotent Stem Cells. Arrhythm. Electrophysiol. Rev. 2019, 8, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Selga, E.; Sendfeld, F.; Martinez-Moreno, R.; Medine, C.N.; Tura-Ceide, O.; Wilmut, S.I.; Pérez, G.J.; Scornik, F.S.; Brugada, R.; Mills, N.L. Sodium channel current loss of function in induced pluripotent stem cell-derived cardiomyocytes from a Brugada syndrome patient. J. Mol. Cell. Cardiol. 2018, 114, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, Z.; Loh, L.J.; Zhao, Y.; Li, G.; Liew, R.; Islam, O.; Wu, J.; Chung, Y.Y.; Teo, W.S.; et al. Identification of an INa-dependent and Ito-mediated proarrhythmic mechanism in cardiomyocytes derived from pluripotent stem cells of a Brugada syndrome patient. Sci. Rep. 2018, 8, 11246. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Gepstein, A.; Arbel, G.; Caspi, O.; Miller, L.; Belhassen, B.; Nof, E.; Glikson, M.; et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J. Am. Coll. Cardiol. 2012, 60, 990–1000. [Google Scholar] [CrossRef]
- Zhang, X.H.; Haviland, S.; Wei, H.; Sarić, T.; Fatima, A.; Hescheler, J.; Cleemann, L.; Morad, M. Ca2+ signaling in human induced pluripotent stem cell-derived cardiomyocytes (iPS-CM) from normal and catecholaminergic polymorphic ventricular tachycardia (CPVT)-afflicted subjects. Cell Calcium 2013, 54, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Marczenke, M.; Fell, J.; Piccini, I.; Röpke, A.; Seebohm, G.; Greber, B. Generation and cardiac subtype-specific differentiation of PITX2-deficient human iPS cell lines for exploring familial atrial fibrillation. Stem Cell Res. 2017, 21, 26–28. [Google Scholar] [CrossRef]
- Marczenke, M.; Piccini, I.; Mengarelli, I.; Fell, J.; Röpke, A.; Seebohm, G.; Verkerk, A.O.; Greber, B. Cardiac Subtype-Specific Modeling of Kv1.5 Ion Channel Deficiency Using Human Pluripotent Stem Cells. Front. Physiol. 2017, 8, 469. [Google Scholar] [CrossRef]
- Shimko, V.F.; Claycomb, W.C. Effect of mechanical loading on three-dimensional cultures of embryonic stem cell-derived cardiomyocytes. Tissue Eng. Part. A 2008, 14, 49–58. [Google Scholar] [CrossRef]
- Chan, Y.C.; Ting, S.; Lee, Y.K.; Ng, K.M.; Zhang, J.; Chen, Z.; Siu, C.W.; Oh, S.K.W.; Tse, H.F. Electrical stimulation promotes maturation of cardiomyocytes derived from human embryonic stem cells. J. Cardiovasc. Transl. Res. 2013, 6, 989–999. [Google Scholar] [CrossRef]
- Kamakura, T.; Makiyama, T.; Sasaki, K.; Yoshida, Y.; Wuriyanghai, Y.; Chen, J.; Hattori, T.; Ohno, S.; Kita, T.; Horie, M.; et al. Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ. J. 2013, 77, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Teixeira, A.; Domian, I.; Serra, M.; Alves, P.M. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci. Rep. 2017, 7, 8590. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, M.D.; Mannhardt, I.; Breckwoldt, K.; Prondzynski, M.; Flenner, F.; Ulmer, B.; Hirt, M.N.; Neuber, C.; Horváth, A.; Kloth, B.; et al. Human iPSC-derived cardiomyocytes cultured in 3D engineered heart tissue show physiological upstroke velocity and sodium current density. Sci. Rep. 2017, 7, 5464. [Google Scholar] [CrossRef]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef]
- Ulmer, B.M.; Stoehr, A.; Schulze, M.L.; Patel, S.; Gucek, M.; Mannhardt, I.; Funcke, S.; Murphy, E.; Eschenhagen, T.; Hansen, A. Contractile Work Contributes to Maturation of Energy Metabolism in hiPSC-Derived Cardiomyocytes. Stem Cell Rep. 2018, 10, 834–847. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lodrini, A.M.; Barile, L.; Rocchetti, M.; Altomare, C. Human Induced Pluripotent Stem Cells Derived from a Cardiac Somatic Source: Insights for an In-Vitro Cardiomyocyte Platform. Int. J. Mol. Sci. 2020, 21, 507. https://doi.org/10.3390/ijms21020507
Lodrini AM, Barile L, Rocchetti M, Altomare C. Human Induced Pluripotent Stem Cells Derived from a Cardiac Somatic Source: Insights for an In-Vitro Cardiomyocyte Platform. International Journal of Molecular Sciences. 2020; 21(2):507. https://doi.org/10.3390/ijms21020507
Chicago/Turabian StyleLodrini, Alessandra Maria, Lucio Barile, Marcella Rocchetti, and Claudia Altomare. 2020. "Human Induced Pluripotent Stem Cells Derived from a Cardiac Somatic Source: Insights for an In-Vitro Cardiomyocyte Platform" International Journal of Molecular Sciences 21, no. 2: 507. https://doi.org/10.3390/ijms21020507
APA StyleLodrini, A. M., Barile, L., Rocchetti, M., & Altomare, C. (2020). Human Induced Pluripotent Stem Cells Derived from a Cardiac Somatic Source: Insights for an In-Vitro Cardiomyocyte Platform. International Journal of Molecular Sciences, 21(2), 507. https://doi.org/10.3390/ijms21020507