Intraarticular Administration Effect of Hydrogen Sulfide on an In Vivo Rat Model of Osteoarthritis

 , and
, and
Abstract
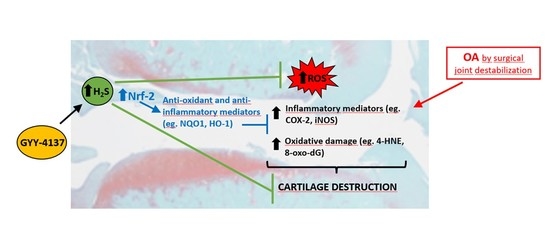
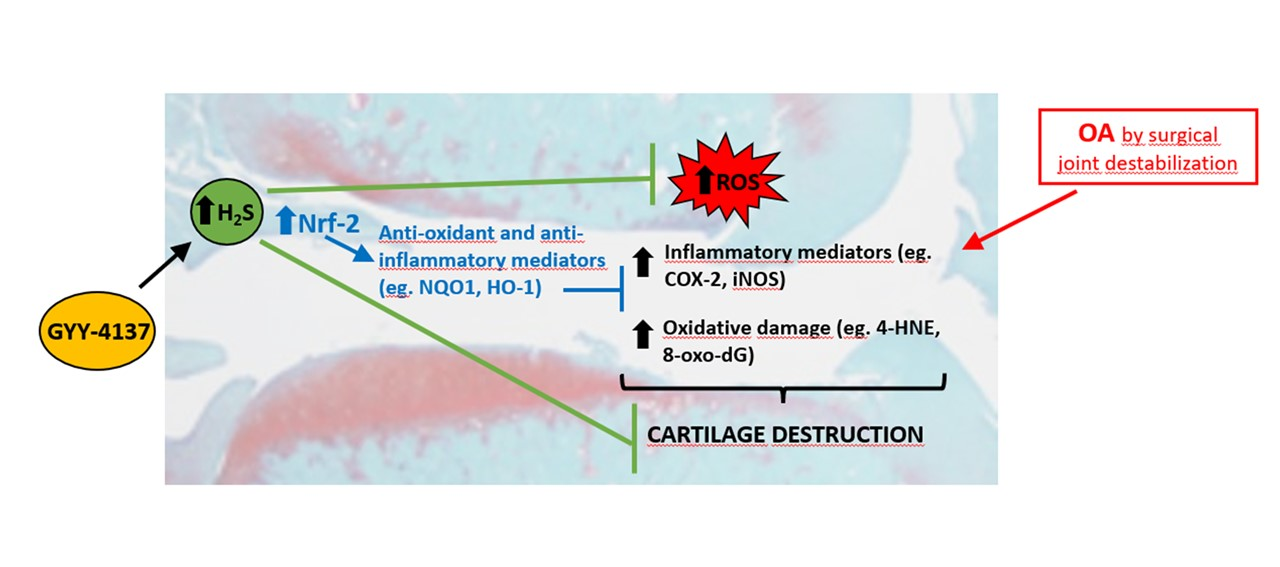
1. Introduction
2. Results
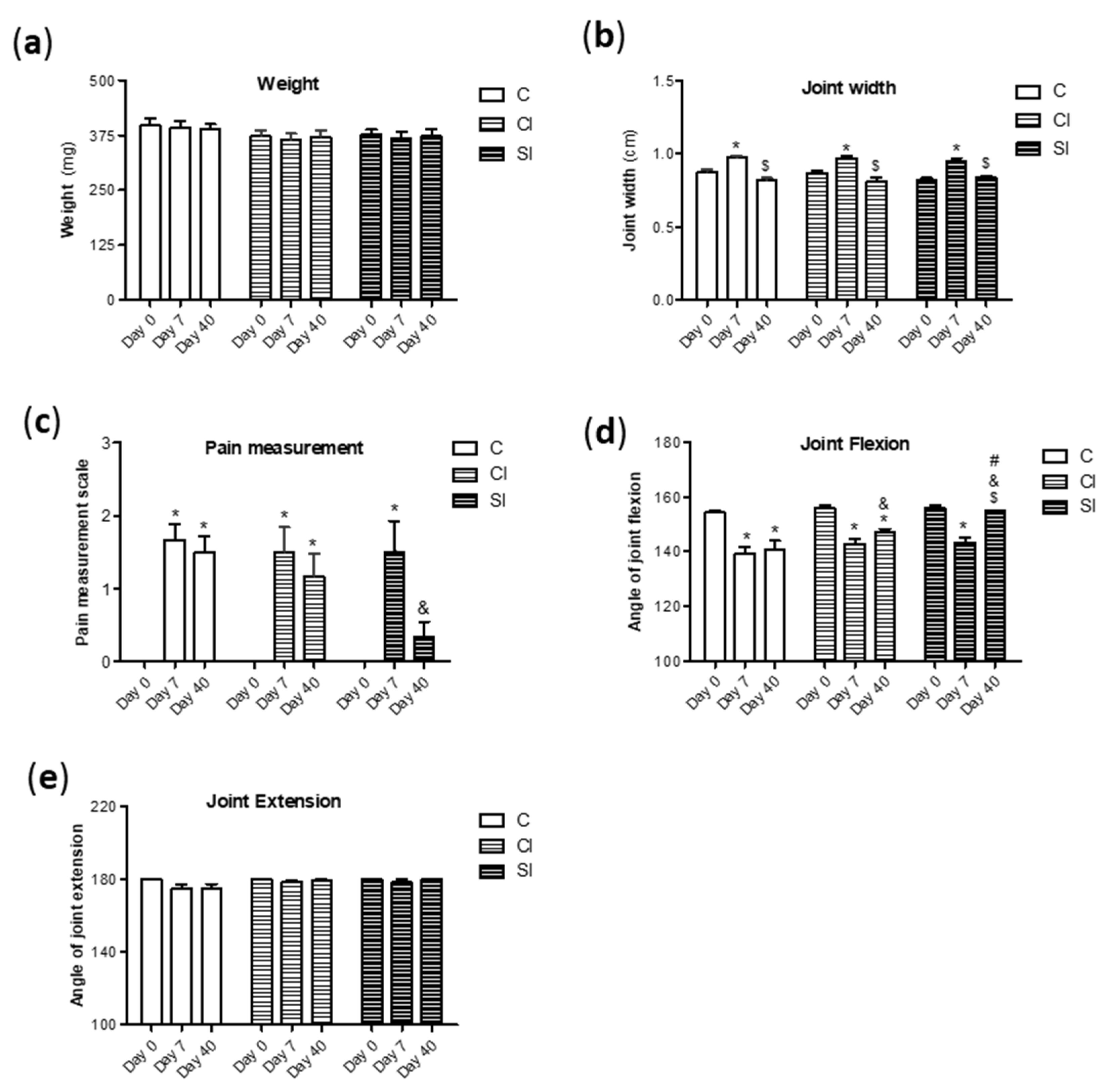
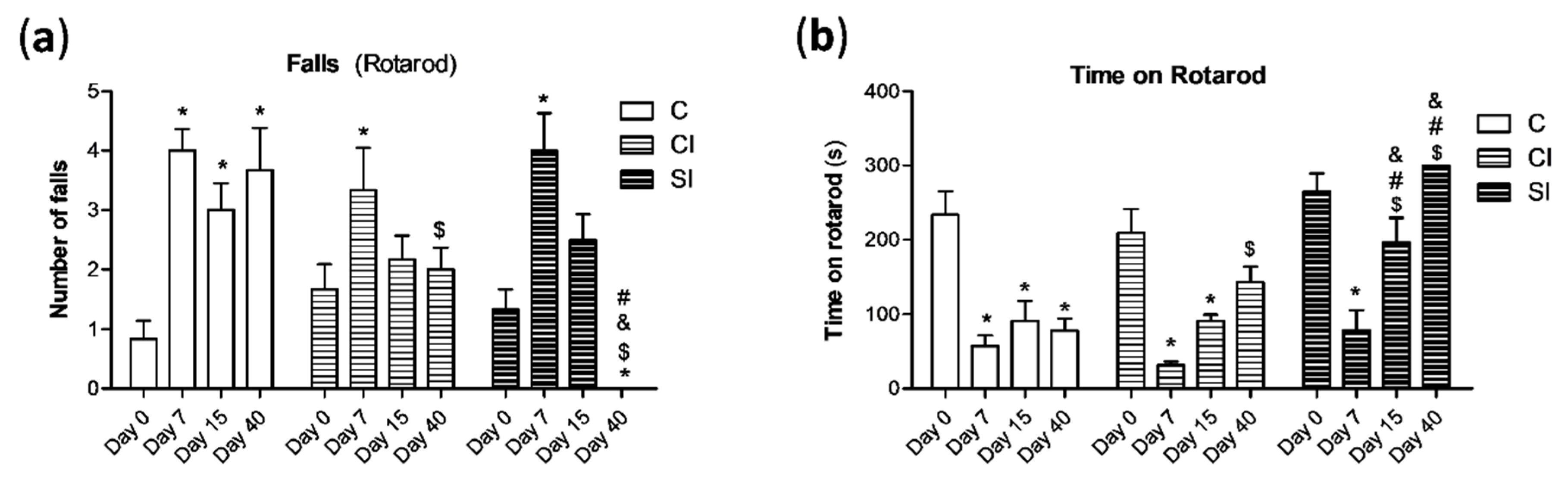
2.1. Intraarticular Administration of H2S Attenuates Joint Pain and Protects against Motor Dysfunction after Experimental OA
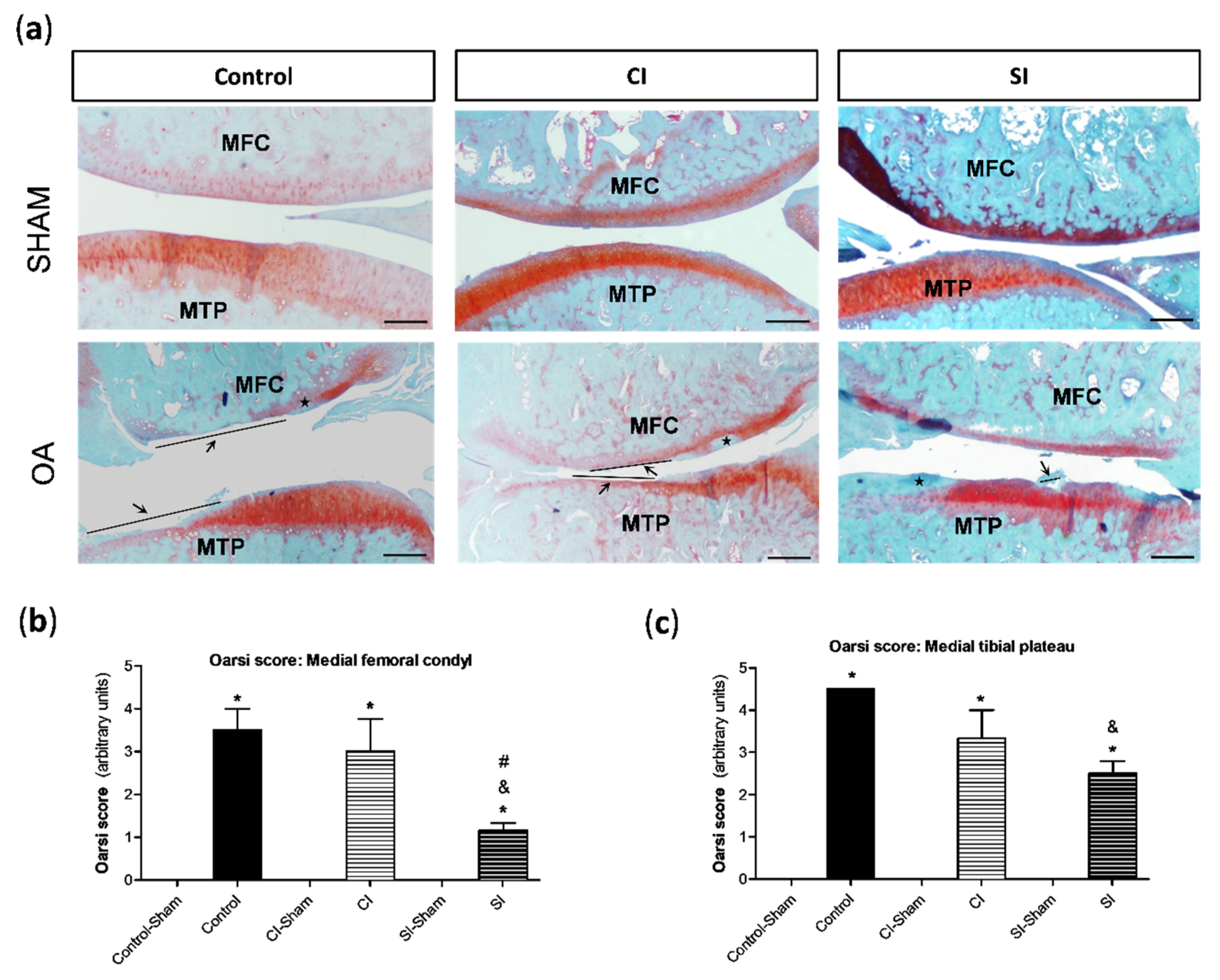
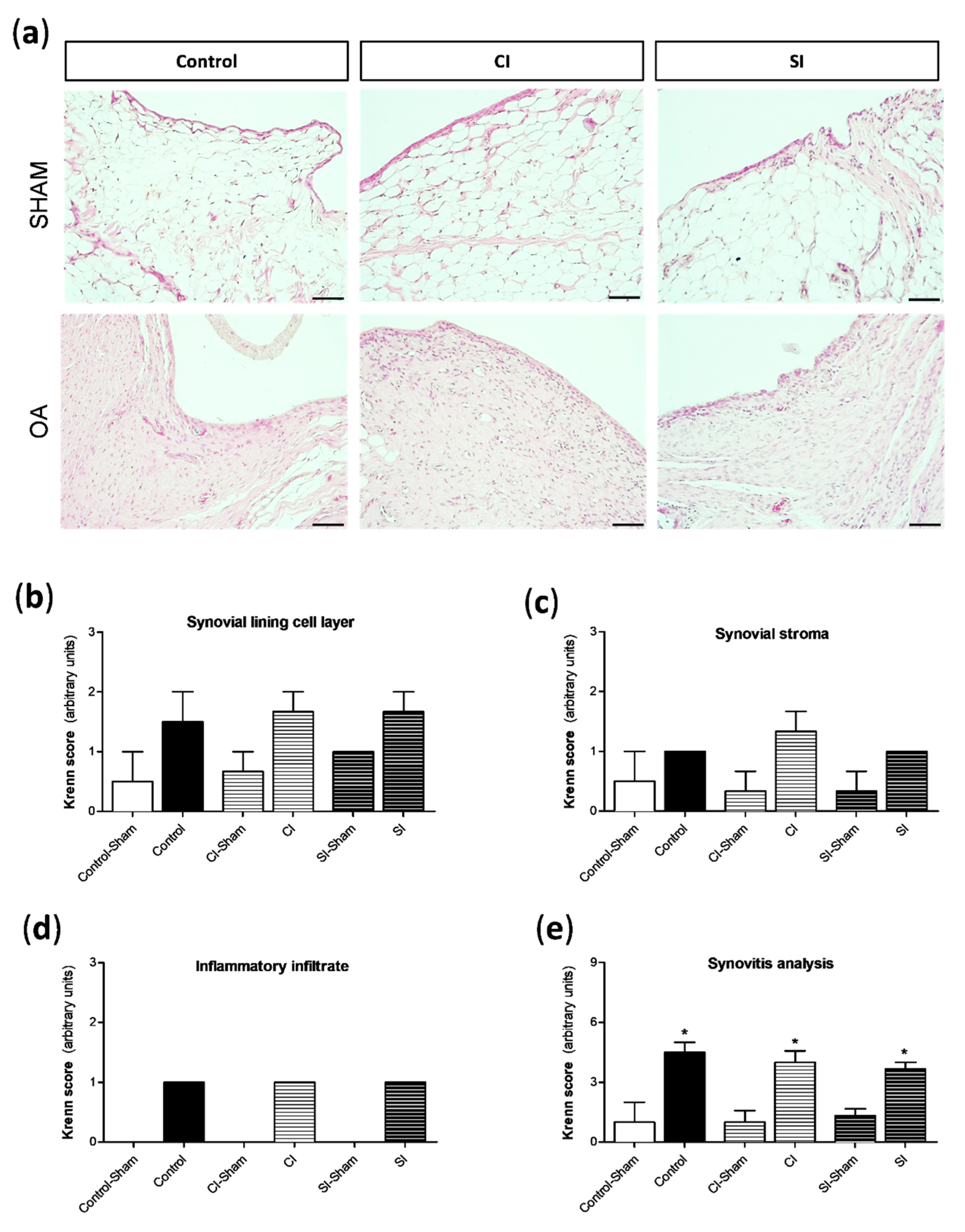
2.2. Intraarticular Administration of H2S Protects against Cartilage Destruction Induced by Joint Surgical Destabilization
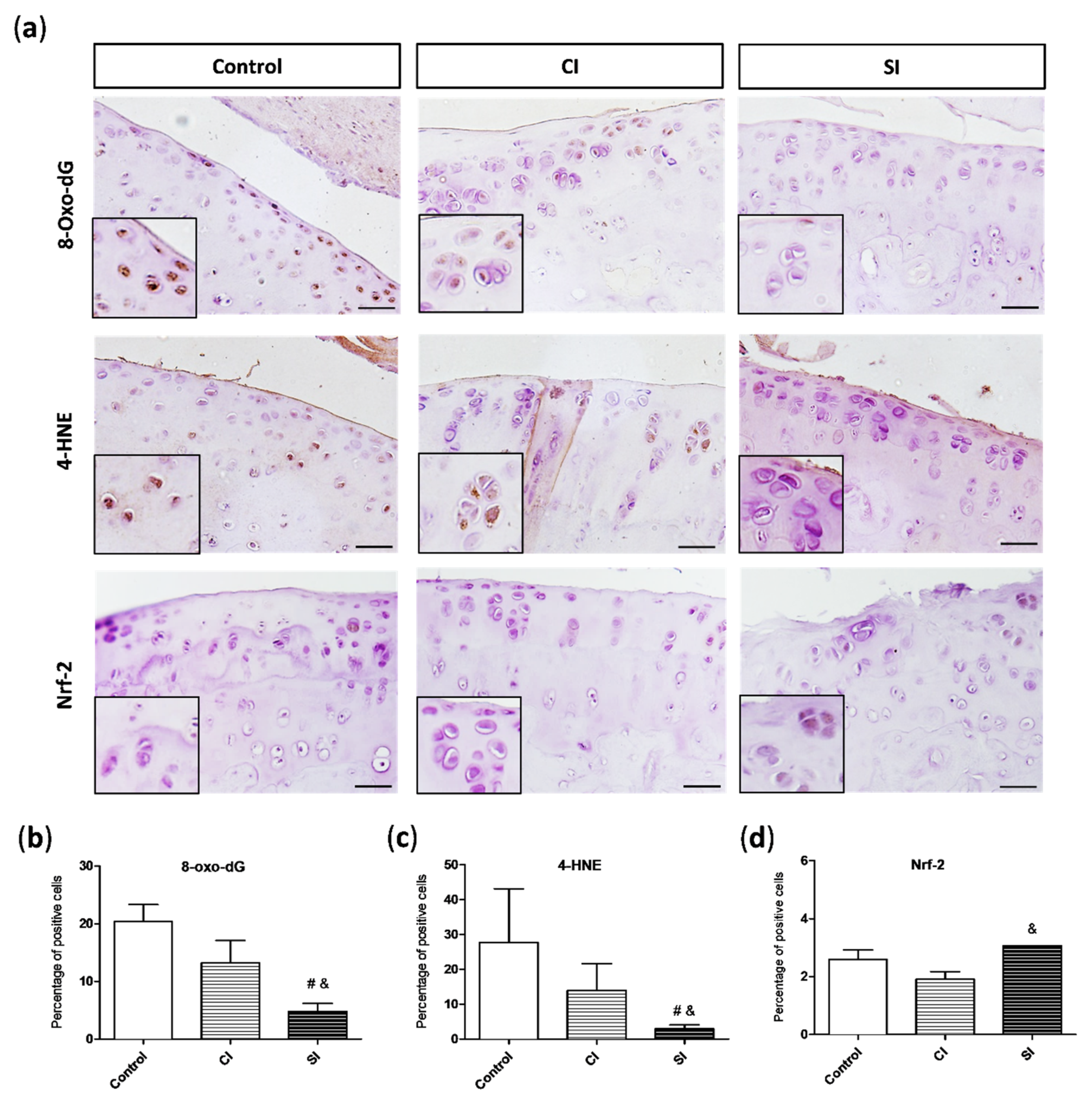
2.3. Oxidative Damage Is Attenuated by Intraarticular Administration of H2S
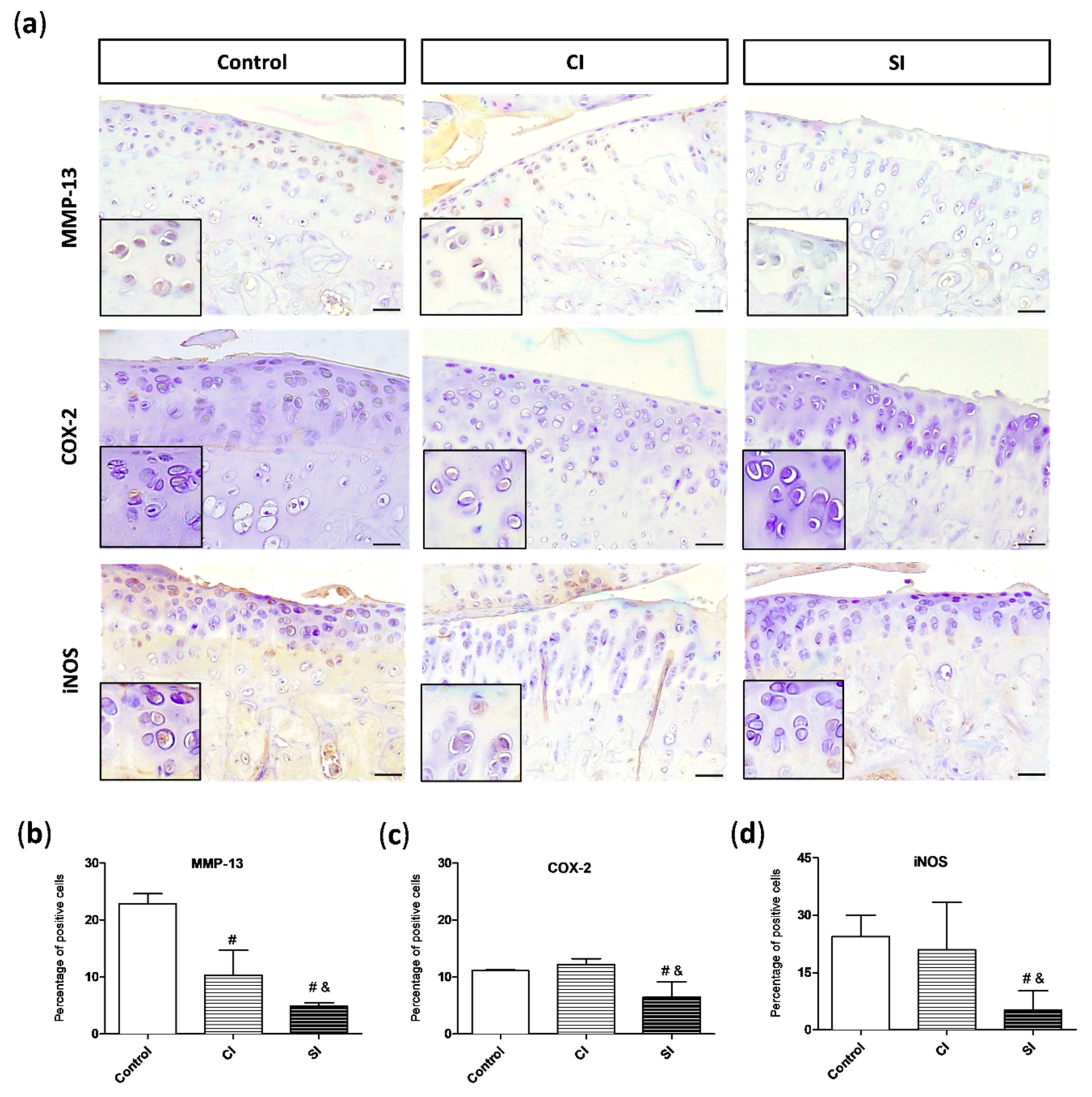
2.4. Intraarticular Injection of H2S Reduces Levels of Pro-catabolic Mediators in the Cartilage
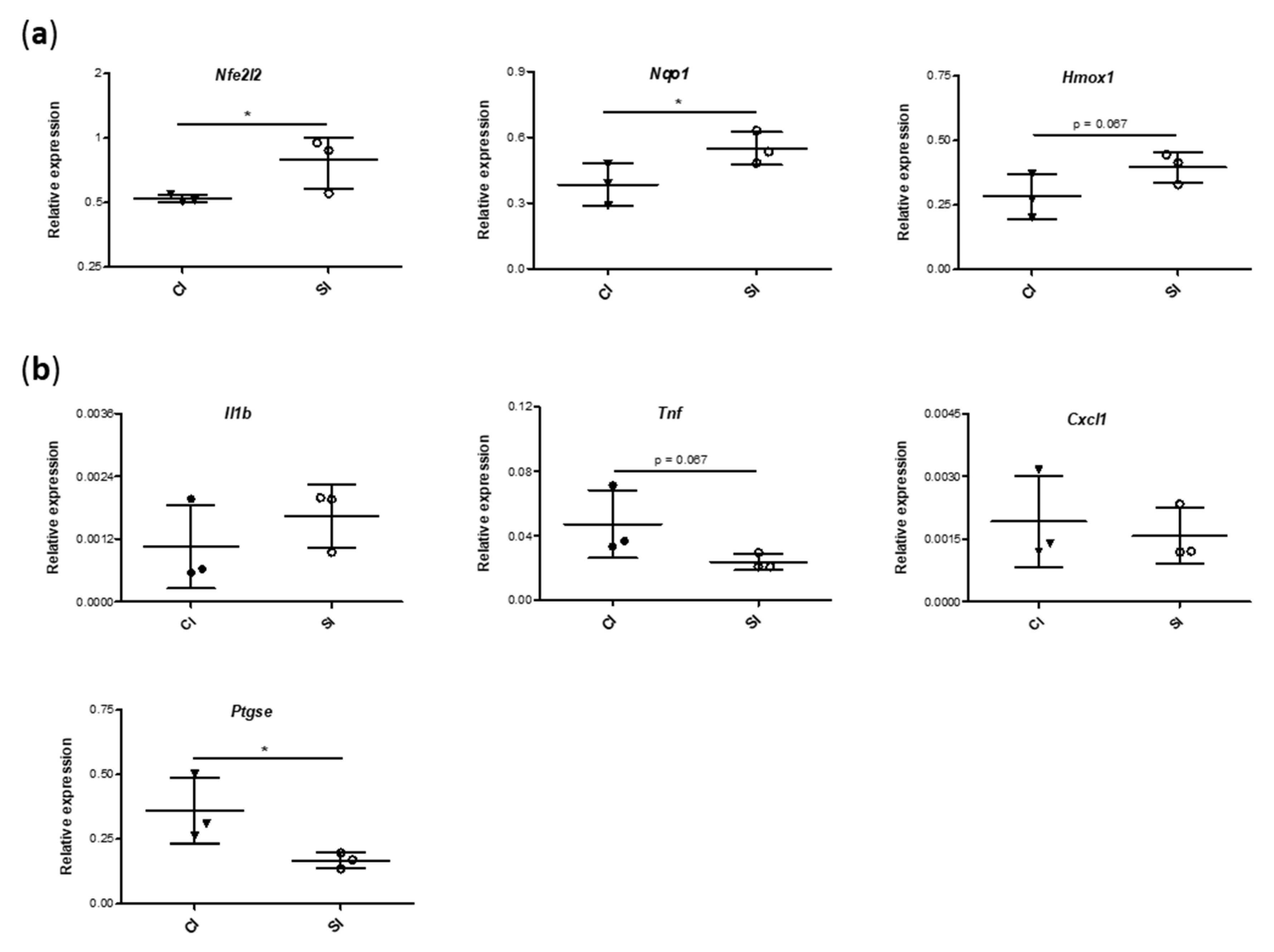
2.5. Blood Gene Expression of Mediators Involved in Antioxidant and Inflammatory Responses Is Modulated by the Intraarticular Administration of H2S
3. Discussion
4. Materials and Methods
4.1. Experimental Osteoarthritis in Rats
4.2. Macroscopic and Clinical Evaluation
4.3. Rotarod Performance Test
4.4. Histological Analysis
4.5. Immunohistochemistry
4.6. RNA Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| OA | osteoarthritis |
| H2S | hydrogen sulfide |
| Nrf-2 | nuclear factor-erythroid 2-related factor 2 |
| PGs | prostaglandins |
| MMPs | metalloproteinases |
| COX-2 | inducible isoform of cyclooxygenase |
| ROS | reactive oxygen species |
| NO | nitric oxide |
| iNOS | inducible NO synthase |
| ECM | extracellular matrix |
| C | group under non-treatment |
| CI | group treated with intraarticular injection of saline |
| SI | group treated with intraarticular injection of H2S donor |
| MTP | medial compartment from tibial plateau |
| MFC | medial compartment from femoral condyl |
| 8-oxo-dG | 8-Hydroxy-2′-deoxyguanosine |
| 4-HNE | 4-hidroxi-2-nonenal |
| Nfe2l2 | nrf-2 gene |
| Nqo1 | NADPH:quinone oxidoreductase gene |
| Hmox1 | heme oxygenase-1 gene |
| Tnf | tumor necrosis factor-α gene |
| Ptgse | COX-2 gene |
| Il1b | interleukin-1β gene |
| Cxcl1 | cytokine-induced neutrophil chemoattractant-1 gene |
| 3-MPST | 3-Mercaptopyruvate sulfurtransferase |
| CSE | cystathionine γ-lyase |
| CBS | cystathionine β-synthetase |
| MIA | monosodium iodoacetate |
| NaSH | sodium hydrosulfide |
| TNF-α | tumor necrosis factor-α |
| NF-κB | nuclear factor-κB |
| NQO1 | NADPH:quinone oxidoreductase |
| HO-1 | heme oxygenase-1 |
| SEM | standard error of the mean |
References
- Kraus, V.B.; Blanco, F.J.; Englund, M.; Karsdal, M.A.; Lohmander, L.S. Call for standardized definitions of osteoarthritis and risk stratification for clinical trials and clinical use. Osteoarthr. Cartil. 2015, 23, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Bultink, I.E.; Vis, M.; Van der Horst-Bruinsma, I.E.; Lems, W.F. Inflammatory rheumatic disorders and bone. Curr. Rheumatol. Rep. 2012, 14, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Vaamonde-García, C.; López-Armada, M.J. Role of mitochondrial dysfunction on rheumatic diseases. Biochem. Pharmacol. 2019, 165, 181–195. [Google Scholar] [CrossRef]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta 2016, 1862, 576–591. [Google Scholar] [CrossRef]
- Amin, A.R.; Dave, M.; Attur, M.; Abramson, S.B. COX-2, NO, and cartilage damage and repair. Curr. Rheumatol. Rep. 2000, 2, 447–453. [Google Scholar] [CrossRef]
- Bar-Or, D.; Rael, L.T.; Thomas, G.W.; Brody, E.N. Inflammatory Pathways in Knee Osteoarthritis: Potential Targets for Treatment. Curr. Rheumatol. Rev. 2015, 11, 50–58. [Google Scholar] [CrossRef]
- Biniecka, M.; Fox, E.; Gao, W.; Ng, C.T.; Veale, D.J.; Fearon, U.; O’Sullivan, J. Hypoxia induces mitochondrial mutagenesis and dysfunction in inflammatory arthritis. Arthritis. Rheum. 2011, 63, 2172–2182. [Google Scholar] [CrossRef]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef]
- Abramson, S.B. Osteoarthritis and nitric oxide. Osteoarthr. Cartil. 2008, 16 (Suppl. 2), S15–S20. [Google Scholar] [CrossRef]
- Needleman, P.; Manning, P.T. Interactions between the inducible cyclooxygenase (COX-2) and nitric oxide synthase (iNOS) pathways: Implications for therapeutic intervention in osteoarthritis. Osteoarthr. Cartil. 1999, 7, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Lacey, J.E.; Grabowski, P.S.; Skerry, T.M. Nitric oxide activates matrix metalloproteinases indirectly in human articular chondrocytes. Int. J. Exp. Pathol. 2000, 81, A17. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Q. Salidroside attenuates interleukin-1β-induced inflammation in human osteoarthritis chondrocytes. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chanalaris, A.; Troeberg, L. ADAMTS and ADAM metalloproteinases in osteoarthritis—looking beyond the ’usual suspects’. Osteoarthr. Cartil. 2017, 25, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Okano, H.; Miyagawa, W.; Visse, R.; Shitomi, Y.; Santamaria, S.; Dudhia, J.; Troeberg, L.; Strickland, D.K.; Hirohata, S.; et al. MMP-13 is constitutively produced in human chondrocytes and co-endocytosed with ADAMTS-5 and TIMP-3 by the endocytic receptor LRP1. Matrix Biol. 2016, 56, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Waldron, A.L.; Schroder, P.A.; Bourgon, K.L.; Bolduc, J.K.; Miller, J.L.; Pellegrini, A.D.; Dubois, A.L.; Blaszkiewicz, M.; Townsend, K.L.; Rieger, S. Oxidative stress-dependent MMP-13 activity underlies glucose neurotoxicity. J. Diabetes Complicat. 2018, 32, 249–257. [Google Scholar] [CrossRef]
- Young, D.A.; Barter, M.J.; Wilkinson, D.J. Recent advances in understanding the regulation of metalloproteinases. F1000Res 2019, 8. [Google Scholar] [CrossRef]
- Wu, Y.; Goh, E.L.; Wang, D.; Ma, S. Novel treatments for osteoarthritis: An update. Open Access Rheumatol. 2018, 10, 135–140. [Google Scholar] [CrossRef]
- Burguera, E.F.; Vela-Anero, A.; Magalhães, J.; Meijide-Faílde, R.; Blanco, F.J. Effect of hydrogen sulfide sources on inflammation and catabolic markers on interleukin 1β-stimulated human articular chondrocytes. Osteoarthr. Cartil. 2014, 22, 1026–1035. [Google Scholar] [CrossRef]
- Vela-Anero, Á.; Hermida-Gómez, T.; Gato-Calvo, L.; Vaamonde-García, C.; Díaz-Prado, S.; Meijide-Faílde, R.; Blanco, F.J.; Burguera, E.F. Long-term effects of hydrogen sulfide on the anabolic-catabolic balance of articular cartilage in vitro. Nitric Oxide 2017, 70, 42–50. [Google Scholar] [CrossRef]
- Ha, C.; Tian, S.; Sun, K.; Wang, D.; Lv, J.; Wang, Y. Hydrogen sulfide attenuates IL-1β-induced inflammatory signaling and dysfunction of osteoarthritic chondrocytes. Int. J. Mol. Med. 2015, 35, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.P.; Cao, Y.P.; Wen, L.C.; Chai, W.B.; DU, J.B.; Jin, H.F.; Liu, J.; Yang, X.; Meng, Z.C.; Liu, H.; et al. Hydrogen sulfide in cartilage and its inhibitory effect on matrix metalloproteinase 13 expression in chondrocytes induced by interlukin-1β. Beijing Da Xue Xue Bao Yi Xue Ban 2016, 48, 194–202. [Google Scholar] [PubMed]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants (Basel) 2018, 7, 129. [Google Scholar] [CrossRef]
- Burguera, E.F.; Vela-Anero, Á.; Gato-Calvo, L.; Vaamonde-García, C.; Meijide-Faílde, R.; Blanco, F.J. Hydrogen sulfide biosynthesis is impaired in the osteoarthritic joint. Int. J. Biometeorol. 2019. [Google Scholar] [CrossRef]
- Fox, B.; Schantz, J.T.; Haigh, R.; Wood, M.E.; Moore, P.K.; Viner, N.; Spencer, J.P.; Winyard, P.G.; Whiteman, M. Inducible hydrogen sulfide synthesis in chondrocytes and mesenchymal progenitor cells: Is H2S a novel cytoprotective mediator in the inflamed joint? J. Cell. Mol. Med. 2012, 16, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Aytekin, K.; Erhan, S.; Erişgin, Z.; Esenyel, C.Z.; Takır, S. Intra-articular injection of hydrogen sulfide decreased the progression of gonarthrosis. Can. J. Physiol. Pharmacol. 2019, 97, 47–54. [Google Scholar] [CrossRef]
- Batallé, G.; Cabarga, L.; Pol, O. The Inhibitory Effects of Slow-Releasing Hydrogen Sulfide Donors in the Mechanical Allodynia, Grip Strength Deficits, and Depressive-Like Behaviors Associated with Chronic Osteoarthritis Pain. Antioxidants (Basel) 2019, 9, 31. [Google Scholar] [CrossRef]
- Vaamonde-García, C.; Vela-Anero, Á.; Hermida-Gómez, T.; Fernández-Burguera, E.; Filgueira-Fernández, P.; Goyanes, N.; Blanco, F.J.; Meijide-Faílde, R. Effect of balneotherapy in sulfurous water on an in vivo murine model of osteoarthritis. Int. J. Biometeorol. 2020, 64, 307–318. [Google Scholar] [CrossRef]
- Wallace, J.L.; Vaughan, D.; Dicay, M.; MacNaughton, W.K.; De Nucci, G. Hydrogen Sulfide-Releasing Therapeutics: Translation to the Clinic. Antioxid. Redox Signal. 2018, 28, 1533–1540. [Google Scholar] [CrossRef]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef]
- Abramoff, B.; Caldera, F.E. Osteoarthritis: Pathology, Diagnosis, and Treatment Options. Med. Clin. N. Am. 2020, 104, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Jones, I.A.; Togashi, R.; Wilson, M.L.; Heckmann, N.; Vangsness, C.T. Intra-articular treatment options for knee osteoarthritis. Nat. Rev. Rheumatol. 2019, 15, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Burguera, E.F.; Meijide-Failde, R.; Blanco, F.J. Hydrogen Sulfide and Inflammatory Joint Diseases. Curr. Drug Targets 2017, 18, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Nasi, S.; Ehirchiou, D.; Chatzianastasiou, A.; Nagahara, N.; Papapetropoulos, A.; Bertrand, J.; Cirino, G.; So, A.; Busso, N. The protective role of the 3-mercaptopyruvate sulfurtransferase (3-MST)-hydrogen sulfide (H2S). Arthritis Res. Ther. 2020, 22, 49. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zhao, J.; Tang, C.S.; Geng, B. Effect of synthesized GYY4137, a slowly releasing hydrogen sulfide donor, on cell viability and distribution of hydrogen sulfide in mice. Beijing Da Xue Xue Bao Yi Xue Ban 2010, 42, 493–497. [Google Scholar]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef]
- Nandi, S.S.; Mishra, P.K. H2S and homocysteine control a novel feedback regulation of cystathionine beta synthase and cystathionine gamma lyase in cardiomyocytes. Sci. Rep. 2017, 7, 3639. [Google Scholar] [CrossRef]
- Damba, T.; Zhang, M.; Buist-Homan, M.; Van Goor, H.; Faber, K.N.; Moshage, H. Hydrogen sulfide stimulates activation of hepatic stellate cells through increased cellular bio-energetics. Nitric Oxide 2019, 92, 26–33. [Google Scholar] [CrossRef]
- Perry, M.M.; Hui, C.K.; Whiteman, M.; Wood, M.E.; Adcock, I.; Kirkham, P.; Michaeloudes, C.; Chung, K.F. Hydrogen sulfide inhibits proliferation and release of IL-8 from human airway smooth muscle cells. Am. J. Respir. Cell. Mol. Biol. 2011, 45, 746–752. [Google Scholar] [CrossRef]
- Bar-Or, D.; Rael, L.T.; Brody, E.N. Use of Saline as a Placebo in Intra-articular Injections in Osteoarthritis: Potential Contributions to Nociceptive Pain Relief. Open Rheumatol. J. 2017, 11, 16–22. [Google Scholar] [CrossRef]
- Lucarini, E.; Micheli, L.; Martelli, A.; Testai, L.; Calderone, V.; Ghelardini, C.; Di Cesare Mannelli, L. Efficacy of isothiocyanate-based compounds on different forms of persistent pain. J. Pain Res. 2018, 11, 2905–2913. [Google Scholar] [CrossRef] [PubMed]
- Neogi, T. The epidemiology and impact of pain in osteoarthritis. Osteoarthr. Cartil. 2013, 21, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Pinals, R.S. Mechanisms of joint destruction, pain and disability in osteoarthritis. Drugs 1996, 52 (Suppl. 3), 14–20. [Google Scholar] [CrossRef]
- Li, L.; Fox, B.; Keeble, J.; Salto-Tellez, M.; Winyard, P.G.; Wood, M.E.; Moore, P.K.; Whiteman, M. The complex effects of the slow-releasing hydrogen sulfide donor GYY4137 in a model of acute joint inflammation and in human cartilage cells. J. Cell. Mol. Med. 2013, 17, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Kloesch, B.; Liszt, M.; Broell, J. H2S transiently blocks IL-6 expression in rheumatoid arthritic fibroblast-like synoviocytes and deactivates p44/42 mitogen-activated protein kinase. Cell. Biol. Int. 2010, 34, 477–484. [Google Scholar] [CrossRef]
- Wu, W.J.; Jia, W.W.; Liu, X.H.; Pan, L.L.; Zhang, Q.Y.; Yang, D.; Shen, X.Y.; Liu, L.; Zhu, Y.Z. S-propargyl-cysteine attenuates inflammatory response in rheumatoid arthritis by modulating the Nrf2-ARE signaling pathway. Redox Biol. 2016, 10, 157–167. [Google Scholar] [CrossRef]
- Jackson, M.T.; Moradi, B.; Zaki, S.; Smith, M.M.; McCracken, S.; Smith, S.M.; Jackson, C.J.; Little, C.B. Depletion of protease-activated receptor 2 but not protease-activated receptor 1 may confer protection against osteoarthritis in mice through extracartilaginous mechanisms. Arthritis Rheumatol. 2014, 66, 3337–3348. [Google Scholar] [CrossRef]
- Lieberthal, J.; Sambamurthy, N.; Scanzello, C.R. Inflammation in joint injury and post-traumatic osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1825–1834. [Google Scholar] [CrossRef]
- Herrero-Beaumont, G.; Pérez-Baos, S.; Sánchez-Pernaute, O.; Roman-Blas, J.A.; Lamuedra, A.; Largo, R. Targeting chronic innate inflammatory pathways, the main road to prevention of osteoarthritis progression. Biochem. Pharmacol. 2019, 165, 24–32. [Google Scholar] [CrossRef]
- Krenn, V.; Morawietz, L.; Burmester, G.R.; Kinne, R.W.; Mueller-Ladner, U.; Muller, B.; Haupl, T. Synovitis score: Discrimination between chronic low-grade and high-grade synovitis. Histopathology 2006, 49, 358–364. [Google Scholar] [CrossRef]
- More, A.S.; Kumari, R.R.; Gupta, G.; Lingaraju, M.C.; Balaganur, V.; Pathak, N.N.; Kumar, D.; Sharma, A.K.; Tandan, S.K. Effect of iNOS inhibitor S-methylisothiourea in monosodium iodoacetate-induced osteoathritic pain: Implication for osteoarthritis therapy. Pharmacol. Biochem. Behav. 2013, 103, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Lu, D.; Liu, H.; Xue, E.; Zhang, Y.; Shang, P.; Pan, X. Pharmacological blockade of PCAF ameliorates osteoarthritis development via dual inhibition of TNF-α-driven inflammation and ER stress. EBioMedicine 2019, 50, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Rocha, F.A.; Silva, F.S.; Leite, A.C.; Leite, A.K.; Girão, V.C.; Castro, R.R.; Cunha, F.Q. Tadalafil analgesia in experimental arthritis involves suppression of intra-articular TNF release. Br. J. Pharmacol. 2011, 164, 828–835. [Google Scholar] [CrossRef]
- Dief, A.E.; Mostafa, D.K.; Sharara, G.M.; Zeitoun, T.H. Hydrogen sulfide releasing naproxen offers better anti-inflammatory and chondroprotective effect relative to naproxen in a rat model of zymosan induced arthritis. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1537–1546. [Google Scholar]
- Yurinskaya, M.M.; Krasnov, G.S.; Kulikova, D.A.; Zatsepina, O.G.; Vinokurov, M.G.; Chuvakova, L.N.; Rezvykh, A.P.; Funikov, S.Y.; Morozov, A.V.; Evgen’ev, M.B. H2S counteracts proinflammatory effects of LPS through modulation of multiple pathways in human cells. Inflamm. Res. 2020, 69, 481–495. [Google Scholar] [CrossRef]
- Wallace, J.L.; Wang, R. Hydrogen sulfide-based therapeutics: Exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015, 14, 329–345. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Cai, D.; Yin, S.; Yang, J.; Jiang, Q.; Cao, W. Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Arthritis Res. Ther. 2015, 17, 269. [Google Scholar] [CrossRef]
- Vaamonde-Garcia, C.; Courties, A.; Pigenet, A.; Laiguillon, M.C.; Sautet, A.; Houard, X.; Kerdine-Römer, S.; Meijide, R.; Berenbaum, F.; Sellam, J. The nuclear factor-erythroid 2-related factor/heme oxygenase-1 axis is critical for the inflammatory features of type 2 diabetes-associated osteoarthritis. J. Biol. Chem. 2017, 292, 14505–14515. [Google Scholar] [CrossRef]
- Vaamonde-García, C.; Loureiro, J.; Valcárcel-Ares, M.N.; Riveiro-Naveira, R.R.; Ramil-Gómez, O.; Hermida-Carballo, L.; Centeno, A.; Meijide-Failde, R.; Blanco, F.J.; López-Armada, M.J. The mitochondrial inhibitor oligomycin induces an inflammatory response in the rat knee joint. BMC Musculoskelet. Disord. 2017, 18, 254. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.M.; Ahmad, I.; Haqqi, T.M. Nrf2/ARE pathway attenuates oxidative and apoptotic response in human osteoarthritis chondrocytes by activating ERK1/2/ELK1-P70S6K-P90RSK signaling axis. Free Radic. Biol. Med. 2018, 116, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, A.; Jafari, D.; Kamarul, T.; Bagheri, A.; Sharifi, A.M. Evaluating the Protective Effects and Mechanisms of Diallyl Disulfide on Interlukin-1β-Induced Oxidative Stress and Mitochondrial Apoptotic Signaling Pathways in Cultured Chondrocytes. J. Cell. Biochem. 2017, 118, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, X.; Li, L.; Wei, G.; Zhao, M. Hydrogen Sulfide Protects against Paraquat-Induced Acute Liver Injury in Rats by Regulating Oxidative Stress, Mitochondrial Function, and Inflammation. Oxid. Med. Cell. Longev. 2020, 2020, 6325378. [Google Scholar] [CrossRef] [PubMed]
- Gerwin, N.; Bendele, A.M.; Glasson, S.; Carlson, C.S. The OARSI histopathology initiative—recommendations for histological assessments of osteoarthritis in the rat. Osteoarthr. Cartil. 2010, 18 (Suppl. 3), S24–S34. [Google Scholar] [CrossRef]
- Ekundi-Valentim, E.; Mesquita, F.P.; Santos, K.T.; De Paula, M.A.; Florenzano, J.; Zanoni, C.I.; Rodrigues, L.; De Nucci, G.; Teixeira, S.A.; Ferreira, H.H.; et al. A comparative study on the anti-inflammatory effects of single oral doses of naproxen and its hydrogen sulfide (H2S)-releasing derivative ATB-346 in rats with carrageenan-induced synovitis. Med. Gas Res. 2013, 3, 24. [Google Scholar] [CrossRef]
- Wallace, J.; Buret, A.; Nagy, P.; Muscara, M.; Nucci, G.d. THU0464 phase 2 clinical trial of the Gl safety of a hydrogen sulfide-releasing anti-inflammatory drug (ATB-346). Ann. Rheum. Dis. 2019, 78, 522. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Forward | Reverse |
|---|---|---|
| Interleukin-1β (Il1b) | gctgacagaccccaaaagat | agctggatgctctcatctgg |
| Tumor necrosis factor-α (Tnf) | gcccagaccctcacactc | ccactccagctgctcctct |
| Cytokine-induced neutrophil chemoattractant-1 (Cxcl1) | cacactccaacagagcacca | tgacagcgcagctcattg |
| Cyclooxygenase 2 (Ptgse) | atgacgagcgactgttcca | tcaggtgttgcacgtagtcttc |
| Nuclear factor erythroid-derived 2-like 2 (Nfe2l2) | acgtgatgaggatgggaaac | gctcttgggaacaaggaaca |
| Heme oxygenase-1 (Hmox1) | gaaagcttttggggttcctc | gcctctaccgaccacagttc |
| NADPH:quinone oxidoreductase (Nqo1) | ctttctgtgggccatcattt | gaggcccctaatctgacctc |
| Hypoxanthine guanine phosphoribosyltransferase 1 (Hprt1) | gaccggttctgtcatgtcg | acctggttcatcatcactaatac |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaamonde-García, C.; Burguera, E.F.; Vela-Anero, Á.; Hermida-Gómez, T.; Filgueira-Fernández, P.; Fernández-Rodríguez, J.A.; Meijide-Faílde, R.; Blanco, F.J. Intraarticular Administration Effect of Hydrogen Sulfide on an In Vivo Rat Model of Osteoarthritis. Int. J. Mol. Sci. 2020, 21, 7421. https://doi.org/10.3390/ijms21197421
Vaamonde-García C, Burguera EF, Vela-Anero Á, Hermida-Gómez T, Filgueira-Fernández P, Fernández-Rodríguez JA, Meijide-Faílde R, Blanco FJ. Intraarticular Administration Effect of Hydrogen Sulfide on an In Vivo Rat Model of Osteoarthritis. International Journal of Molecular Sciences. 2020; 21(19):7421. https://doi.org/10.3390/ijms21197421
Chicago/Turabian StyleVaamonde-García, Carlos, Elena F. Burguera, Ángela Vela-Anero, Tamara Hermida-Gómez, Purificación Filgueira-Fernández, Jennifer A. Fernández-Rodríguez, Rosa Meijide-Faílde, and Francisco J. Blanco. 2020. "Intraarticular Administration Effect of Hydrogen Sulfide on an In Vivo Rat Model of Osteoarthritis" International Journal of Molecular Sciences 21, no. 19: 7421. https://doi.org/10.3390/ijms21197421
APA StyleVaamonde-García, C., Burguera, E. F., Vela-Anero, Á., Hermida-Gómez, T., Filgueira-Fernández, P., Fernández-Rodríguez, J. A., Meijide-Faílde, R., & Blanco, F. J. (2020). Intraarticular Administration Effect of Hydrogen Sulfide on an In Vivo Rat Model of Osteoarthritis. International Journal of Molecular Sciences, 21(19), 7421. https://doi.org/10.3390/ijms21197421