An Injectable Hyaluronan–Methylcellulose (HAMC) Hydrogel Combined with Wharton’s Jelly-Derived Mesenchymal Stromal Cells (WJ-MSCs) Promotes Degenerative Disc Repair

 ,
, 
 ,
, 

Abstract
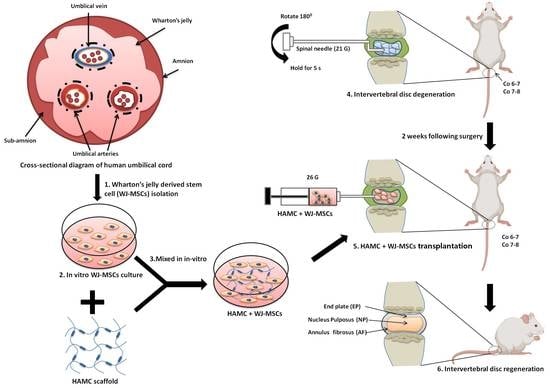
1. Introduction
2. Results
2.1. Quality Control of WJ-MSCs
2.2. HAMC Promotes 3D In Vitro Survival of WJ-MSCs
2.3. HAMC Supports Improved Post-Injection WJ-MSCs Viability Compared to TissueFill
2.4. HAMC and WJ-MSCs Transplantation Restores the Disc Anatomy and Water Content of IVDs Following IVD Degeneration
2.5. HAMC and WJ-MSCs Maintains the Proteoglycan Content and Disc Structure
2.6. HAMC and WJ-MSCs Restore the Matrix Proteins and Downregulate Catabolic Enzymes
2.7. HAMC and WJ-MSCs Inhibit the mRNA Expression of Pro-Inflammatory Cytokines and Matrix-Degrading Proteases
3. Discussion
4. Materials and Methods
4.1. Preparation of Hydrogels
4.2. Preparation and Quality Control of WJ-MSCs
4.3. In Vitro 3D Hydrogel Cell Survival
4.4. In Vitro Post-Injection Cell Viability
4.5. Injury-Induced Disc Degeneration Rat Model
4.6. Experimental Design in a Rat Disc Degeneration Model
4.7. Magnetic Resonance Imaging (MRI)
4.8. RNA Isolation and Real Time RT-PCR
4.9. Safranin-O Staining
4.10. Immunohistochemistry
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IVD | intervertebral disc |
| WJ-MSCs | Wharton’s jelly-derived mesenchymal stromal cell |
| HAMC | hyaluronan (HA) and methyl cellulo |
| MRI | magnetic resonance imaging |
| LBP | low back pain |
| ECMs | extracellular matricess |
| NP | nucleus pulposus |
| AF | annulus fibrosus |
| CNS | central nervous system |
| aCSF | artificial cerebrospinal fluid |
| cNEPs | cortically specified neuroepithelial progenitors |
| CCK-8 | cell counting kit-8 |
| Co | coccygeal |
| IVDD | intervertebral disc degeneration |
| PBS | phosphate buffer saline |
| RT-qPCR | reverse transcriptase polymerase chain reaction |
| iNOS | inducible nitric oxide synthase |
| MMP-13 | matrix metalloproteinase-13 |
| Adamts4 | A disintegrin and metalloproteinase with thrombospondin motifs 4 |
| Cox-2 | Cycloxygenase-2 |
References
- Kumar, H.; Ha, D.-H.; Lee, E.-J.; Park, J.H.; Shim, J.H.; Ahn, T.-K.; Kim, K.-T.; Ropper, A.E.; Sohn, S.; Kim, C.-H. Safety and tolerability of intradiscal implantation of combined autologous adipose-derived mesenchymal stem cells and hyaluronic acid in patients with chronic discogenic low back pain: 1-year follow-up of a phase I study. Stem Cell Res. Ther. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Takeoka, Y.; Yurube, T.; Nishida, K. Gene therapy approach for intervertebral disc degeneration: An update. Neurospine 2020, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Han, I.; Ropper, A.E.; Konya, D.; Kabatas, S.; Toktas, Z.; Aljuboori, Z.; Zeng, X.; Chi, J.H.; Zafonte, R.; Teng, Y.D. Biological approaches to treating intervertebral disk degeneration: Devising stem cell therapies. Cell Transplant. 2015, 24, 2197–2208. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Park, E.-M.; Kim, B.J.; Kim, J.-S.; Choi, B.; Lee, S.-H.; Han, I. Transplantation of human Wharton’s jelly-derived mesenchymal stem cells highly expressing TGFβ receptors in a rabbit model of disc degeneration. Stem Cell Res. Ther. 2015, 6, 190. [Google Scholar] [CrossRef] [PubMed]
- Meisel, H.-J.; Agarwal, N.; Hsieh, P.C.; Skelly, A.; Park, J.-B.; Brodke, D.; Wang, J.C.; Yoon, S.T.; Buser, Z. Cell therapy for treatment of intervertebral disc degeneration: A systematic review. Glob. Spine J. 2019, 9, 39S–52S. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.J.; Corte-Real, J.V.; Ferreira, J.R.; Cunha, C.; Gonçalves, M.; Barbosa, M.A.; Santos, S.G.; Gonçalves, R.M. Macrophages down-regulate gene expression of intervertebral disc degenerative markers under a pro-inflammatory microenvironment. Front. Immunol. 2019, 10, 1508. [Google Scholar] [CrossRef]
- Kraemer, J. Natural course and prognosis of intervertebral disc diseases. International Society for the Study of the Lumbar Spine Seattle, Washington, June 1994. Spine 1995, 20, 635–639. [Google Scholar]
- Antoniou, J.; Steffen, T.; Nelson, F.; Winterbottom, N.; Hollander, A.P.; Poole, R.A.; Aebi, M.; Alini, M. The human lumbar intervertebral disc: Evidence for changes in the biosynthesis and denaturation of the extracellular matrix with growth, maturation, ageing, and degeneration. J. Clin. Investig. 1996, 98, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.W., III; Levicoff, E.A.; Gilbertson, L.G.; Kang, J.D. Biologic modification of animal models of intervertebral disc degeneration. JBJS 2006, 88 Suppl. 2, 83–87. [Google Scholar] [CrossRef]
- Peng, B.G. Pathophysiology, diagnosis, and treatment of discogenic low back pain. World J. Orthop. 2013, 4, 42–52. [Google Scholar] [CrossRef]
- Wang, F.; Cai, F.; Shi, R.; Wang, X.H.; Wu, X.T. Aging and age related stresses: A senescence mechanism of intervertebral disc degeneration. Osteoarthr. Cartil. 2016, 24, 398–408. [Google Scholar] [CrossRef]
- Isa, I.L.M.; Abbah, S.A.; Kilcoyne, M.; Sakai, D.; Dockery, P.; Finn, D.P.; Pandit, A. Implantation of hyaluronic acid hydrogel prevents the pain phenotype in a rat model of intervertebral disc injury. Sci. Adv. 2018, 4, eaaq0597. [Google Scholar]
- Sakai, D.; Andersson, G.B. Stem cell therapy for intervertebral disc regeneration: Obstacles and solutions. Nat. Rev. Rheumatol. 2015, 11, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Nesti, L.J.; Li, W.-J.; Shanti, R.M.; Jiang, Y.J.; Jackson, W.; Freedman, B.A.; Kuklo, T.R.; Giuliani, J.R.; Tuan, R.S. Intervertebral disc tissue engineering using a novel hyaluronic acid–nanofibrous scaffold (HANFS) amalgam. Tissue Eng. Part A 2008, 14, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Nekanti, U.; Rao, V.B.; Bahirvani, A.G.; Jan, M.; Totey, S.; Ta, M. Long-term expansion and pluripotent marker array analysis of Wharton’s jelly-derived mesenchymal stem cells. Stem Cells Dev. 2010, 19, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hou, K.D.; Yuan, M.; Peng, J.; Zhang, L.; Sui, X.; Zhao, B.; Xu, W.; Wang, A.; Lu, S. Characteristics of mesenchymal stem cells derived from Wharton’s jelly of human umbilical cord and for fabrication of non-scaffold tissue-engineered cartilage. J. Biosci. Bioeng. 2014, 117, 229–235. [Google Scholar] [CrossRef]
- Mallis, P.; Michalopoulos, E.; Chatzistamatiou, T.; Stavropoulos-Giokas, C. Mesenchymal stromal cells as potential immunomodulatory players in severe acute respiratory distress syndrome induced by SARS-CoV-2 infection. World J. Stem Cells 2020, 12, 731–751. [Google Scholar] [CrossRef]
- K Batsali, A.; Kastrinaki, M.-C.; A Papadaki, H.; Pontikoglou, C. Mesenchymal stem cells derived from Wharton’s Jelly of the umbilical cord: Biological properties and emerging clinical applications. Curr. Stem Cell Res. Ther. 2013, 8, 144–155. [Google Scholar] [CrossRef]
- Jyothi Prasanna, S.; Sowmya Jahnavi, V. Wharton’s jelly mesenchymal stem cells as off-the-shelf cellular therapeutics: A closer look into their regenerative and immunomodulatory properties. Open Tissue Eng. Regen. Med. J. 2011, 4, 28–38. [Google Scholar] [CrossRef]
- Marino, L.; Castaldi, M.A.; Rosamilio, R.; Ragni, E.; Vitolo, R.; Fulgione, C.; Castaldi, S.G.; Serio, B.; Bianco, R.; Guida, M. Mesenchymal Stem Cells from the Wharton’s Jelly of the Human Umbilical Cord: Biological Properties and Therapeutic Potential. Int. J. Stem Cells 2019, 12, 218. [Google Scholar] [CrossRef]
- Prasanna, S.J.; Gopalakrishnan, D.; Shankar, S.R.; Vasandan, A.B. Pro-inflammatory cytokines, IFNγ and TNFα, influence immune properties of human bone marrow and Wharton jelly mesenchymal stem cells differentially. PLoS ONE 2010, 5, e9016. [Google Scholar] [CrossRef]
- Kim, D.-W.; Staples, M.; Shinozuka, K.; Pantcheva, P.; Kang, S.-D.; Borlongan, C.V. Wharton’s jelly-derived mesenchymal stem cells: Phenotypic characterization and optimizing their therapeutic potential for clinical applications. Int. J. Mol. Sci. 2013, 14, 11692–11712. [Google Scholar] [CrossRef] [PubMed]
- Caicco, M.J.; Zahir, T.; Mothe, A.J.; Ballios, B.G.; Kihm, A.J.; Tator, C.H.; Shoichet, M.S. Characterization of hyaluronan–methylcellulose hydrogels for cell delivery to the injured spinal cord. J. Biomed. Mater. Res. Part A 2013, 101, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.L.; Tuladhar, A.; Obermeyer, J.M.; Varga, B.V.; Teal, C.J.; Morshead, C.M.; Nagy, A.; Shoichet, M.S. Initial cell maturity changes following transplantation in a hyaluronan-based hydrogel and impacts therapeutic success in the stroke-injured rodent brain. Biomaterials 2019, 192, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Mitrousis, N.; Hacibekiroglu, S.; Ho, M.T.; Sauvé, Y.; Nagy, A.; van der Kooy, D.; Shoichet, M.S. Hydrogel-mediated co-transplantation of retinal pigmented epithelium and photoreceptors restores vision in an animal model of advanced retinal degeneration. Biomaterials 2020, 257, 120233. [Google Scholar] [CrossRef]
- Ho, M.T.; Teal, C.J.; Shoichet, M.S. A hyaluronan/methylcellulose-based hydrogel for local cell and biomolecule delivery to the central nervous system. Brain Res. Bull. 2019, 148, 46–54. [Google Scholar] [CrossRef]
- Cooke, M.J.; Wang, Y.; Morshead, C.M.; Shoichet, M.S. Controlled epi-cortical delivery of epidermal growth factor for the stimulation of endogenous neural stem cell proliferation in stroke-injured brain. Biomaterials 2011, 32, 5688–5697. [Google Scholar] [CrossRef]
- Kang, C.E.; Poon, P.C.; Tator, C.H.; Shoichet, M.S. A new paradigm for local and sustained release of therapeutic molecules to the injured spinal cord for neuroprotection and tissue repair. Tissue Eng. Part A 2009, 15, 595–604. [Google Scholar] [CrossRef]
- Ballios, B.G.; Cooke, M.J.; van der Kooy, D.; Shoichet, M.S. A hydrogel-based stem cell delivery system to treat retinal degenerative diseases. Biomaterials 2010, 31, 2555–2564. [Google Scholar] [CrossRef]
- Austin, J.W.; Kang, C.E.; Baumann, M.D.; DiDiodato, L.; Satkunendrarajah, K.; Wilson, J.R.; Stanisz, G.J.; Shoichet, M.S.; Fehlings, M.G. The effects of intrathecal injection of a hyaluronan-based hydrogel on inflammation, scarring and neurobehavioural outcomes in a rat model of severe spinal cord injury associated with arachnoiditis. Biomaterials 2012, 33, 4555–4564. [Google Scholar] [CrossRef]
- Wang, Y.; Lapitsky, Y.; Kang, C.E.; Shoichet, M.S. Accelerated release of a sparingly soluble drug from an injectable hyaluronan–methylcellulose hydrogel. J. Control. Release 2009, 140, 218–223. [Google Scholar] [CrossRef]
- Aguado, B.A.; Mulyasasmita, W.; Su, J.; Lampe, K.J.; Heilshorn, S.C. Improving viability of stem cells during syringe needle flow through the design of hydrogel cell carriers. Tissue Eng. Part A 2012, 18, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.; McGourty, K.; Newport, D. The in vitro inertial positions and viability of cells in suspension under different in vivo flow conditions. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, T.; Nicholls, F.; Modo, M. Intracerebral cell implantation: Preparation and characterization of cell suspensions. Cell Transplant. 2016, 25, 645–664. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, B.; Ghuman, H.; Liu, J.R.; Modo, M. Ex vivo biomechanical characterization of syringe-needle ejections for intracerebral cell delivery. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.L.; Anandakumaran, P.N.; Varga, B.V.; Morshead, C.M.; Nagy, A.; Shoichet, M.S. In vitro maturation of human iPSC-derived neuroepithelial cells influences transplant survival in the stroke-injured rat brain. Tissue Eng. Part A 2018, 24, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Takeoka, Y.; Yurube, T.; Morimoto, K.; Kunii, S.; Kanda, Y.; Tsujimoto, R.; Kawakami, Y.; Fukase, N.; Takemori, T.; Omae, K. Reduced nucleotomy-induced intervertebral disc disruption through spontaneous spheroid formation by the Low Adhesive Scaffold Collagen (LASCol). Biomaterials 2020, 235, 119781. [Google Scholar] [CrossRef] [PubMed]
- Le Maitre, C.L.; Freemont, A.J.; Hoyland, J.A. Accelerated cellular senescence in degenerate intervertebral discs: A possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Res. Ther. 2007, 9, R45. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Huang, D.; Liu, S.; Li, J.; Qing, X.; Shao, Z. Biomaterials-Induced Stem Cells Specific Differentiation Into Intervertebral Disc Lineage Cells. Front. Bioeng. Biotechnol. 2020, 8, 56. [Google Scholar] [CrossRef]
- Yim, R.L.-H.; Lee, J.T.-Y.; Bow, C.H.; Meij, B.; Leung, V.; Cheung, K.M.; Vavken, P.; Samartzis, D. A systematic review of the safety and efficacy of mesenchymal stem cells for disc degeneration: Insights and future directions for regenerative therapeutics. Stem Cells Dev. 2014, 23, 2553–2567. [Google Scholar] [CrossRef]
- Ballios, B.G.; Cooke, M.J.; Donaldson, L.; Coles, B.L.; Morshead, C.M.; van der Kooy, D.; Shoichet, M.S. A hyaluronan-based injectable hydrogel improves the survival and integration of stem cell progeny following transplantation. Stem Cell Rep. 2015, 4, 1031–1045. [Google Scholar] [CrossRef]
- Gupta, D.; Tator, C.H.; Shoichet, M.S. Fast-gelling injectable blend of hyaluronan and methylcellulose for intrathecal, localized delivery to the injured spinal cord. Biomaterials 2006, 27, 2370–2379. [Google Scholar] [CrossRef] [PubMed]
- Kazezian, Z.; Sakai, D.; Pandit, A. Hyaluronic acid microgels modulate inflammation and key matrix molecules toward a regenerative signature in the injured annulus fibrosus. Adv. Biosyst. 2017, 1, 1700077. [Google Scholar] [CrossRef] [PubMed]
- Sobajima, S.; Vadala, G.; Shimer, A.; Kim, J.S.; Gilbertson, L.G.; Kang, J.D. Feasibility of a stem cell therapy for intervertebral disc degeneration. Spine J. 2008, 8, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Mochida, J.; Yamamoto, Y.; Nomura, T.; Okuma, M.; Nishimura, K.; Nakai, T.; Ando, K.; Hotta, T. Transplantation of mesenchymal stem cells embedded in Atelocollagen® gel to the intervertebral disc: A potential therapeutic model for disc degeneration. Biomaterials 2003, 24, 3531–3541. [Google Scholar] [CrossRef]
- Gao, F.; Chiu, S.; Motan, D.; Zhang, Z.; Chen, L.; Ji, H.; Tse, H.; Fu, Q.-L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, e2062. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Tseng, W.-C.; Yang, C.-Y.; Tarng, D.-C. The anti-inflammatory, anti-oxidative, and anti-apoptotic benefits of stem cells in acute ischemic kidney injury. Int. J. Mol. Sci. 2019, 20, 3529. [Google Scholar] [CrossRef]
- Su, J.; Chen, X.; Huang, Y.; Li, W.; Li, J.; Cao, K.; Cao, G.; Zhang, L.; Li, F.; Roberts, A. Phylogenetic distinction of iNOS and IDO function in mesenchymal stem cell-mediated immunosuppression in mammalian species. Cell Death Differ. 2014, 21, 388–396. [Google Scholar] [CrossRef]
- Rowart, P.; Erpicum, P.; Detry, O.; Weekers, L.; Grégoire, C.; Lechanteur, C.; Briquet, A.; Beguin, Y.; Krzesinski, J.-M.; Jouret, F. Mesenchymal stromal cell therapy in ischemia/reperfusion injury. J. Immunol. Res. 2015, 2015, 602597. [Google Scholar] [CrossRef]
- Bianchi, F.; Sala, E.; Donadei, C.; Capelli, I.; La Manna, G. Potential advantages of acute kidney injury management by mesenchymal stem cells. World J. Stem Cells 2014, 6, 644. [Google Scholar] [CrossRef]
- Hua, J.; Liang, C.; Yu, R.; Zhang, M. In The relationship between MRI and histology in a rat model of intervertebral disc degeneration. Proc. Intl. Soc. Mag. Reson. Med 2014, 22, 1253. [Google Scholar]
- Tam, V.; Chan, W.C.W.; Leung, V.Y.L.; Cheah, K.S.E.; Cheung, K.M.C.; Sakai, D.; McCann, M.R.; Bedore, J.; Séguin, C.A.; Chan, D. Histological and reference system for the analysis of mouse intervertebral disc. J. Orthop. Res. 2018, 36, 233–243. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Direction | Sequences |
|---|---|---|
| Adamts4 | Forward | 5′-CGTGGTGTGTGTGTGTGT-3′ |
| Reverse | 5′- AGAGGAAAGTAGGGCAGGT-3′ | |
| Cox-2 | Forward | 5′-TGTATGCTACCATCTGGCTTCGG-3′ |
| Reverse | 5′-GTTTGGAACAFTCGCTCGTCATC-3′ | |
| MMP-13 | Forward | 5′-TGGTCCCTGCCCCTTCCCTA-3′ |
| Reverse | 5′-CCGCAAGAGTCACAGGATGGTAGTA-3′ | |
| iNOS | Forward | 5′-CTGCAGGTCTTTGACGCTCGGAG -3′ |
| Reverse | 5′-GTGGAACACAGGGGTGATGCTCC-3′ | |
| GAPDH | Forward | 5′-CAACTCCCTCAAGATTGTCAGCAA-3′ |
| Reverse | 5′-GGCATGGACTGTGGTCATGA-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, U.Y.; Joshi, H.P.; Payne, S.; Kim, K.T.; Kyung, J.W.; Choi, H.; Cooke, M.J.; Kwon, S.Y.; Roh, E.J.; Sohn, S.; et al. An Injectable Hyaluronan–Methylcellulose (HAMC) Hydrogel Combined with Wharton’s Jelly-Derived Mesenchymal Stromal Cells (WJ-MSCs) Promotes Degenerative Disc Repair. Int. J. Mol. Sci. 2020, 21, 7391. https://doi.org/10.3390/ijms21197391
Choi UY, Joshi HP, Payne S, Kim KT, Kyung JW, Choi H, Cooke MJ, Kwon SY, Roh EJ, Sohn S, et al. An Injectable Hyaluronan–Methylcellulose (HAMC) Hydrogel Combined with Wharton’s Jelly-Derived Mesenchymal Stromal Cells (WJ-MSCs) Promotes Degenerative Disc Repair. International Journal of Molecular Sciences. 2020; 21(19):7391. https://doi.org/10.3390/ijms21197391
Chicago/Turabian StyleChoi, Un Yong, Hari Prasad Joshi, Samantha Payne, Kyoung Tae Kim, Jae Won Kyung, Hyemin Choi, Michael J. Cooke, Su Yeon Kwon, Eun Ji Roh, Seil Sohn, and et al. 2020. "An Injectable Hyaluronan–Methylcellulose (HAMC) Hydrogel Combined with Wharton’s Jelly-Derived Mesenchymal Stromal Cells (WJ-MSCs) Promotes Degenerative Disc Repair" International Journal of Molecular Sciences 21, no. 19: 7391. https://doi.org/10.3390/ijms21197391
APA StyleChoi, U. Y., Joshi, H. P., Payne, S., Kim, K. T., Kyung, J. W., Choi, H., Cooke, M. J., Kwon, S. Y., Roh, E. J., Sohn, S., Shoichet, M. S., & Han, I. (2020). An Injectable Hyaluronan–Methylcellulose (HAMC) Hydrogel Combined with Wharton’s Jelly-Derived Mesenchymal Stromal Cells (WJ-MSCs) Promotes Degenerative Disc Repair. International Journal of Molecular Sciences, 21(19), 7391. https://doi.org/10.3390/ijms21197391