Dendritic Cell and T Cell Crosstalk in Liver Fibrogenesis and Hepatocarcinogenesis: Implications for Prevention and Therapy of Liver Cancer



Abstract
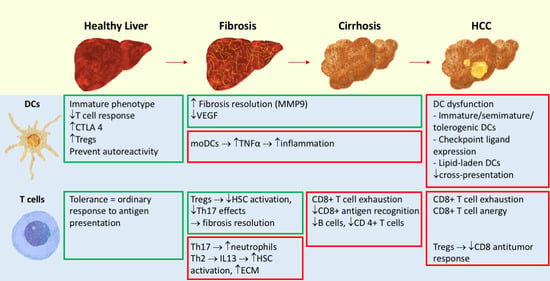
1. Introduction
2. Crosstalk of DCs and T Cells
2.1. DCs in the Liver
2.2. T Cells in the Liver
2.3. DC and T Cell Interaction in the Liver
3. Fibrosis and Cirrhosis
3.1. DCs in Liver Fibrosis
3.2. T Cells in Liver Fibrosis
4. Carcinogenesis and HCC
4.1. DCs in HCC
4.2. Tumor Microenvironment
4.3. Tregs
4.4. Lymphocyte Function in HCC
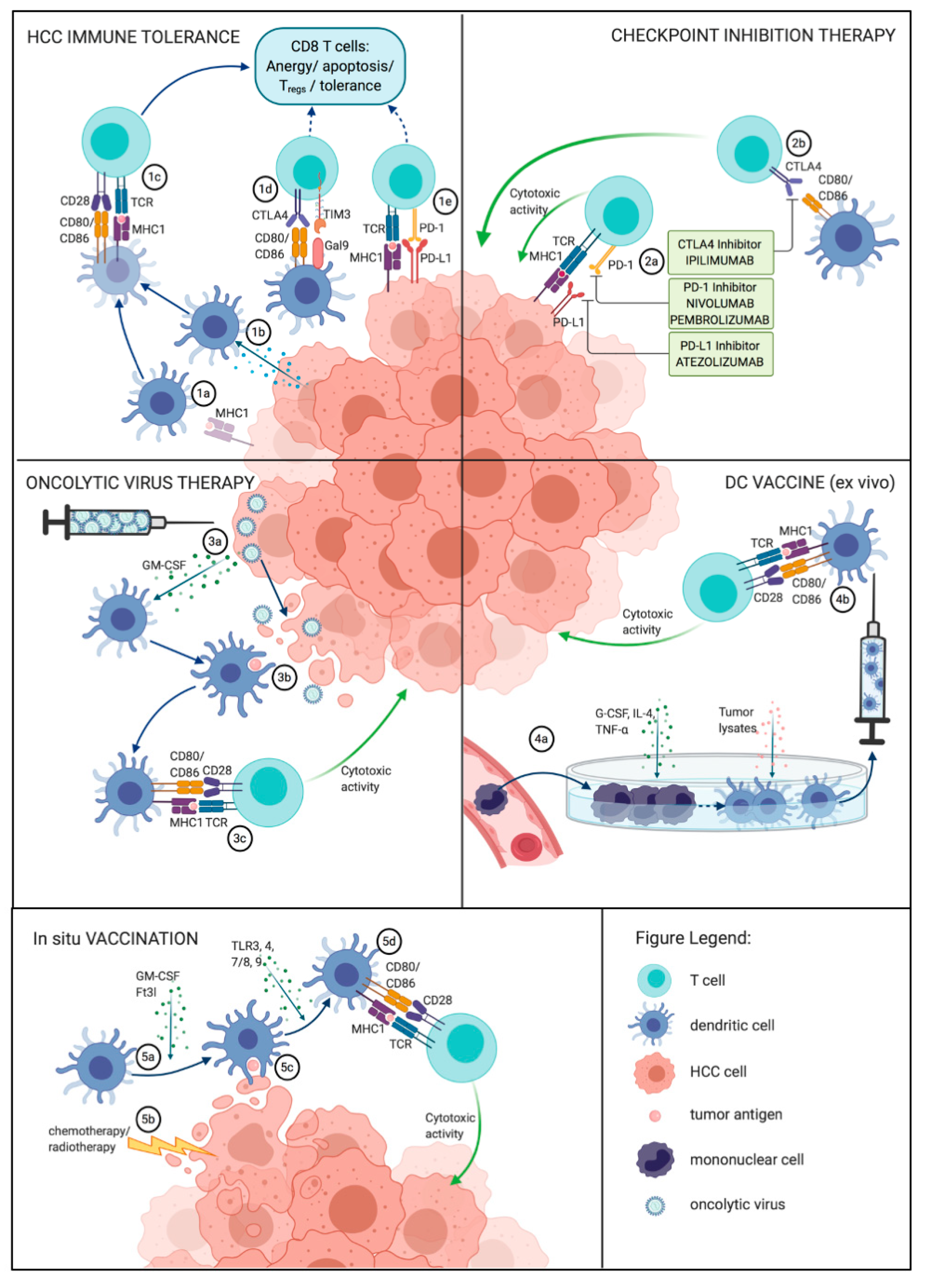
5. Immunological Therapeutic Approaches
5.1. Immune Checkpoint Inhibition
5.2. DC-Based Vaccines
5.3. Oncolytic Immunotherapies
5.4. Combination of Checkpoint Inhibitors
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALD | Alcohol-related liver disease |
| APC | Antigen-presenting cell |
| BDL | Bile duct ligation |
| CAF | Cancer-associated fibroblasts |
| CCL | Chemokine (C-C motif) ligand |
| CCl4 | Carbon tetrachloride |
| CCR | C-C chemokine receptor type |
| cDC | Conventional dendritic cell |
| CTLA | Cytotoxic T-lymphocyte antigen |
| DC | Dendritic cell |
| DFS | Disease-free survival |
| DTR | Diphtheria toxin receptor |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| FDA | Food and Drug Administration |
| Flt3L | Fms-like tyrosine kinase 3 ligand |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GRO-α | Growth-related oncogene α |
| HCC | Hepatocellular carcinoma |
| HLA | Human leukocyte antigen |
| HSC | Hepatic stellate cell |
| ICAM | Intercellular adhesion molecule |
| ICOS-L | Inducible co-stimulatory ligand |
| IDO | Indoleamine 2,3-dioxygenase |
| IL | Interleukin |
| infDC | Inflammatory dendritic cell |
| LAG3 | Lymphocyte-activation gene 3 |
| LPS | Lipopolysaccharides |
| LSEC | Liver sinusoidal cell |
| MCD | Methionine/choline-deficient |
| MDSC | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| MMP | Matrix metalloproteinase |
| moDC | Monocyte-derived dendritic cell |
| NASH | Nonalcoholic steatohepatitis |
| NK | Natural killer |
| OS | Overall survival |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death 1 ligand 1 |
| pDC | Plasmacytoid dendritic cell |
| PDGF | Platelet-derived growth factor |
| ROS | Reactive oxygen species |
| sFlt-1 | Soluble vascular endothelial growth factor receptor 1 |
| TA | Tumor-associated antigen |
| TAP | Transporter associated with antigen processing |
| TGF | Transforming growth factor |
| TAM | Tumor-associated macrophage |
| TIM1 | T cell immunoglobulin and mucin-domain containing-3 |
| TIMP1 | Tissue inhibitor of metalloproteinases |
| TLR | Toll-like receptor |
| TME | Tumor microenvironment |
| TNF | Tumor necrosis factor |
| Tregs | Regulatory T cells |
| VAP1 | Vascular adhesion protein 1 |
| VCAM | Vascular cellular adhesion molecule |
| VEGF | Vascular endothelial growth factor |
References
- Crispe, I.N. Hepatic T cells and liver tolerance. Nat. Rev. Immunol. 2003, 3, 51–62. [Google Scholar] [CrossRef]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Tacke, F. Immunology in the liver—From homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Jeong, W.-I.; Gao, B. Innate immunity and alcoholic liver fibrosis. J. Gastroenterol. Hepatol. 2008, 23, S112–S118. [Google Scholar] [CrossRef]
- O’Beirne, J.P.; Mitchell, J.; Farzaneh, F.; Harrison, P.M. Inhibition of major histocompatibility complex Class I antigen presentation by hepatitis C virus core protein in myeloid dendritic cells. Virology 2009, 389, 1–7. [Google Scholar] [CrossRef][Green Version]
- Fèvre, E.M.; Finucane, M.M.; Flaxman, S.; Flood, L.; Foreman, K.; Forou, M.H.; Fowkes, F.G.R.; Fransen, M.; Freeman, M.K.; Gabbe, B.J.; et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2197–2223. [Google Scholar] [CrossRef]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar] [PubMed]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Bosetti, C.; Turati, F.; La Vecchia, C. Hepatocellular carcinoma epidemiology. Best Pr. Res. Clin. Gastroenterol. 2014, 28, 753–770. [Google Scholar] [CrossRef]
- Lin, S.; Hoffmann, K.; Schemmer, P. Treatment of hepatocellular carcinoma: A systematic review. Liver Cancer 2012, 1, 144–158. [Google Scholar] [CrossRef]
- Lurje, I.; Czigany, Z.; Bednarsch, J.; Roderburg, C.; Isfort, P.; Neumann, U.P.; Lurje, G. Treatment Strategies for Hepatocellular Carcinoma – a Multidisciplinary Approach. Int. J. Mol. Sci. 2019, 20, 1465. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Schwartz, L.; Ricci, S.; Amadori, D.; Santoro, A.; Figer, A.; De Greve, J.; Douillard, J.-Y.; Lathia, C.; Schwartz, B.; et al. Phase II Study of Sorafenib in Patients With Advanced Hepatocellular Carcinoma. J. Clin. Oncol. 2006, 24, 4293–4300. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Yau, T.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Kang, Y.-K.; Hou, M.-M.; Numata, K.; Yeo, W.; Chopra, A.; Ikeda, M.; et al. Nivolumab in advanced hepatocellular carcinoma: Sorafenib-experienced Asian cohort analysis. J. Hepatol. 2019, 71, 543–552. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Ann. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef]
- Tacke, F.; Randolph, G.J. Migratory fate and differentiation of blood monocyte subsets. Immunobiol. 2006, 211, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Jenne, C.N. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef]
- Hilligan, K.L.; Ronchese, F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell. Mol. Immunol. 2020, 17, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Bamboat, Z.M.; Stableford, J.A.; Plitas, G.; Burt, B.M.; Nguyen, H.M.; Welles, A.P.; Gonen, M.; Young, J.W.; DeMatteo, R.P. Human Liver Dendritic Cells Promote T Cell Hyporesponsiveness. J. Immunol. 2009, 182, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-J. IPC: Professional Type 1 Interferon-Producing Cells and Plasmacytoid Dendritic Cell Precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human Inflammatory Dendritic Cells Induce Th17 Cell Differentiation. Immunology 2013, 38, 336–348. [Google Scholar] [CrossRef]
- Dutertre, C.-A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; Hoogen, L.L.V.D.; Leong, J.Y.; Lee, B.; et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunology 2019, 51, 573–589.e8. [Google Scholar] [CrossRef]
- Ibrahim, J.; Nguyen, A.H.; Rehman, A.; Ochi, A.; Jamal, M.; Graffeo, C.S.; Henning, J.R.; Zambirinis, C.P.; Fallon, N.C.; Barilla, R.; et al. Dendritic Cell Populations With Different Concentrations of Lipid Regulate Tolerance and Immunity in Mouse and Human Liver. Gastroenterology 2012, 143, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Manh, T.-P.V.; Bertho, N.; Hosmalin, A.; Schwartz-Cornil, I.; Dalod, M. Investigating Evolutionary Conservation of Dendritic Cell Subset Identity and Functions. Front. Immunol. 2015, 6, 260. [Google Scholar] [CrossRef]
- Nakano, H.; Yanagita, M.; Gunn, M.D. Cd11c+B220+Gr-1+ Cells in Mouse Lymph Nodes and Spleen Display Characteristics of Plasmacytoid Dendritic Cells. J. Exp. Med. 2001, 194, 1171–1178. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ripoche, A.-C.; Matheoud, D.; Nascimbeni, M.; Escriou, N.; Lebon, P.; Heshmati, F.; Guillet, J.-G.; Gannagé, M.; Caillat-Zucman, S.; et al. Antigen Crosspresentation by Human Plasmacytoid Dendritic Cells. Immunology 2007, 27, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Cheong, C.; Matos, I.; Choi, J.-H.; Dandamudi, D.B.; Shrestha, E.; Longhi, M.P.; Jeffrey, K.L.; Anthony, R.M.; Kluger, C.; Nchinda, G.; et al. Microbial Stimulation Fully Differentiates Monocytes to DC-SIGN/CD209+ Dendritic Cells for Immune T Cell Areas. Cell 2010, 143, 416–429. [Google Scholar] [CrossRef]
- Norris, S.; Collins, C.; Doherty, D.G.; Smith, F.; McEntee, G.; Traynor, O.; Nolan, N.; Hegarty, J.; O’Farrelly, C. Resident human hepatitis lymphocytes are phenotypically different from circulating lymphocytes. J. Hepatol. 1998, 28, 84–90. [Google Scholar] [CrossRef]
- McNab, G.; Reeves, J.; Salmi, M.; Hubscher, S.; Jalkanen, S.; Adams, D.; Adams, D.H. Vascular adhesion protein 1 mediates binding of T cells to human hepatic endothelium. Gastroenterol. 1996, 110, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Ezaki, T.; Kudo, S.; Uehara, Y. A life stage of particle-laden rat dendritic cells in vivo: Their terminal division, active phagocytosis, and translocation from the liver to the draining lymph. J. Exp. Med. 1996, 183, 1865–1878. [Google Scholar] [CrossRef]
- Uwatoku, R.; Suematsu, M.; Ezaki, T.; Saiki, T.; Tsuiji, M.; Irimura, T.; Kawada, N.; Suganuma, T.; Naito, M.; Ando, M.; et al. Kupffer cell–mediated recruitment of rat dendritic cells to the liver: Roles of N-acetylgalactosamine–specific sugar receptors. Gastroenterology 2001, 121, 1460–1472. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; Crispe, I.N. Presentation of hepatocellular antigens. Cell. Mol. Immunol. 2016, 13, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Knoll, P.; Schlaak, J.; Uhrig, A.; Kempf, P.; Büschenfelde, K.-H.M.Z.; Gerken, G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J. Hepatol. 1995, 22, 226–229. [Google Scholar] [CrossRef]
- Steinbrink, K.; Graulich, E.; Kubsch, S.; Knop, J.; Enk, A. CD4+ and CD8+ anergic T cells induced by interleukin-10–treated human dendritic cells display antigen-specific suppressor activity. Blood 2002, 99, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Jürgens, B.; Hainz, U.; Fuchs, D.; Felzmann, T.; Heitger, A. Interferon-γ–triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood 2009, 114, 3235–3243. [Google Scholar] [CrossRef]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011, 12, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; Van Rooijen, N.; Ochando, J.C.; Randolph, G.J.; et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 2015, 62, 279–291. [Google Scholar] [CrossRef]
- Wiegand, J.; Berg, T. The Etiology, Diagnosis and Prevention of Liver Cirrhosis. Dtsch. Aerzteblatt Online 2013, 110, 85–91. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; Lario, M.; Álvarez-Mon, M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef]
- Jiao, J.; Sastre, D.; Fiel, M.I.; Lee, U.E.; Ghiassi-Nejad, Z.; Ginhoux, F.; Vivier, E.; Friedman, S.L.; Merad, M.; Aloman, C. Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatoloy 2011, 55, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Lukacs-Kornek, V.; Schuppan, D. Dendritic cells in liver injury and fibrosis: Shortcomings and promises. J. Hepatol. 2013, 59, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Sutti, S.; Bruzzì, S.; Heymann, F.; Liepelt, A.; Krenkel, O.; Toscani, A.; Ramavath, N.N.; Cotella, D.; Albano, E.; Tacke, F. CX3CR1 Mediates the Development of Monocyte-Derived Dendritic Cells during Hepatic Inflammation. Cells 2019, 8, 1099. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Locatelli, I.; Bruzzi’, S.; Jindal, A.; Vacchiano, M.; Bozzola, C.; Albano, E. CX3CR1-expressing inflammatory dendritic cells contribute to the progression of steatohepatitis. Clin. Sci. 2015, 129, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.K.; Bedrosian, A.S.; Clair, J.M.-S.; Mitchell, A.P.; Ibrahim, J.; Stroud, A.; Pachter, H.L.; Bar-Sagi, D.; Frey, A.B.; Miller, G. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J. Clin. Investig. 2009, 119, 3213–3225. [Google Scholar] [CrossRef]
- Heymann, F.; Trautwein, C.; Tacke, F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm. Allergy-Drug Targets 2009, 8, 307–318. [Google Scholar]
- Pradere, J.-P.; Kluwe, J.; De Minicis, S.; Jiao, J.-J.; Gwak, G.-Y.; Dapito, D.H.; Jang, M.-K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef]
- Blois, S.M.; Piccioni, F.; Freitag, N.; Tirado-Gonzalez, I.; Moschansky, P.; Lloyd, R.; Hensel-Wiegel, K.; Rose, M.; Garcia, M.G.; Alaniz, L.; et al. Dendritic cells regulate angiogenesis associated with liver fibrogenesis. Angiogenesis 2013, 17, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Möckel, D.; Baeck, C.; Hittatiya, K.; Eulberg, D.; Luedde, T.; et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Schrammen, P.L.; Möckel, D.; Govaere, O.; Liepelt, A.; Krenkel, O.; Ergen, C.; McCain, M.V.; Eulberg, D.; Luedde, T.; et al. The CCR2+ Macrophage Subset Promotes Pathogenic Angiogenesis for Tumor Vascularization in Fibrotic Livers. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 371–390. [Google Scholar] [CrossRef]
- Gao, B.; Radaeva, S. Natural killer and natural killer T cells in liver fibrosis. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1832, 1061–1069. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. T Cells in Fibrosis and Fibrotic Diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2019, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Inverso, D.; Sironi, L.; Di Lucia, P.; Fioravanti, J.; Ganzer, L.; Fiocchi, A.; Vacca, M.; Aiolfi, R.; Sammicheli, S.; et al. Immunosurveillance of the Liver by Intravascular Effector CD8 + T Cells. Cell 2015, 161, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Kawashima, K.; Sheikh, K.; Tanaka, Y.; Isogawa, M. Intrahepatic Cross-Presentation and Hepatocellular Antigen Presentation Play Distinct Roles in the Induction of Hepatitis B Virus-Specific CD8+ T Cell Responses. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Bénéchet, A.P.; De Simone, G.; Di Lucia, P.; Cilenti, F.; Barbiera, G.; Le Bert, N.; Fumagalli, V.; Lusito, E.; Moalli, F.; Bianchessi, V.; et al. Dynamics and genomic landscape of CD8+ T cells undergoing hepatic priming. Nat. Cell Biol. 2019, 574, 200–205. [Google Scholar] [CrossRef]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Imm. 2004, 4, 583–594. [Google Scholar] [CrossRef]
- Gieseck, R.L.; Ramalingam, T.R.; Hart, K.M.; Vannella, K.M.; Cantu, D.A.; Lu, W.-Y.; Ferreira-González, S.; Forbes, S.J.; Vallier, L.; Wynn, T.A. Interleukin-13 Activates Distinct Cellular Pathways Leading to Ductular Reaction, Steatosis, and Fibrosis. Immunology 2016, 45, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.F.; Cheever, A.W.; Wynn, T.A. IL-10 and the Dangers of Immune Polarization: Excessive Type 1 and Type 2 Cytokine Responses Induce Distinct Forms of Lethal Immunopathology in Murine Schistosomiasis. J. Immunol. 2000, 164, 6406–6416. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, M.G.; Donaldson, D.D.; Cheever, A.W.; Wynn, T.A. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2–dominated inflammatory response. J. Clin. Investig. 1999, 104, 777–785. [Google Scholar] [CrossRef]
- Lee, C.G.; Homer, R.J.; Zhu, Z.; Lanone, S.; Wang, X.; Koteliansky, V.; Shipley, J.M.; Gotwals, P.; Noble, P.; Chen, Q.; et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J. Exp Med. 2001, 194, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Fabre, T.; Molina, M.F.; Soucy, G.; Goulet, J.-P.; Willems, B.; Villeneuve, J.-P.; Bilodeau, M.; Shoukry, N.H. Type 3 cytokines IL-17A and IL-22 drive TGF-β–dependent liver fibrosis. Sci. Immunol. 2018, 3, eaar7754. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, A.; Moreno, C.; Gustot, T.; Maréchal, R.; Degré, D.; Demetter, P.; De Nadai, P.; Geerts, A.; Quertinmont, E.; Vercruysse, V.; et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 2008, 49, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qiu, S.-J.; She, W.-M.; Wang, F.-P.; Gao, H.; Li, L.; Tu, C.-T.; Wang, J.-Y.; Shen, X.-Z.; Jiang, W. Significance of the Balance between Regulatory T (Treg) and T Helper 17 (Th17) Cells during Hepatitis B Virus Related Liver Fibrosis. PLoS ONE 2012, 7, e39307. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Heymann, F.; Tacke, F. Role of IL-17 and Th17 Cells in Liver Diseases. Clin. Dev. Immunol. 2010, 2011, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 Signaling in Inflammatory, Kupffer Cells, and Hepatic Stellate Cells Exacerbates Liver Fibrosis in Mice. Gastroenterology 2012, 143, 765–776.e3. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M. Regulatory T Cells in Autoimmmunity. Annu. Rev. Immunol. 2000, 18, 423–449. [Google Scholar] [CrossRef] [PubMed]
- Breous, E.; Somanathan, S.; Vandenberghe, L.H.; Wilson, J.M. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology 2009, 50, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Feng, M.; Liu, X.; Bai, L.; Kong, M.; Chen, Y.; Zheng, S.; Liu, S.; Wan, Y.-J.Y.; Duan, Z.; et al. Persistence of cirrhosis is maintained by intrahepatic regulatory T cells that inhibit fibrosis resolution by regulating the balance of tissue inhibitors of metalloproteinases and matrix metalloproteinases. Transl. Res. 2016, 169, 67–79.e2. [Google Scholar] [CrossRef]
- Zhang, X.; Lou, J.; Bai, L.; Chen, Y.; Zheng, S.; Duan, Z. Immune Regulation of Intrahepatic Regulatory T Cells in Fibrotic Livers of Mice. Med. Sci. Monit. 2017, 23, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine Receptor CXCR6-Dependent Hepatic NK T Cell Accumulation Promotes Inflammation and Liver Fibrosis. J. Immunol. 2013, 190, 5226–5236. [Google Scholar] [CrossRef] [PubMed]
- Mossanen, J.C.; Kohlhepp, M.; Wehr, A.; Krenkel, O.; Liepelt, A.; Roeth, A.A.; Möckel, D.; Heymann, F.; Lammers, T.; Gassler, N.; et al. CXCR6 Inhibits Hepatocarcinogenesis by Promoting Natural Killer T- and CD4+ T-Cell–Dependent Control of Senescence. Gastroenterology 2019, 156, 1877–1889.e4. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic Activation of Intrahepatic CD8+ T Cells and NKT Cells Causes Nonalcoholic Steatohepatitis and Liver Cancer via Cross-Talk with Hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef]
- Tacke, F.; Yoneyama, H. From NAFLD to NASH to fibrosis to HCC: Role of dendritic cell populations in the liver. Hepatology 2013, 58, 494–496. [Google Scholar] [CrossRef]
- Lai, W.K.; Curbishley, S.M.; Goddard, S.; Alabraba, E.; Shaw, J.; Youster, J.; McKeating, J.; Adams, D.H. Hepatitis C is associated with perturbation of intrahepatic myeloid and plasmacytoid dendritic cell function. J. Hepatol. 2007, 47, 338–347. [Google Scholar] [CrossRef]
- Shin, E.-C.; Sung, P.S.; Park, S. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Bernsmeier, C.; Van Der Merwe, S.; Périanin, A. Innate immune cells in cirrhosis. J. Hepatol. 2020, 73, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Laso, F.J.; Madruga, J.I.; Lopez, A.; Ciudad, J.; Alvarez-Mon, M.; San-Miguel, J.; Orfao, A. Distribution of Peripheral Blood Lymphoid Subsets in Alcoholic Liver Cirrhosis: Influence of Ethanol Intake. Alcohol. Clin. Exp. Res. 1996, 20, 1564–1568. [Google Scholar] [CrossRef]
- Tangye, S.G.; Good, K.L. Human IgM+CD27+B Cells: Memory B Cells or “Memory” B Cells? J. Immunol. 2007, 179, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Iyer, T.K.; Carpenter, E.; Li, H.; Chang, K.-M.; Vonderheide, R.H.; Kaplan, D.E. Dysfunctional B-cell activation in cirrhosis resulting from hepatitis C infection associated with disappearance of CD27-Positive B-cell population. Hepatology 2012, 55, 709–719. [Google Scholar] [CrossRef]
- McGovern, B.H.; Golan, Y.; Lopez, M.; Pratt, D.; Lawton, A.; Moore, G.; Epstein, M.; Knox, T.A. The Impact of Cirrhosis on CD4+ T Cell Counts in HIV-Seronegative Patients. Clin. Infect. Dis. 2007, 44, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Lario, M.; Muñoz, L.; Ubeda, M.; Borrero, M.-J.; Martínez, J.; Monserrat, J.; Diaz, D.; Álvarez-Mon, M.; Albillos, A. Defective thymopoiesis and poor peripheral homeostatic replenishment of T-helper cells cause T-cell lymphopenia in cirrhosis. J. Hepatol. 2013, 59, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.-I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8, 2122. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Tyurin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J.; Celis, E.; et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Imm. 2014, 192, 2920–2931. [Google Scholar]
- Harimoto, H.; Shimizu, M.; Nakagawa, Y.; Nakatsuka, K.; Wakabayashi, A.; Sakamoto, C.; Takahashi, H. Inactivation of tumor-specific CD8+ CTLs by tumor-infiltrating tolerogenic dendritic cells. Imm. Cell Bio. 2013, 91, 545–555. [Google Scholar]
- Dudek, A.M.; Martin, S.; Garg, A.D.; Agostinis, P. Immature, Semi-Mature, and Fully Mature Dendritic Cells: Toward a DC-Cancer Cells Interface That Augments Anticancer Immunity. Front. Immunol. 2013, 4, 438. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Steinman, R.M. Antigen-bearing immature dendritic cells induce peptide-specific CD8+ regulatory T cells in vivo in humans. Blood 2002, 100, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.J.; Boldajipour, B.; Beemiller, P.; Pandurangi, P.; Sorensen, C.; Werb, Z.; Egeblad, M.; Krummel, M.F. Marginating Dendritic Cells of the Tumor Microenvironment Cross-Present Tumor Antigens and Stably Engage Tumor-Specific T Cells. Cancer Cell 2012, 21, 402–417. [Google Scholar] [CrossRef]
- Lundqvist, A.; Palmborg, A.; Pavlenko, M.; Levitskaya, J.; Pisa, P. Mature Dendritic Cells Induce Tumor-Specific Type 1 Regulatory T Cells. J. Immunother. 2005, 28, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Gonzalez, A.; Zhou, G.; Vargas-Mendez, E.; Boor, P.P.; Mancham, S.; Verhoef, C.; Polak, W.G.; Grünhagen, D.; Pan, Q.; La Janssen, H.; et al. Tumor-infiltrating plasmacytoid dendritic cells promote immunosuppression by Tr1 cells in human liver tumors. OncoImmunology 2015, 4, e1008355. [Google Scholar] [CrossRef] [PubMed]
- Gagliani, N.; Magnani, C.F.; Huber, S.; E Gianolini, M.; Pala, M.; Licona-Limon, P.; Guo, B.; Herbert, D.R.; Bulfone, A.; Trentini, F.; et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Sprengers, D.; Boor, P.P.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; De Jonge, J.; Gaspersz, M.; et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119.e10. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Tacke, F. Emerging roles of myeloid derived suppressor cells in hepatic inflammation and fibrosis. World J. Gastrointest. Pathophysiol. 2015, 6, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Poschke, I.; Mao, Y.; Adamson, L.; Salazar-Onfray, F.; Masucci, G.; Kiessling, R. Myeloid-derived suppressor cells impair the quality of dendritic cell vaccines. Cancer Immunol. Immunother. 2011, 61, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Res. 2009, 70, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, A.; Bronte, V.; Visintin, A.; Spitzer, J.H.; Apolloni, E.; Serafini, P.; Zanovello, P.; Segal, D.M. Myeloid Suppressor Lines Inhibit T Cell Responses by an NO-Dependent Mechanism. J. Immunol. 2002, 168, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Krüger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef]
- Gao, Q.; Qiu, S.-J.; Fan, J.; Zhou, J.; Wang, X.-Y.; Xiao, Y.-S.; Xu, Y.; Li, Y.-W.; Tang, Z.-Y. Intratumoral Balance of Regulatory and Cytotoxic T Cells Is Associated With Prognosis of Hepatocellular Carcinoma After Resection. J. Clin. Oncol. 2007, 25, 2586–2593. [Google Scholar] [CrossRef]
- Chen, K.-J.; Lin, S.-Z.; Zhou, L.; Xie, H.-Y.; Zhou, W.-H.; Taki-Eldin, A.; Zheng, S.-S. Selective Recruitment of Regulatory T Cell through CCR6-CCL20 in Hepatocellular Carcinoma Fosters Tumor Progression and Predicts Poor Prognosis. PLoS ONE 2011, 6, e24671. [Google Scholar] [CrossRef]
- Flecken, T.; Schmidt, N.; Hild, S.; Gostick, E.; Drognitz, O.; Zeiser, R.; Schemmer, P.; Bruns, H.; Eiermann, T.; A Price, D.; et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 2014, 59, 1415–1426. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef]
- Han, Y.; Chen, Z.; Yang, Y.; Jiang, Z.; Gu, Y.; Liu, Y.; Lin, C.; Pan, Z.; Yu, Y.; Jiang, M.; et al. Human CD14+CTLA-4+regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 2013, 59, 567–579. [Google Scholar] [CrossRef]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nat. Cell Biol. 1998, 392, 86–89. [Google Scholar] [CrossRef]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.S.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Schreiber, R.D.; et al. Batf3 Deficiency Reveals a Critical Role for CD8 + Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef]
- Pedroza-Gonzalez, A.; Verhoef, C.; Ijzermans, J.N.; Peppelenbosch, M.P.; Kwekkeboom, J.; Verheij, J.; Janssen, H.L.A.; Sprengers, D. Activated tumor-infiltrating CD4+ regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology 2012, 57, 183–194. [Google Scholar] [CrossRef]
- Budhu, A.; Forgues, M.; Ye, Q.-H.; Jia, H.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.-X.; Tang, Z.-Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Tauber, C.; Schultheiss, M.; De Luca, R.; Buettner, N.; Llewellyn-Lacey, S.; Emmerich, F.; Zehe, S.; Price, D.A.; Neumann-Haefelin, C.; Schmitt-Graeff, A.; et al. Inefficient induction of circulating TAA-specific CD8+ T-cell responses in hepatocellular carcinoma. Oncotarget 2019, 10, 5194–5206. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. From bench to bed: The tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 1–21. [Google Scholar] [CrossRef]
- Ye, L.; Chen, W.; Bai, X.-L.; Xu, X.-Y.; Zhang, Q.; Xia, X.-F.; Sun, X.; Li, G.-G.; Hu, Q.; Fu, Q.-H.; et al. Hypoxia-Induced Epithelial-to-Mesenchymal Transition in Hepatocellular Carcinoma Induces an Immunosuppressive Tumor Microenvironment to Promote Metastasis. Cancer Res. 2016, 76, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, Y.; Okano, S.; Matsumoto, Y.; Nakagawara, H.; Matono, R.; Yoshiya, S.; Yamashita, Y.-I.; Yoshizumi, T.; Ikegami, T.; Soejima, Y.; et al. Prognostic impact of programmed cell death 1 ligand 1 expression in human leukocyte antigen class I-positive hepatocellular carcinoma after curative hepatectomy. J. Gastroenterol. 2014, 50, 65–75. [Google Scholar] [CrossRef]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar]
- Chen, C.; Ma, Y.H.; Zhang, Y.T.; Zhang, F.; Zhou, N.; Wang, X.; Liu, T.; Li, Y.M. Effect of dendritic cell-based immunotherapy on hepatocellular carcinoma: A systematic review and meta-analysis. Cytotherapy 2018, 20, 975–989. [Google Scholar] [PubMed]
- AB, T. N. M. The Nobel Prize in Physiology or Medicine 2018. Available online: https://www.nobelprize.org/prizes/medicine/2018/summary/ (accessed on 28 August 2020).
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [PubMed]
- O’Day, S.J.; Hamid, O.; Urba, W.J. Targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4). Cancer 2007, 110, 2614–2627. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.K.; Saleh, S.N. Checkpoint inhibitors for malignant melanoma: A systematic review and meta-analysis. Clin. Cosmet. Investig. Dermatol. 2017, 10, 325–339. [Google Scholar] [CrossRef]
- Administration, U. S. F. a. D. FDA Grants Accelerated Approval to Nivolumab for HCC Previously Treated with Sorafenib. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-nivolumab-hcc-previously-treated-sorafenib (accessed on 28 August 2020).
- Ji, R.R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Imm. Immunothe. 2012, 61, 1019–1031. [Google Scholar]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Chow, L.Q.; Haddad, R.; Gupta, S.; Mahipal, A.; Mehra, R.; Tahara, M.; Berger, R.; Eder, J.P.; Burtness, B.; Lee, S.-H.; et al. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results From the Phase Ib KEYNOTE-012 Expansion Cohort. J. Clin. Oncol. 2016, 34, 3838–3845. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; De Moura, M.C.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterol. 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Sánchez-Paulete, A.R.; Cueto, F.J.; Martínez-López, M.; Labiano, S.; Morales-Kastresana, A.; Rodríguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Quetglas, J.I.; Sancho, D.; et al. Abstract 4908: Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires Batf3-dependent dendritic cells. Immunology 2016, 76, 4908. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015, 13, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2017, 24, 1260–1270. [Google Scholar] [CrossRef]
- Johnston, M.P.; Khakoo, S.I. Immunotherapy for hepatocellular carcinoma: Current and future. World J. Gastroenterol. 2019, 25, 2977–2989. [Google Scholar] [CrossRef] [PubMed]
- Schuler-Thurner, B.; Schultz, E.S.; Berger, T.G.; Weinlich, G.; Ebner, S.; Woerl, P.; Bender, A.; Feuerstein, B.; Fritsch, P.O.; Romani, N.; et al. Rapid Induction of Tumor-specific Type 1 T Helper Cells in Metastatic Melanoma Patients by Vaccination with Mature, Cryopreserved, Peptide-loaded Monocyte-derived Dendritic Cells. J. Exp. Med. 2002, 195, 1279–1288. [Google Scholar] [CrossRef]
- Rizell, M.; Eilard, M.S.; Andersson, M.; Andersson, B.; Karlsson-Parra, A.; Suenaert, P. Phase 1 Trial With the Cell-Based Immune Primer Ilixadencel, Alone, and Combined With Sorafenib, in Advanced Hepatocellular Carcinoma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Formenti, S.C.; DeMaria, S. Radiation Therapy to Convert the Tumor Into an In Situ Vaccine. Int. J. Radiat. Oncol. 2012, 84, 879–880. [Google Scholar] [CrossRef]
- Van Elsas, A.; Hurwitz, A.A.; Allison, J.P. Combination Immunotherapy of B16 Melanoma Using Anti–Cytotoxic T Lymphocyte–Associated Antigen 4 (Ctla-4) and Granulocyte/Macrophage Colony-Stimulating Factor (Gm-Csf)-Producing Vaccines Induces Rejection of Subcutaneous and Metastatic Tumors Accompanied by Autoimmune Depigmentation. J. Exp. Med. 1999, 190, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.; Heo, J.; Lee, H.; Tak, W.; Chao, Y.; Paik, S.; Yim, H.; Byun, K.; Baron, A.; Ungerechts, G.; et al. Vaccinia-based oncolytic immunotherapy Pexastimogene Devacirepvec in patients with advanced hepatocellular carcinoma after sorafenib failure: A randomized multicenter Phase IIb trial (TRAVERSE). OncoImmunology 2019, 8, 1615817. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.H.; Thorne, S.H. Targeted and armed oncolytic poxviruses: A novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 2009, 9, 64–71. [Google Scholar] [CrossRef]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Galle, P.R.; Chao, Y.; Brown, K.T.; Heo, J.; Borad, M.J.; Luca, A.; Pelusio, A.; Agathon, D.; Lusky, M.; et al. PHOCUS: A phase 3 randomized, open-label study comparing the oncolytic immunotherapy Pexa-Vec followed by sorafenib (SOR) vs SOR in patients with advanced hepatocellular carcinoma (HCC) without prior systemic therapy. J. Clin. Oncol. 2016, 34, TPS4146. [Google Scholar] [CrossRef]
- News., G. E. B. Pexa-Vec/Nexavar Combination Fails Phase III Trial in Liver Cancer. Available online: https://www.genengnews.com/news/pexa-vec-nexavar-combination-fails-phase-iii-trial-in-liver-cancer/ (accessed on 29 August 2020).
- Roderburg, C.; Özdirik, B.; Wree, A.; Demir, M.; Tacke, F. Systemic treatment of hepatocellular carcinoma: From sorafenib to combination therapies. Hepatic Oncol. 2020, 7, HEP20. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; Bossche, B.V.D.; De Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2015, 90, 1025–1048. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2019, 37, 4012. [Google Scholar] [CrossRef]
- Cheng, H.; Sun, G.; Chen, H.; Li, Y.; Han, Z.; Li, Y.; Zhang, P.; Yang, L.; Li, Y. Trends in the treatment of advanced hepatocellular carcinoma: Immune checkpoint blockade immunotherapy and related combination therapies. Am. J. Cancer Res. 2019, 9, 1536–1545. [Google Scholar] [PubMed]
- Wei, S.; Sharma, R.; Anang, N.; Levine, J.; Zhao, Y.; Mancuso, J.; Setty, M.; Sharma, P.; Wang, J.; Pe’Er, D.; et al. Negative Co-stimulation Constrains T Cell Differentiation by Imposing Boundaries on Possible Cell States. immuneACCESS 2019, 50, 1084. [Google Scholar] [CrossRef]
- Kurtulus, S.; Madi, A.; Escobar, G.; Klapholz, M.; Nyman, J.; Christian, E.; Pawlak, M.; Dionne, D.; Xia, J.; Rozenblatt-Rosen, O.; et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1−CD8+ Tumor-Infiltrating T Cells. Immunology 2019, 50, 181–194.e6. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nat. Cell Biol. 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Wei, S.C.; Anang, N.-A.A.S.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.; Wargo, J.A.; Pe’Er, D.; et al. Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar]


{kind=link}
{kind=link}
| Population | Marker | Properties/Function | ||
|---|---|---|---|---|
| Human | Mice | |||
| cDC | Type 1 | CD141, CD8, BATF3, IRF8, Clec9a, XCR1, TLR3 | CD103, CD8, BATF3, IRF8, Clec9a, XCR1, TLR3 | CD8+ T cell activation and cross-presentation [30], TLR2, TLR4 |
| Type 2 | CD1c, CD11b | CD11b, IRF4 | T helper cell priming with polarization toward Th2 or Th17 and promotion of humoral immunity [30]. Overall rare, but most frequent DC type in the human liver [25]. Secrete IL-10 upon TLR4 stimulation and induce T cell hyporesponsiveness [25]. Further subsets can be distinguished based on CD5, CD163 and CD14 expression, one of them is a circulating inflammatory (CD5-CD163+CD14+) subtype [28] | |
| Pdc (precursors) | CD303, CD304, CD4, CD123high, TLR-7, TLR9 | CD11c+ B220+ Gr-1+, TLR7, CD45Rbhigh [26,31] | Antiviral innate immunity: antiviral response with abundant type 1 IFN production, stimulation of B cells, NK cells and T cells, differentiate into mature dendritic cells with intense T cell interaction [26] and capable of cross-presentation [32]. | |
| moDC | DC-SIGN(+) | Monocytes adopt a dendritic function and morphology in the presence of lipopolysaccharides or Gram-negative bacteria [33]. Used in vitro to model the DC function. | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lurje, I.; Hammerich, L.; Tacke, F. Dendritic Cell and T Cell Crosstalk in Liver Fibrogenesis and Hepatocarcinogenesis: Implications for Prevention and Therapy of Liver Cancer. Int. J. Mol. Sci. 2020, 21, 7378. https://doi.org/10.3390/ijms21197378
Lurje I, Hammerich L, Tacke F. Dendritic Cell and T Cell Crosstalk in Liver Fibrogenesis and Hepatocarcinogenesis: Implications for Prevention and Therapy of Liver Cancer. International Journal of Molecular Sciences. 2020; 21(19):7378. https://doi.org/10.3390/ijms21197378
Chicago/Turabian StyleLurje, Isabella, Linda Hammerich, and Frank Tacke. 2020. "Dendritic Cell and T Cell Crosstalk in Liver Fibrogenesis and Hepatocarcinogenesis: Implications for Prevention and Therapy of Liver Cancer" International Journal of Molecular Sciences 21, no. 19: 7378. https://doi.org/10.3390/ijms21197378
APA StyleLurje, I., Hammerich, L., & Tacke, F. (2020). Dendritic Cell and T Cell Crosstalk in Liver Fibrogenesis and Hepatocarcinogenesis: Implications for Prevention and Therapy of Liver Cancer. International Journal of Molecular Sciences, 21(19), 7378. https://doi.org/10.3390/ijms21197378