Chrysin Inhibits NF-κB-Dependent CCL5 Transcription by Targeting IκB Kinase in the Atopic Dermatitis-Like Inflammatory Microenvironment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
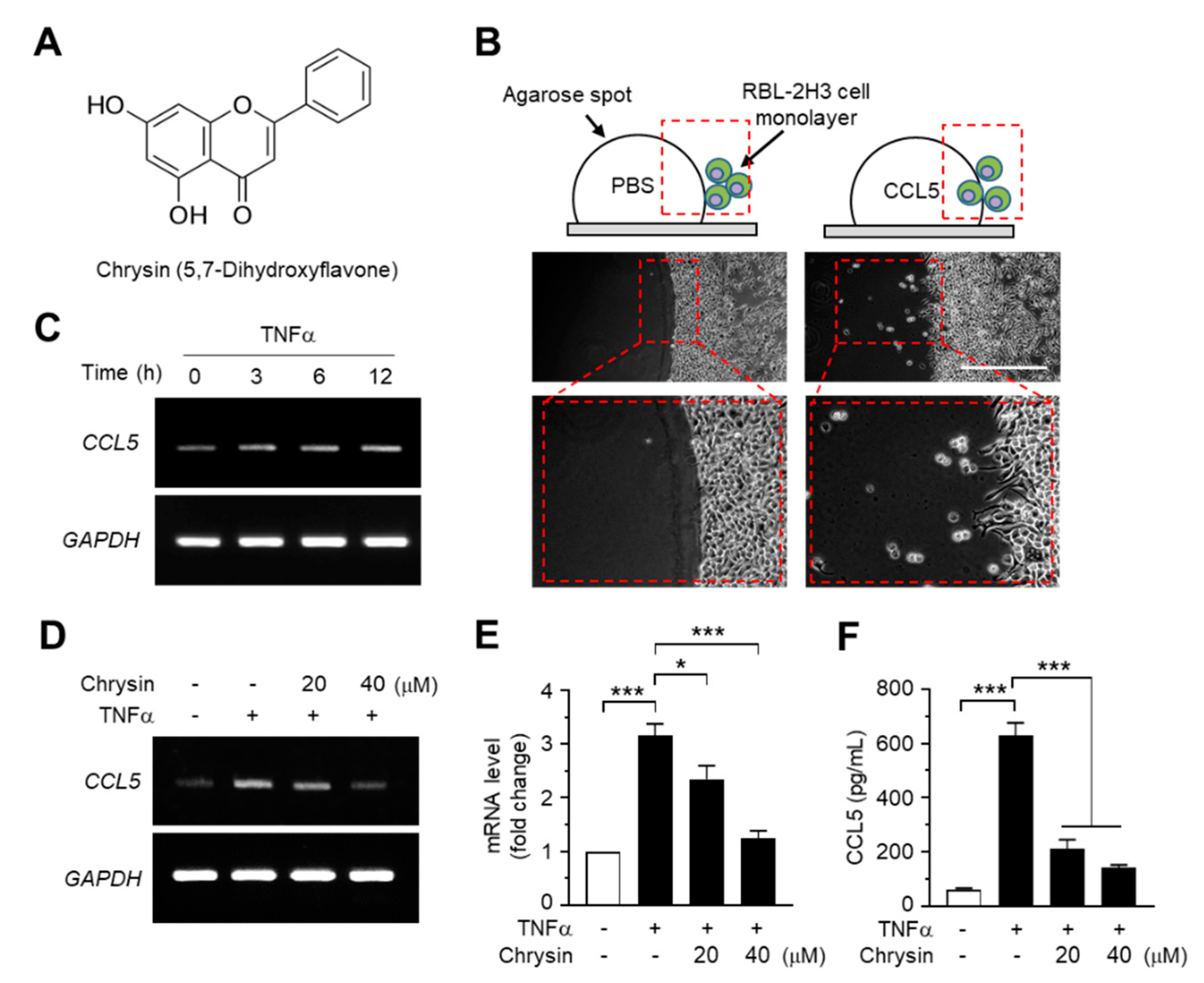
2.1. Effect of Chrysin on the Suppression of TNFα-Induced CCL5 Expression in HaCaT Cells
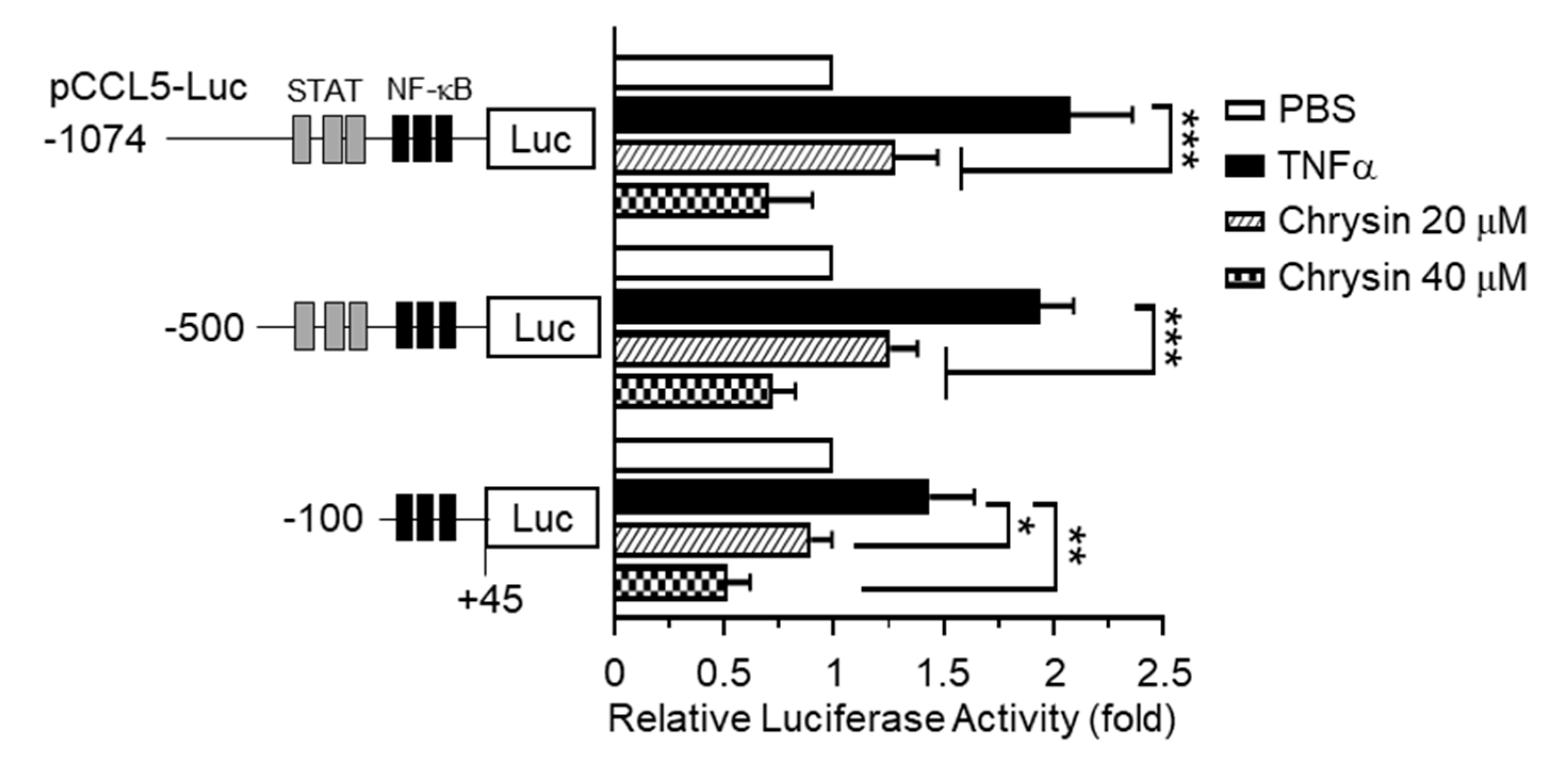
2.2. Effect of Chrysin on the Inhibition of TNFα-Induced CCL5 Promoter Activity
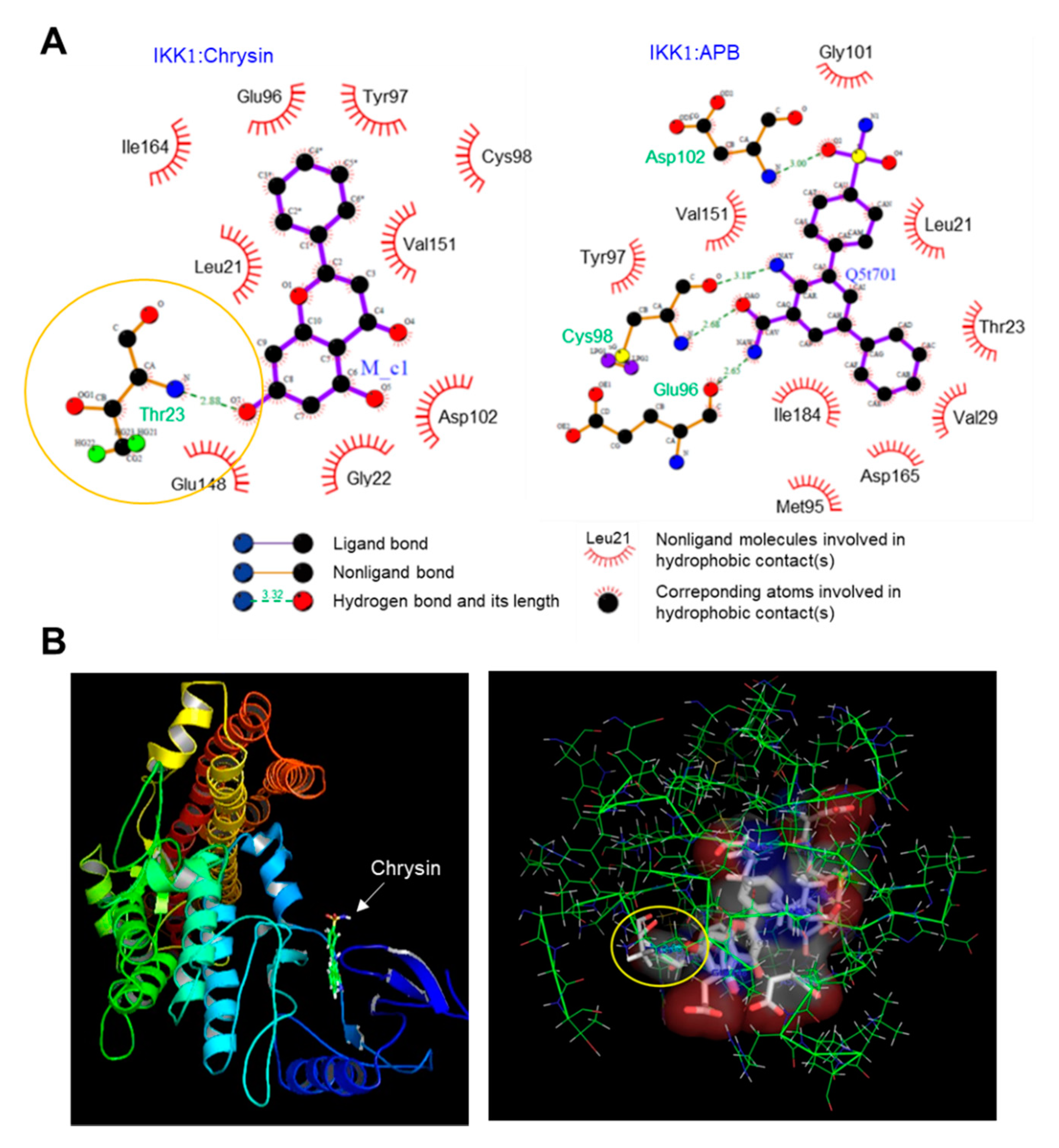
2.3. Binding of Chrysin to the ATP-Binding Pocket of IKK
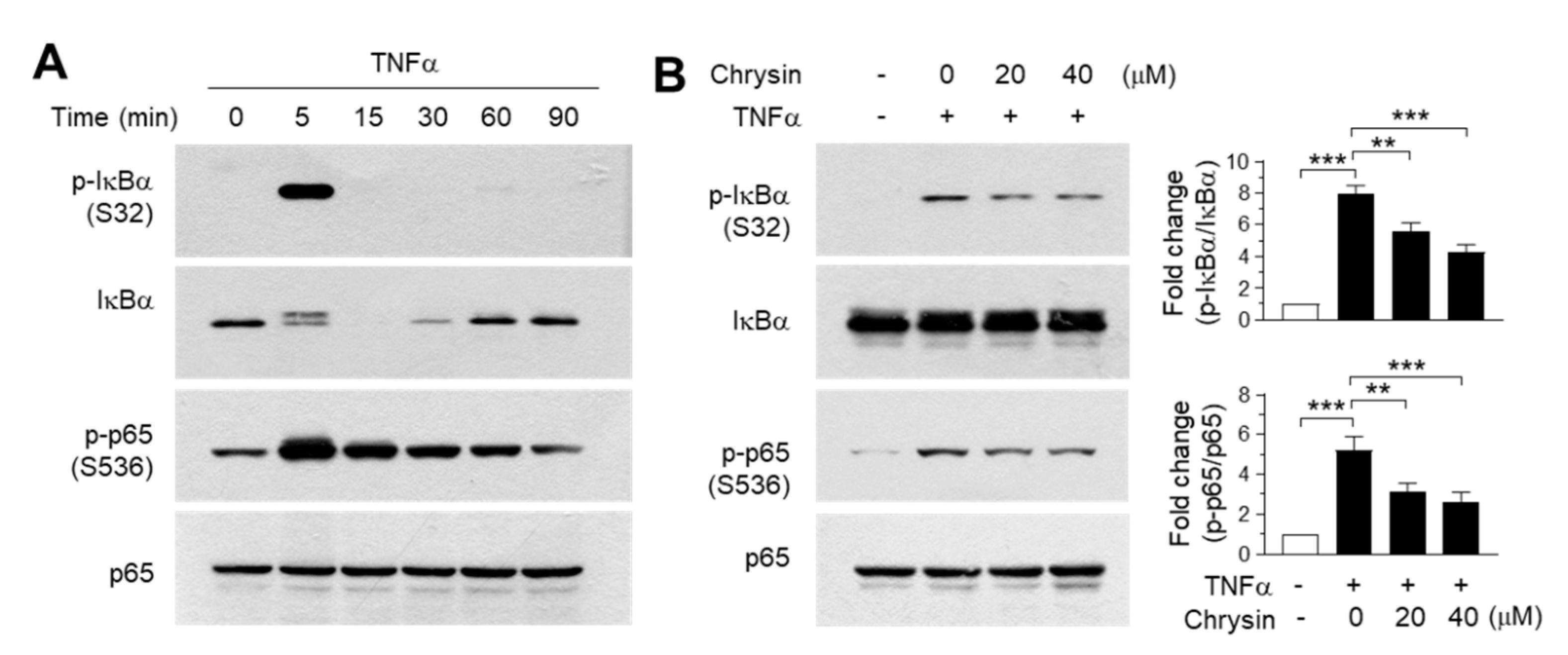
2.4. Effect of Chrysin on the Inhibition of the IKK Downstream Pathway
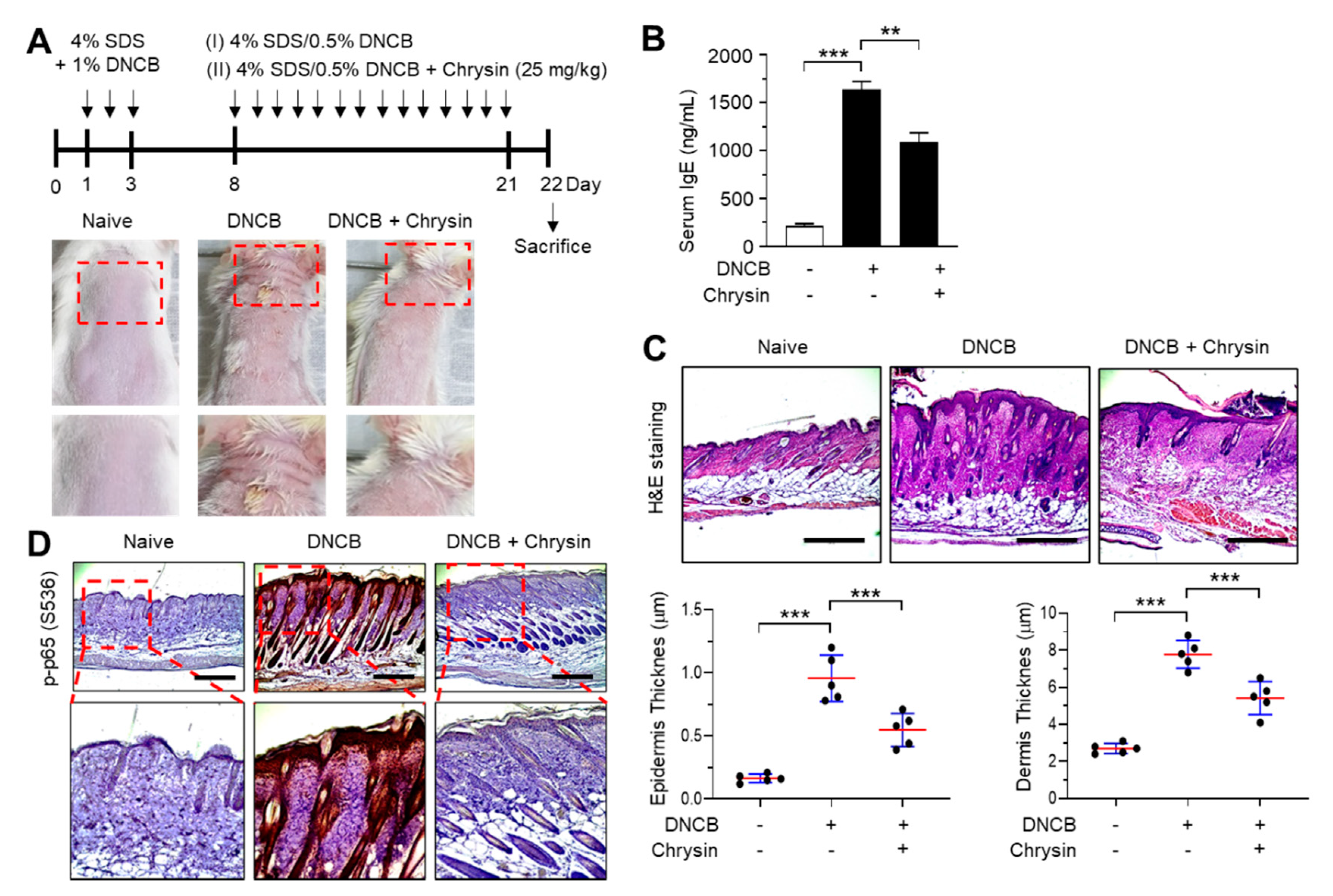
2.5. Effect of Chrysin on the Inhibition of NF-κB in 2,4-Dinitrochlorobenzene (DNCB)-Induced Skin Lesions
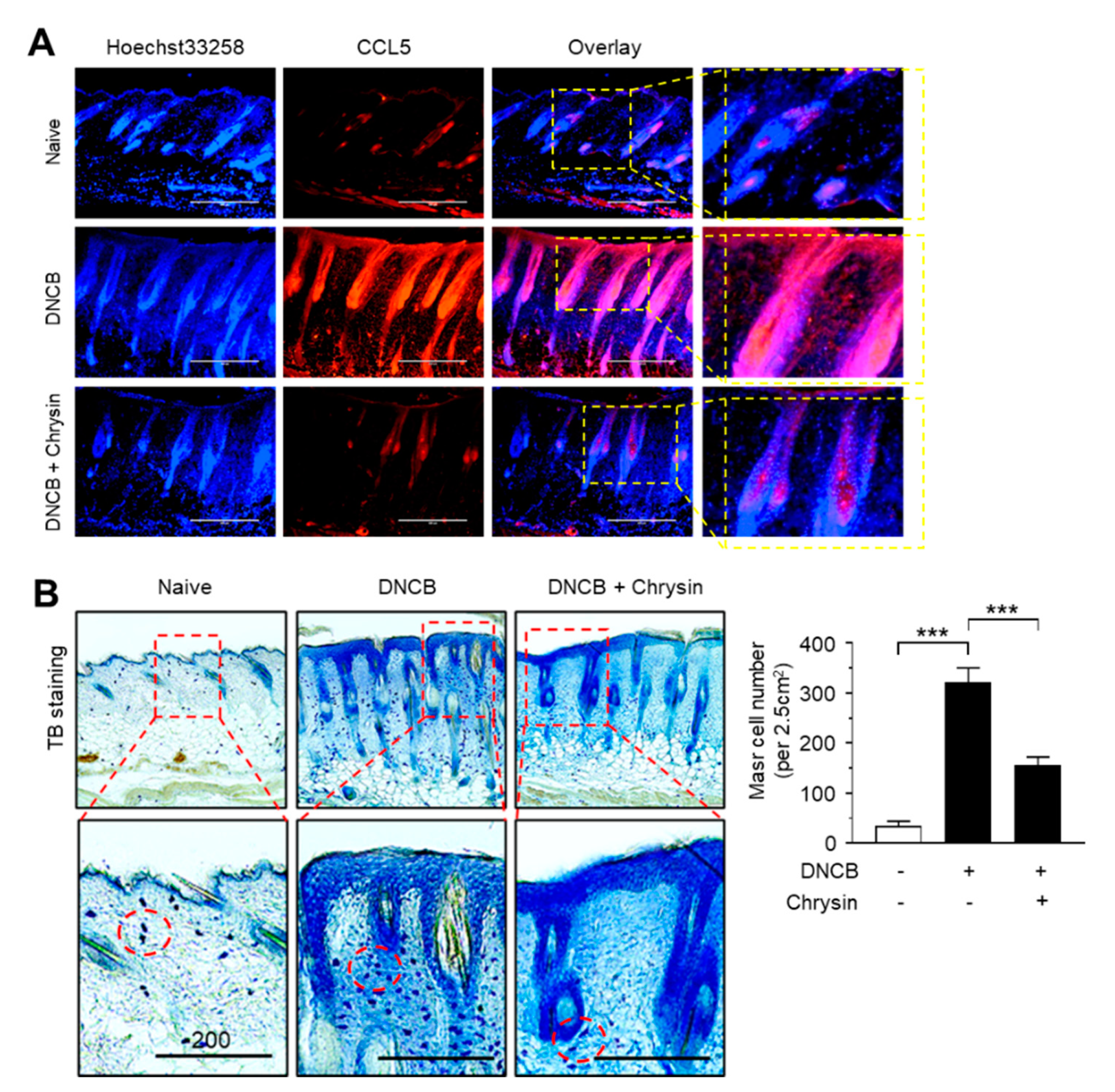
2.6. Effect of Chrysin on the Suppression of CCL5 Expression and Infiltration of Mast Cells in DNCB-Induced Skin Lesions
3. Materials and Methods
3.1. Materials
3.2. Cells and Cell Culture
3.3. Reverse Transcription-PCR (RT-PCR)
- CCL5 forward, 5′-ACA GGT ACC ATG AAG GTC TC–3′,
- CCL5 reverse, 5′–GCA AAT TTG TGT AAG TTC AGG–3,
- Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward, 5′–CCA AGG AGT AAG AAA CCC TGG AC–3′, and
- GAPDH reverse, 5′–GGG CCG AGT TGG GAT AGG G–3′.
3.4. Quantitative Real-Time PCR (Q-PCR)
3.5. Agarose Spot Migration Assay
3.6. Enzyme-Linked Immunosorbent Assay (ELISA)
3.7. Construction of Human CCL5 Promoter-Reporter Constructs
3.8. Luciferase Promoter-Reporter Assay
3.9. Immunoblot Analysis
3.10. Molecular Docking
3.11. Mice
3.12. Induction of Atopic Dermatitis-like Skin Lesions in Mice
3.13. Tissue Preparation And Histopathological Analysis
3.14. Immunohistochemical and Immunofluorescence Stainings
3.15. Measurement of Serum IgE Levels
3.16. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | atopic dermatitis |
| APB | 2-azanyl-5-phenyl-3-(4-sulfamoylphenyl)benzamide |
| CCL5 | C-C motif chemokine ligand 5 |
| DNCB | 2,4-dinitrochlorobenzene |
| ELISA | enzyme-linked immunosorbent assay |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| H&E | hematoxylin and eosin |
| H-bond | hydrogen bond |
| IgE | immunoglobulin E |
| IκB | inhibitor of κB |
| IKK | inhibitor of κB kinase |
| IL | interleukin |
| NF-κB | nuclear factor kappa B |
| Q-PCR | quantitative real-time PCR |
| RANTES | regulated on activation, normal T cell expressed and secreted |
| RelA | v-rel Avian reticuloendotheliosis viral oncogene homolog A |
| RT-PCR | reverse-transcription polymerase chain reaction |
| TB | toluidine blue |
| Th2 | T-helper cell (Th2) |
| TNFα | tumor necrosis factor-alpha |
References
- Ahn, S.; Lim, Y.; Koh, D. Crystal structure of 2-(2,3-di-meth-oxy-naphthalen-1-yl)-3-hy-droxy-6-meth-oxy-4H-chromen-4-one. Acta Crystallogr. E Crystallogr. Commun. 2015, 71, o842–o843. [Google Scholar] [CrossRef] [PubMed]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor beta, and TNF-alpha: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef] [PubMed]
- Trier, A.M.; Kim, B.S. Cytokine modulation of atopic itch. Curr. Opin. Immunol. 2018, 54, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, A.; Bieber, T. Atopic dermatitis: From the genes to skin lesions. Allergy 2000, 55, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Ando, T.; Kimura, M.; Wilson, B.S.; Kawakami, Y. Mast cells in atopic dermatitis. Curr. Opin. Immunol. 2009, 21, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Sehra, S.; Serezani, A.P.M.; Ocana, J.A.; Travers, J.B.; Kaplan, M.H. Mast Cells Regulate Epidermal Barrier Function and the Development of Allergic Skin Inflammation. J. Investig. Dermatol. 2016, 136, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Kabashima, K.; Yoshiki, R.; Sugita, K.; Shiraishi, N.; Onoue, A.; Kuroda, E.; Kobayashi, M.; Yamashita, U.; Tokura, Y. Cutaneous hypersensitivities to hapten are controlled by IFN-gamma-upregulated keratinocyte Th1 chemokines and IFN-gamma-downregulated langerhans cell Th2 chemokines. J. Investig. Dermatol. 2008, 128, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Rebane, A.; Zimmermann, M.; Aab, A.; Baurecht, H.; Koreck, A.; Karelson, M.; Abram, K.; Metsalu, T.; Pihlap, M.; Meyer, N.; et al. Mechanisms of IFN-gamma-induced apoptosis of human skin keratinocytes in patients with atopic dermatitis. J. Allergy Clin. Immunol 2012, 129, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Canavese, M.; Altruda, F.; Silengo, L. Therapeutic efficacy and immunological response of CCL5 antagonists in models of contact skin reaction. PLoS ONE 2010, 5, e8725. [Google Scholar] [CrossRef] [PubMed]
- Wickremasinghe, M.I.; Thomas, L.H.; O’Kane, C.M.; Uddin, J.; Friedland, J.S. Transcriptional mechanisms regulating alveolar epithelial cell-specific CCL5 secretion in pulmonary tuberculosis. J. Biol. Chem. 2004, 279, 27199–27210. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Ahn, Y.T.; McPherson, L.; Clayberger, C.; Krensky, A.M. Interaction of PRP4 with Kruppel-like factor 13 regulates CCL5 transcription. J. Immunol. 2007, 178, 7081–7087. [Google Scholar] [CrossRef] [PubMed]
- Zeinali, M.; Rezaee, S.A.; Hosseinzadeh, H. An overview on immunoregulatory and anti-inflammatory properties of chrysin and flavonoids substances. Biomed. Pharm. 2017, 92, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.; Lee, S.; Kim, S.H. Chrysin suppresses mast cell-mediated allergic inflammation: Involvement of calcium, caspase-1 and nuclear factor-kappaB. Toxicol. Appl. Pharm. 2011, 254, 56–64. [Google Scholar] [CrossRef]
- Shin, E.K.; Kwon, H.S.; Kim, Y.H.; Shin, H.K.; Kim, J.K. Chrysin, a natural flavone, improves murine inflammatory bowel diseases. Biochem. Biophys. Res. Commun. 2009, 381, 502–507. [Google Scholar] [CrossRef]
- Choi, J.K.; Jang, Y.H.; Lee, S.; Lee, S.R.; Choi, Y.A.; Jin, M.; Choi, J.H.; Park, J.H.; Park, P.H.; Choi, H.; et al. Chrysin attenuates atopic dermatitis by suppressing inflammation of keratinocytes. Food Chem. Toxicol. 2017, 110, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Schottelius, A.J.; Zugel, U.; Docke, W.D.; Zollner, T.M.; Rose, L.; Mengel, A.; Buchmann, B.; Becker, A.; Grutz, G.; Naundorf, S.; et al. The role of mitogen-activated protein kinase-activated protein kinase 2 in the p38/TNF-alpha pathway of systemic and cutaneous inflammation. J. Investig. Derm. 2010, 130, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Werfel, T. The role of leukocytes, keratinocytes, and allergen-specific IgE in the development of atopic dermatitis. J. Investig. Dermatol. 2009, 129, 1878–1891. [Google Scholar] [CrossRef] [PubMed]
- Buyanravjikh, S.; Han, S.; Lee, S.; Jeong, A.L.; Ka, H.I.; Park, J.Y.; Boldbaatar, A.; Lim, J.S.; Lee, M.S.; Yang, Y. Cryptotanshinone inhibits IgEmediated degranulation through inhibition of spleen tyrosine kinase and tyrosineprotein kinase phosphorylation in mast cells. Mol. Med. Rep. 2018, 18, 1095–1103. [Google Scholar] [CrossRef]
- Lee, Y.; Choi, H.K.; N’Deh, K.P.U.; Choi, Y.J.; Fan, M.; Kim, E.K.; Chung, K.H.; An, A.J.H. Inhibitory Effect of Centella asiatica Extract on DNCB-Induced Atopic Dermatitis in HaCaT Cells and BALB/c Mice. Nutrients 2020, 12, 411. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, H.; Rappoport, J. An agarose spot assay for chemotactic invasion. Biotechniques 2010, 48, 121–124. [Google Scholar] [CrossRef]
- Polley, S.; Passos, D.O.; Huang, D.B.; Mulero, M.C.; Mazumder, A.; Biswas, T.; Verma, I.M.; Lyumkis, D.; Ghosh, G. Structural Basis for the Activation of IKK1/alpha. Cell Rep. 2016, 17, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins 1999, 37, 228–241. [Google Scholar] [CrossRef]
- Shin, S.Y.; Lee, D.H.; Gil, H.N.; Kim, B.S.; Choe, J.S.; Kim, J.B.; Lee, Y.H.; Lim, Y. Agerarin, identified from Ageratum houstonianum, stimulates circadian CLOCK-mediated aquaporin-3 gene expression in HaCaT keratinocytes. Sci. Rep. 2017, 7, 11175. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Chang, J.H.; Jin, J. Regulation of nuclear factor-kappaB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.A.; Birbach, A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB)—A key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev. 2008, 19, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Hatada, E.N.; Krappmann, D.; Scheidereit, C. NF-kappaB and the innate immune response. Curr. Opin. Immunol. 2000, 12, 52–58. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Gaynor, R.B. IkappaB kinases: Key regulators of the NF-kappaB pathway. Trends Biochem. Sci. 2004, 29, 72–79. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, H.; Lee, Y.H.; Koh, D.; Lim, Y.; Shin, S.Y. Chrysin Inhibits NF-κB-Dependent CCL5 Transcription by Targeting IκB Kinase in the Atopic Dermatitis-Like Inflammatory Microenvironment. Int. J. Mol. Sci. 2020, 21, 7348. https://doi.org/10.3390/ijms21197348
Yeo H, Lee YH, Koh D, Lim Y, Shin SY. Chrysin Inhibits NF-κB-Dependent CCL5 Transcription by Targeting IκB Kinase in the Atopic Dermatitis-Like Inflammatory Microenvironment. International Journal of Molecular Sciences. 2020; 21(19):7348. https://doi.org/10.3390/ijms21197348
Chicago/Turabian StyleYeo, Hyunjin, Young Han Lee, Dongsoo Koh, Yoongho Lim, and Soon Young Shin. 2020. "Chrysin Inhibits NF-κB-Dependent CCL5 Transcription by Targeting IκB Kinase in the Atopic Dermatitis-Like Inflammatory Microenvironment" International Journal of Molecular Sciences 21, no. 19: 7348. https://doi.org/10.3390/ijms21197348
APA StyleYeo, H., Lee, Y. H., Koh, D., Lim, Y., & Shin, S. Y. (2020). Chrysin Inhibits NF-κB-Dependent CCL5 Transcription by Targeting IκB Kinase in the Atopic Dermatitis-Like Inflammatory Microenvironment. International Journal of Molecular Sciences, 21(19), 7348. https://doi.org/10.3390/ijms21197348