Pharmacological Inhibition of WEE1 Potentiates the Antitumoral Effect of the dl922-947 Oncolytic Virus in Malignant Mesothelioma Cell Lines

 ,
,  ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
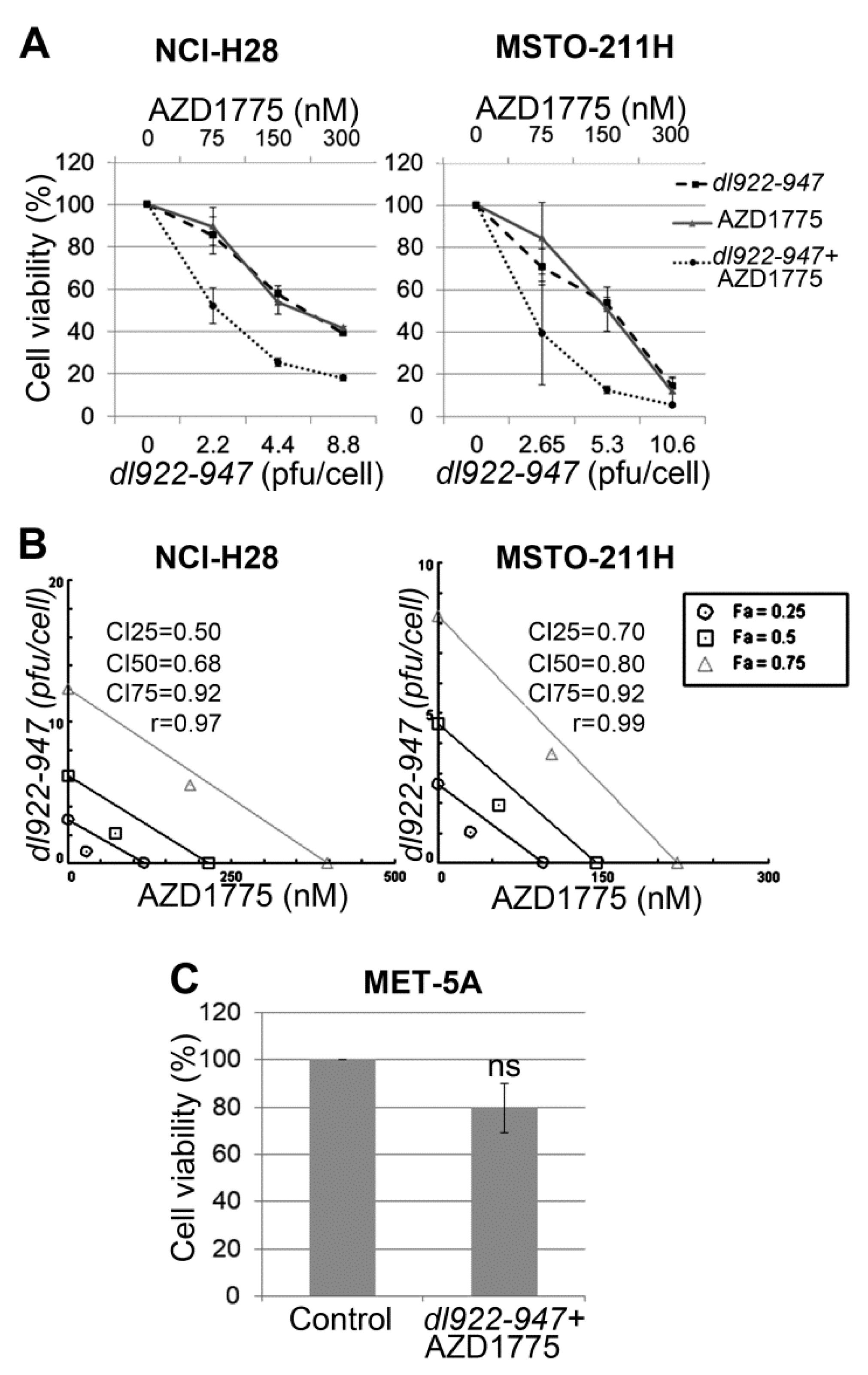
2.1. AZD1775 Synergizes with dl922-947 in MM Cell Lines
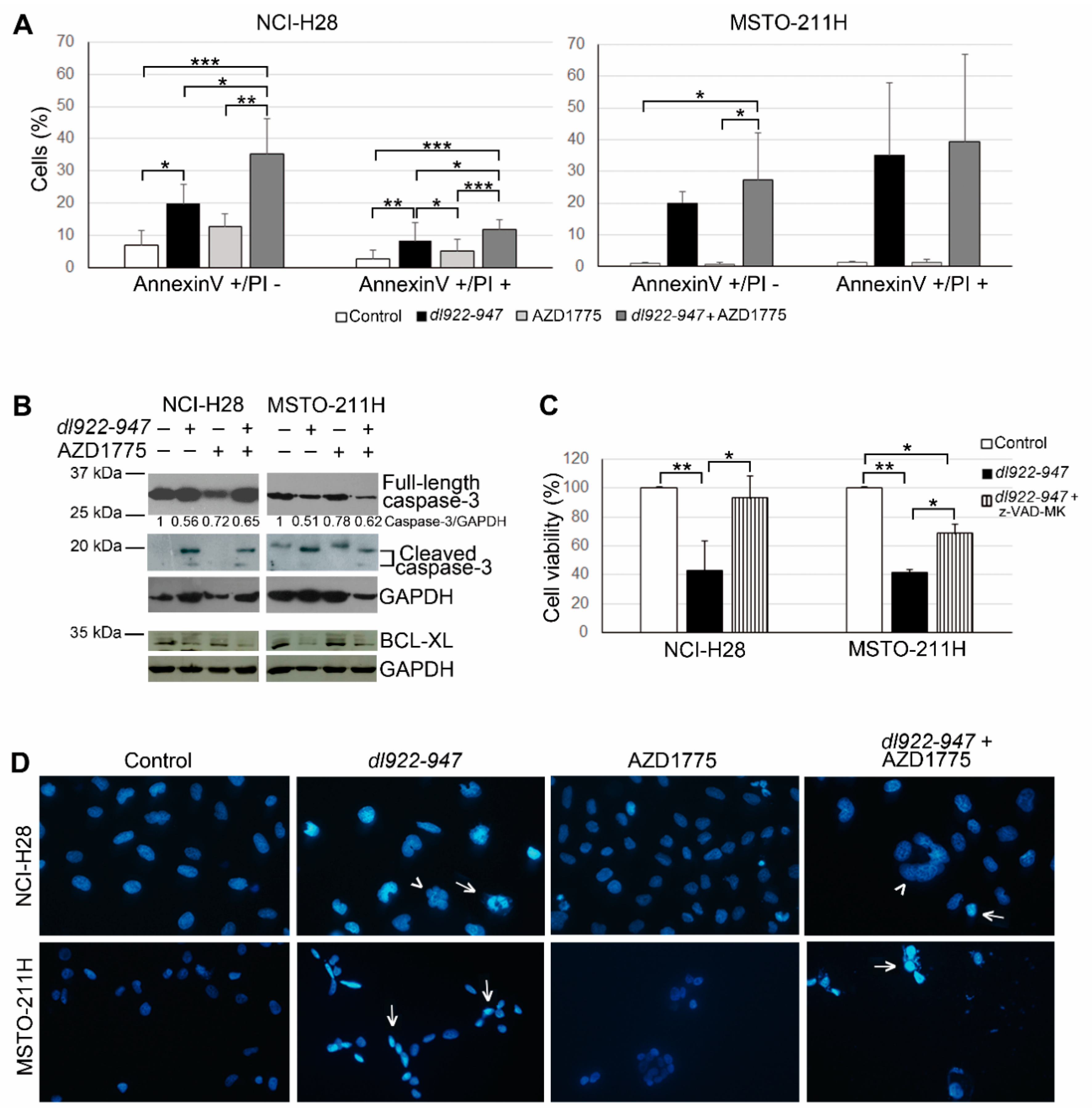
2.2. AZD1775 Increases the dl922-947-Induced Cell Death in MM Cells
2.3. AZD1775 Inactivates the DNA Damage Checkpoint Induced by dl922-947 in MM Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Adenovirus Preparation
4.2. Drug Combination, Sulforhodamine B (SRB) Assay, and Synergism Analysis
4.3. Apoptosis Analysis
4.4. DAPI Staining
4.5. Western Blotting Analysis
4.6. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3 related |
| CDK | Cyclin-dependent kinase |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CHK1 | Checkpoint kinase 1 |
| CI | Combination index |
| COSMIC | Catalogue of Somatic Mutations in Cancer |
| DDR | DNA damage response |
| Fa | Fraction affected |
| FBS | Fetal bovine serum |
| IC50 | Half maximal inhibitory concentration |
| MM | Malignant mesothelioma |
| OV | Oncolytic virus |
| PARP | Poly(ADP-ribose) polymerase |
| PI | Propidium iodide |
| RB | Retinoblastoma |
| RPA | Replication protein A |
| SRB | Sulforhodamine B |
| γ-H2AX | Phosphorylated histone H2AX |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Baumann, F.; Carbone, M. Environmental risk of mesothelioma in the United States: An emerging concern-epidemiological issues. J. Toxicol. Environ. Health B Crit. Rev. 2016, 19, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H. Mesothelioma: Recent highlights. Ann. Transl. Med. 2017, 5, 238. [Google Scholar] [CrossRef] [PubMed]
- The Lancet Respiratory Medicine. Pleural mesothelioma: Tackling a deadly cancer. Lancet Respir. Med. 2019, 7, 99. [Google Scholar] [CrossRef]
- Scherpereel, A.; Opitz, I.; Berghmans, T.; Psallidas, I.; Glatzer, M.; Rigau, D.; Astoul, P.; Bölükbas, S.; Boyd, J.; Coolen, J.; et al. ERS/ESTS/EACTS/ESTRO guidelines for the management of malignant pleural mesothelioma. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef]
- Robinson, B.W.; Musk, A.W.; Lake, R.A. Malignant mesothelioma. Lancet 2005, 366, 397–408. [Google Scholar] [CrossRef]
- Hinz, T.K.; Heasley, L.E. Translating mesothelioma molecular genomics and dependencies into precision oncology-based therapies. Semin. Cancer Biol. 2020, 61, 11–22. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef]
- Malfitano, A.M.; Di Somma, S.; Iannuzzi, C.A.; Pentimalli, F.; Portella, G. Virotherapy: From single agents to combinatorial treatments. Biochem. Pharmacol. 2020, 177, 113986. [Google Scholar] [CrossRef]
- Pease, D.F.; Kratzke, R.A. Oncolytic Viral Therapy for Mesothelioma. Front. Oncol. 2017, 7, 179. [Google Scholar] [CrossRef]
- Di Somma, S.; Iannuzzi, C.A.; Passaro, C.; Forte, I.M.; Iannone, R.; Gigantino, V.; Indovina, P.; Botti, G.; Giordano, A.; Formisano, P.; et al. The Oncolytic Virus dl922-947 Triggers Immunogenic Cell Death in Mesothelioma and Reduces Xenograft Growth. Front. Oncol. 2019, 9, 564. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, M.; Francis, J.; Eddouadi, A.; Lemoine, N.R.; Hallden, G. An oncolytic adenovirus defective in pRb-binding (dl922-947) can efficiently eliminate pancreatic cancer cells and tumors in vivo in combination with 5-FU or gemcitabine. Cancer Gene Ther. 2011, 18, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Lockley, M.; Fernandez, M.; Wang, Y.; Li, N.F.; Conroy, S.; Lemoine, N.; McNeish, I. Activity of the adenoviral E1A deletion mutant dl922-947 in ovarian cancer: Comparison with E1A wild-type viruses, bioluminescence monitoring, and intraperitoneal delivery in icodextrin. Cancer Res. 2006, 66, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Botta, G.; Perruolo, G.; Libertini, S.; Cassese, A.; Abagnale, A.; Beguinot, F.; Formisano, P.; Portella, G. PED/PEA-15 modulates coxsackievirus-adenovirus receptor expression and adenoviral infectivity via ERK-mediated signals in glioma cells. Hum. Gene Ther. 2010, 21, 1067–1076. [Google Scholar] [CrossRef]
- Libertini, S.; Iacuzzo, I.; Perruolo, G.; Scala, S.; Ierano, C.; Franco, R.; Hallden, G.; Portella, G. Bevacizumab increases viral distribution in human anaplastic thyroid carcinoma xenografts and enhances the effects of E1A-defective adenovirus dl922-947. Clin. Cancer Res. 2008, 14, 6505–6514. [Google Scholar] [CrossRef]
- Malfitano, A.M.; Somma, S.D.; Prevete, N.G.; Portella, G. Virotherapy as a Potential Therapeutic Approach for the Treatment of Aggressive Thyroid Cancer. Cancers 2019, 11, 1532. [Google Scholar] [CrossRef]
- Heise, C.; Hermiston, T.; Johnson, L.; Brooks, G.; Sampson-Johannes, A.; Williams, A.; Hawkins, L.; Kirn, D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 2000, 6, 1134–1139. [Google Scholar] [CrossRef]
- Forte, I.M.; Giordano, A.; Pentimalli, F. Molecular markers of mesothelioma aiding in diagnostic challenges: The combined use of p16 and BAP1. In Malignant Pleural Mesothelioma: A Guide for Clinicians; Giordano, A., Franco, R., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 109–115. [Google Scholar]
- Prins, J.B.; Williamson, K.A.; Kamp, M.M.; Van Hezik, E.J.; Van der Kwast, T.H.; Hagemeijer, A.; Versnel, M.A. The gene for the cyclin-dependent-kinase-4 inhibitor, CDKN2A, is preferentially deleted in malignant mesothelioma. Int. J. Cancer. 1998, 7, 649–653. [Google Scholar] [CrossRef]
- Howells, A.; Marelli, G.; Lemoine, N.R.; Wang, Y. Oncolytic Viruses-Interaction of Virus and Tumor Cells in the Battle to Eliminate Cancer. Front. Oncol. 2017, 7, 195. [Google Scholar] [CrossRef]
- Bartlett, D.L.; Liu, Z.; Sathaiah, M.; Ravindranathan, R.; Guo, Z.; He, Y.; Guo, Z.S. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer 2013, 12, 103. [Google Scholar] [CrossRef]
- Berkey, S.E.; Thorne, S.H.; Bartlett, D.L. Oncolytic Virotherapy and the Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 157–172. [Google Scholar] [PubMed]
- Passaro, C.; Borriello, F.; Vastolo, V.; Di Somma, S.; Scamardella, E.; Gigantino, V.; Franco, R.; Marone, G.; Portella, G. The oncolytic virus dl922-947 reduces IL-8/CXCL8 and MCP-1/CCL2 expression and impairs angiogenesis and macrophage infiltration in anaplastic thyroid carcinoma. Oncotarget 2016, 7, 1500–1515. [Google Scholar] [CrossRef] [PubMed]
- Connell, C.M.; Wheatley, S.P.; McNeish, I.A. Nuclear survivin abrogates multiple cell cycle checkpoints and enhances viral oncolysis. Cancer Res. 2008, 68, 7923–7931. [Google Scholar] [CrossRef] [PubMed]
- Passaro, C.; Volpe, M.; Botta, G.; Scamardella, E.; Perruolo, G.; Gillespie, D.; Libertini, S.; Portella, G. PARP inhibitor olaparib increases the oncolytic activity of dl922-947 in in vitro and in vivo model of anaplastic thyroid carcinoma. Mol. Oncol. 2015, 9, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Connell, C.M.; Shibata, A.; Tookman, L.A.; Archibald, K.M.; Flak, M.B.; Pirlo, K.J.; Lockley, M.; Wheatley, S.P.; McNeish, I.A. Genomic DNA damage and ATR-Chk1 signaling determine oncolytic adenoviral efficacy in human ovarian cancer cells. J. Clin. Investig. 2011, 121, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Passaro, C.; Abagnale, A.; Libertini, S.; Volpe, M.; Botta, G.; Cella, L.; Pacelli, R.; Hallden, G.; Gillespie, D.; Portella, G. Ionizing radiation enhances dl922-947-mediated cell death of anaplastic thyroid carcinoma cells. Endocr. Relat. Cancer 2013, 20, 633–647. [Google Scholar] [CrossRef]
- Fu, S.; Wang, Y.; Keyomarsi, K.; Meric-Bernstam, F.; Meric-Bernstein, F. Strategic development of AZD1775, a Wee1 kinase inhibitor, for cancer therapy. Expert Opin. Investig. Drug 2018, 27, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef]
- Cole, K.A.; Pal, S.; Kudgus, R.A.; Ijaz, H.; Liu, X.; Minard, C.G.; Pawel, B.R.; Maris, J.M.; Haas-Kogan, D.A.; Voss, S.D.; et al. Phase I Clinical Trial of the Wee1 Inhibitor Adavosertib (AZD1775) with Irinotecan in Children with Relapsed Solid Tumors: A COG Phase I Consortium Report (ADVL1312). Clin. Cancer Res. 2020, 26, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; de Souza, P.; Kim, S.W.; Lickliter, J.D.; Naito, Y.; Park, K.; Kumar, S.; Mugundu, G.M.; Bang, Y.J. Safety, Pharmacokinetics, and Clinical Activity of Adavosertib in Combination with Chemotherapy in Asian Patients with Advanced Solid Tumors: Phase Ib Study. Target. Oncol. 2020, 15, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Estevez-Diz, M.D.P.; Grischke, E.M.; Hall, M.; Marmé, F.; Provencher, D.M.; Uyar, D.S.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A biomarker-enriched, randomized Phase II trial of adavosertib (AZD1775) plus paclitaxel and carboplatin for women with platinum-sensitive TP53-mutant ovarian cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef] [PubMed]
- Indovina, P.; Giordano, A. Targeting the checkpoint kinase WEE1: Selective sensitization of cancer cells to DNA-damaging drugs. Cancer Biol. Ther. 2010, 9, 523–525. [Google Scholar] [CrossRef][Green Version]
- Liang, J.; Zhao, H.; Diplas, B.H.; Liu, S.; Liu, J.; Wang, D.; Lu, Y.; Zhu, Q.; Wu, J.; Wang, W.; et al. Genome-Wide CRISPR-Cas9 Screen Reveals Selective Vulnerability of ATRX-Mutant Cancers to WEE1 Inhibition. Cancer Res. 2020, 80, 510–523. [Google Scholar] [CrossRef]
- Brunner, A.; Suryo Rahmanto, A.; Johansson, H.; Franco, M.; Viiliäinen, J.; Gazi, M.; Frings, O.; Fredlund, E.; Spruck, C.; Lehtiö, J.; et al. PTEN and DNA-PK determine sensitivity and recovery in response to WEE1 inhibition in human breast cancer. Elife 2020, 9, e57894. [Google Scholar] [CrossRef]
- Young, L.A.; O’Connor, L.O.; de Renty, C.; Veldman-Jones, M.H.; Dorval, T.; Wilson, Z.; Jones, D.R.; Lawson, D.; Odedra, R.; Maya-Mendoza, A.; et al. Differential Activity of ATR and WEE1 Inhibitors in a Highly Sensitive Subpopulation of DLBCL Linked to Replication Stress. Cancer Res. 2019, 79, 3762–3775. [Google Scholar] [CrossRef]
- Lallo, A.; Frese, K.K.; Morrow, C.J.; Sloane, R.; Gulati, S.; Schenk, M.W.; Trapani, F.; Simms, N.; Galvin, M.; Brown, S.; et al. The Combination of the PARP Inhibitor Olaparib and the WEE1 Inhibitor AZD1775 as a New Therapeutic Option for Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 5153–5164. [Google Scholar] [CrossRef]
- Lin, X.; Chen, D.; Zhang, C.; Zhang, X.; Li, Z.; Dong, B.; Gao, J.; Shen, L. Augmented antitumor activity by olaparib plus AZD1775 in gastric cancer through disrupting DNA damage repair pathways and DNA damage checkpoint. J. Exp. Clin. Cancer Res. 2018, 37, 129. [Google Scholar] [CrossRef]
- Haynes, B.; Murai, J.; Lee, J.M. Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat. Rev. 2018, 71, 1–7. [Google Scholar] [CrossRef]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell 2019, 35, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Levin, K.; Rader, J.; Belcastro, L.; Li, Y.; Martinez, D.; Pawel, B.; Shumway, S.D.; Maris, J.M.; Cole, K.A. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013, 73, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Xie, C.; Li, C.; Caldwell, J.T.; Edwards, H.; Taub, J.W.; Wang, Y.; Lin, H.; Ge, Y. CHK1 plays a critical role in the anti-leukemic activity of the wee1 inhibitor MK-1775 in acute myeloid leukemia cells. J. Hematol. Oncol. 2014, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, L.; Vincelette, N.D.; Koh, B.D.; Naylor, R.M.; Flatten, K.S.; Peterson, K.L.; McNally, A.; Gojo, I.; Karp, J.E.; Mesa, R.A.; et al. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica 2014, 99, 688–696. [Google Scholar] [CrossRef]
- Ghelli Luserna Di Rorà, A.; Bocconcelli, M.; Ferrari, A.; Terragna, C.; Bruno, S.; Imbrogno, E.; Beeharry, N.; Robustelli, V.; Ghetti, M.; Napolitano, R.; et al. Synergism Through WEE1 and CHK1 Inhibition in Acute Lymphoblastic Leukemia. Cancers 2019, 11, 1654. [Google Scholar] [CrossRef]
- Deneka, A.Y.; Einarson, M.B.; Bennett, J.; Nikonova, A.S.; Elmekawy, M.; Zhou, Y.; Lee, J.W.; Burtness, B.A.; Golemis, E.A. Synthetic Lethal Targeting of Mitotic Checkpoints in HPV-Negative Head and Neck Cancer. Cancers 2020, 12, 306. [Google Scholar] [CrossRef]
- Jin, J.; Fang, H.; Yang, F.; Ji, W.; Guan, N.; Sun, Z.; Shi, Y.; Zhou, G.; Guan, X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia 2018, 20, 478–488. [Google Scholar] [CrossRef]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef]
- Qi, W.; Xu, X.; Wang, M.; Li, X.; Wang, C.; Sun, L.; Zhao, D.; Sun, L. Inhibition of Wee1 sensitizes AML cells to ATR inhibitor VE-822-induced DNA damage and apoptosis. Biochem. Pharmacol. 2019, 164, 273–282. [Google Scholar] [CrossRef]
- Cozzi, M.; Giorgi, F.; Marcelli, E.; Pentimalli, F.; Forte, I.M.; Schenone, S.; D’Urso, V.; De Falco, G.; Botta, M.; Giordano, A.; et al. Antitumor activity of new pyrazolo[3,4-d]pyrimidine SRC kinase inhibitors in Burkitt lymphoma cell lines and its enhancement by WEE1 inhibition. Cell Cycle 2012, 11, 1029–1039. [Google Scholar] [CrossRef][Green Version]
- Ghelli Luserna Di Rorà, A.; Beeharry, N.; Imbrogno, E.; Ferrari, A.; Robustelli, V.; Righi, S.; Sabattini, E.; Verga Falzacappa, M.V.; Ronchini, C.; Testoni, N.; et al. Targeting WEE1 to enhance conventional therapies for acute lymphoblastic leukemia. J. Hematol. Oncol. 2018, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Caiola, E.; Frapolli, R.; Tomanelli, M.; Valerio, R.; Iezzi, A.; Garassino, M.C.; Broggini, M.; Marabese, M. Wee1 inhibitor MK1775 sensitizes KRAS mutated NSCLC cells to sorafenib. Sci. Rep. 2018, 8, 948. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Parameswaran, J.; Sandoval-Schaefer, T.; Eoh, K.J.; Yang, D.H.; Zhu, F.; Mehra, R.; Sharma, R.; Gaffney, S.G.; Perry, E.B.; et al. Combined Aurora Kinase A (AURKA) and WEE1 Inhibition Demonstrates Synergistic Antitumor Effect in Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2019, 25, 3430–3442. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.R.W.; Langendonk, M.; Reitsma, B.; Herbers, P.; Nijland, M.; Huls, G.; van den Berg, A.; Ammatuna, E.; Visser, L.; van Meerten, T. WEE1 Inhibition Enhances Anti-Apoptotic Dependency as a Result of Premature Mitotic Entry and DNA Damage. Cancers 2019, 11, 1743. [Google Scholar] [CrossRef]
- Corella, A.N.; Cabiliza Ordonio, M.V.A.; Coleman, I.; Lucas, J.M.; Kaipainen, A.; Nguyen, H.M.; Sondheim, D.; Brown, L.G.; True, L.D.; Lee, J.K.; et al. Identification of Therapeutic Vulnerabilities in Small-cell Neuroendocrine Prostate Cancer. Clin. Cancer Res. 2020, 26, 1667–1677. [Google Scholar] [CrossRef]
- Liu, W.; Zeng, X.; Yin, Y.; Li, C.; Yang, W.; Wan, W.; Shi, L.; Wang, G.; Tao, K.; Zhang, P. Targeting the WEE1 kinase strengthens the antitumor activity of imatinib via promoting KIT autophagic degradation in gastrointestinal stromal tumors. Gastric Cancer 2020, 23, 39–51. [Google Scholar] [CrossRef]
- Takashima, Y.; Kikuchi, E.; Kikuchi, J.; Suzuki, M.; Kikuchi, H.; Maeda, M.; Shoji, T.; Furuta, M.; Kinoshita, I.; Dosaka-Akita, H.; et al. Bromodomain and extraterminal domain inhibition synergizes with WEE1-inhibitor AZD1775 effect by impairing nonhomologous end joining and enhancing DNA damage in nonsmall cell lung cancer. Int. J. Cancer 2020, 146, 1114–1124. [Google Scholar] [CrossRef]
- Hu, J.; Wang, T.; Xu, J.; Wu, S.; Wang, L.; Su, H.; Jiang, J.; Yue, M.; Wang, J.; Wang, D.; et al. WEE1 inhibition induces glutamine addiction in T-cell acute lymphoblastic leukemia. Haematologica 2020. [Google Scholar] [CrossRef]
- Xing, L.; Lin, L.; Yu, T.; Li, Y.; Cho, S.F.; Liu, J.; Wen, K.; Hsieh, P.A.; Kinneer, K.; Munshi, N.; et al. A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma. Leukemia 2020, 34, 2150–2162. [Google Scholar] [CrossRef]
- Liang, L.; He, Y.; Wang, H.; Zhou, H.; Xiao, L.; Ye, M.; Kuang, Y.; Luo, S.; Zuo, Y.; Feng, P.; et al. The Wee1 kinase inhibitor MK1775 suppresses cell growth, attenuates stemness and synergises with bortezomib in multiple myeloma. Br. J. Haematol. 2020, 191, 62–67. [Google Scholar] [CrossRef]
- Sun, L.; Moore, E.; Berman, R.; Clavijo, P.E.; Saleh, A.; Chen, Z.; Van Waes, C.; Davies, J.; Friedman, J.; Allen, C.T. WEE1 kinase inhibition reverses G2/M cell cycle checkpoint activation to sensitize cancer cells to immunotherapy. Oncoimmunology 2018, 7, e1488359. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Sun, L.; Robbins, Y.; Clavijo, P.E.; Friedman, J.; Silvin, C.; Van Waes, C.; Cook, J.; Mitchell, J.; Allen, C. Enhancing direct cytotoxicity and response to immune checkpoint blockade following ionizing radiation with Wee1 kinase inhibition. Oncoimmunology 2019, 8, e1638207. [Google Scholar] [CrossRef] [PubMed]
- Hai, J.; Zhang, H.; Zhou, J.; Wu, Z.; Chen, T.; Papadopoulos, E.; Dowling, C.M.; Pyon, V.; Pan, Y.; Liu, J.B.; et al. Generation of Genetically Engineered Mouse Lung Organoid Models for Squamous Cell Lung Cancers Allows for the Study of Combinatorial Immunotherapy. Clin. Cancer Res. 2020, 26, 3431–3442. [Google Scholar] [CrossRef] [PubMed]
- Indovina, P.; Marcelli, E.; Di Marzo, D.; Casini, N.; Forte, I.M.; Giorgi, F.; Alfano, L.; Pentimalli, F.; Giordano, A. Abrogating G(2)/M checkpoint through WEE1 inhibition in combination with chemotherapy as a promising therapeutic approach for mesothelioma. Cancer Biol. Ther. 2014, 15, 380–388. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, D.; Liang, S.Q.; Yang, H.; Bruggmann, R.; Berezowska, S.; Yang, Z.; Marti, T.M.; Hall, S.R.R.; Gao, Y.; Kocher, G.J.; et al. CRISPR Screening Identifies WEE1 as a Combination Target for Standard Chemotherapy in Malignant Pleural Mesothelioma. Mol. Cancer Ther. 2020, 19, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Varin, E.; Denoyelle, C.; Brotin, E.; Meryet-Figuière, M.; Giffard, F.; Abeilard, E.; Goux, D.; Gauduchon, P.; Icard, P.; Poulain, L. Downregulation of Bcl-xL and Mcl-1 is sufficient to induce cell death in mesothelioma cells highly refractory to conventional chemotherapy. Carcinogenesis 2010, 31, 984–993. [Google Scholar] [CrossRef]
- Guertin, A.D.; Martin, M.M.; Roberts, B.; Hurd, M.; Qu, X.; Miselis, N.R.; Liu, Y.; Li, J.; Feldman, I.; Benita, Y.; et al. Unique functions of CHK1 and WEE1 underlie synergistic anti-tumor activity upon pharmacologic inhibition. Cancer Cell Int. 2012, 12, 45. [Google Scholar] [CrossRef]
- Wang, G.; Niu, X.; Zhang, W.; Caldwell, J.T.; Edwards, H.; Chen, W.; Taub, J.W.; Zhao, L.; Ge, Y. Synergistic antitumor interactions between MK-1775 and panobinostat in preclinical models of pancreatic cancer. Cancer Lett. 2015, 356, 656–668. [Google Scholar] [CrossRef]
- Sakurikar, N.; Thompson, R.; Montano, R.; Eastman, A. A subset of cancer cell lines is acutely sensitive to the Chk1 inhibitor MK-8776 as monotherapy due to CDK2 activation in S phase. Oncotarget 2016, 7, 1380–1394. [Google Scholar] [CrossRef]
- Busch, C.J.; Kröger, M.S.; Jensen, J.; Kriegs, M.; Gatzemeier, F.; Petersen, C.; Münscher, A.; Rothkamm, K.; Rieckmann, T. G2-checkpoint targeting and radiosensitization of HPV/p16-positive HNSCC cells through the inhibition of Chk1 and Wee1. Radiother. Oncol. 2017, 122, 260–266. [Google Scholar] [CrossRef]
- Koh, S.B.; Wallez, Y.; Dunlop, C.R.; Bernaldo de Quirós Fernández, S.; Bapiro, T.E.; Richards, F.M.; Jodrell, D.I. Mechanistic Distinctions between CHK1 and WEE1 Inhibition Guide the Scheduling of Triple Therapy with Gemcitabine. Cancer Res. 2018, 78, 3054–3066. [Google Scholar] [CrossRef] [PubMed]
- Hauge, S.; Macurek, L.; Syljuåsen, R.G. p21 limits S phase DNA damage caused by the Wee1 inhibitor MK1775. Cell Cycle 2019, 18, 834–847. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell. Mol. Life Sci. 2013, 70, 4009–4021. [Google Scholar] [CrossRef] [PubMed]
- Baird, S.K.; Aerts, J.L.; Eddaoudi, A.; Lockley, M.; Lemoine, N.R.; McNeish, I.A. Oncolytic adenoviral mutants induce a novel mode of programmed cell death in ovarian cancer. Oncogene 2008, 27, 3081–3090. [Google Scholar] [CrossRef] [PubMed]
- Weigert, M.; Binks, A.; Dowson, S.; Leung, E.Y.L.; Athineos, D.; Yu, X.; Mullin, M.; Walton, J.B.; Orange, C.; Ennis, D.; et al. RIPK3 promotes adenovirus type 5 activity. Cell Death Dis. 2017, 8, 3206. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, D.; Forte, I.M.; Indovina, P.; Di Gennaro, E.; Rizzo, V.; Giorgi, F.; Mattioli, E.; Iannuzzi, C.A.; Budillon, A.; Giordano, A.; et al. Pharmacological targeting of p53 through RITA is an effective antitumoral strategy for malignant pleural mesothelioma. Cell Cycle 2014, 13, 652–665. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Knudsen, E.S.; Wang, J.Y. Targeting the RB-pathway in cancer therapy. Clin. Cancer Res. 2010, 16, 1094–1099. [Google Scholar] [CrossRef]
- Indovina, P.; Pentimalli, F.; Casini, N.; Vocca, I.; Giordano, A. RB1 dual role in proliferation and apoptosis: Cell fate control and implications for cancer therapy. Oncotarget 2015, 6, 17873–17890. [Google Scholar] [CrossRef]
- Indovina, P.; Pentimalli, F.; Conti, D.; Giordano, A. Translating RB1 predictive value in clinical cancer therapy: Are we there yet? Biochem. Pharmacol. 2019, 166, 323–334. [Google Scholar] [CrossRef]
- Pentimalli, F.; Forte, I.M.; Esposito, L.; Indovina, P.; Iannuzzi, C.A.; Alfano, L.; Costa, C.; Barone, D.; Rocco, G.; Giordano, A. RBL2/p130 is a direct AKT target and is required to induce apoptosis upon AKT inhibition in lung cancer and mesothelioma cell lines. Oncogene 2018, 37, 3657–3671. [Google Scholar] [CrossRef] [PubMed]
- Indovina, P.; Giorgi, F.; Rizzo, V.; Khadang, B.; Schenone, S.; Di Marzo, D.; Forte, I.M.; Tomei, V.; Mattioli, E.; D’Urso, V.; et al. New pyrazolo[3,4-d]pyrimidine SRC inhibitors induce apoptosis in mesothelioma cell lines through p27 nuclear stabilization. Oncogene 2012, 31, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Bridges, K.A.; Chen, X.; Liu, H.; Rock, C.; Buchholz, T.A.; Shumway, S.D.; Skinner, H.D.; Meyn, R.E. MK-8776, a novel chk1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Oncotarget 2016, 7, 71660–71672. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, T.N.; Qian, C.; Sugitani, N.; Osmanbeyoglu, H.U.; Bakkenist, C.J. WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and S-phase cells. Proc. Natl. Acad. Sci. USA 2019, 116, 23891–23893. [Google Scholar] [CrossRef]
- Nojima, H.; Homma, H.; Onozato, Y.; Kaida, A.; Harada, H.; Miura, M. Differential properties of mitosis-associated events following CHK1 and WEE1 inhibitor treatments in human tongue carcinoma cells. Exp. Cell Res. 2020, 386, 111720. [Google Scholar] [CrossRef]
- Chung, S.; Vail, P.; Witkiewicz, A.K.; Knudsen, E.S. Coordinately Targeting Cell-Cycle Checkpoint Functions in Integrated Models of Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 2290–2304. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannuzzi, C.A.; Indovina, P.; Forte, I.M.; Di Somma, S.; Malfitano, A.M.; Bruno, M.; Portella, G.; Pentimalli, F.; Giordano, A. Pharmacological Inhibition of WEE1 Potentiates the Antitumoral Effect of the dl922-947 Oncolytic Virus in Malignant Mesothelioma Cell Lines. Int. J. Mol. Sci. 2020, 21, 7333. https://doi.org/10.3390/ijms21197333
Iannuzzi CA, Indovina P, Forte IM, Di Somma S, Malfitano AM, Bruno M, Portella G, Pentimalli F, Giordano A. Pharmacological Inhibition of WEE1 Potentiates the Antitumoral Effect of the dl922-947 Oncolytic Virus in Malignant Mesothelioma Cell Lines. International Journal of Molecular Sciences. 2020; 21(19):7333. https://doi.org/10.3390/ijms21197333
Chicago/Turabian StyleIannuzzi, Carmelina Antonella, Paola Indovina, Iris Maria Forte, Sarah Di Somma, Anna Maria Malfitano, Martina Bruno, Giuseppe Portella, Francesca Pentimalli, and Antonio Giordano. 2020. "Pharmacological Inhibition of WEE1 Potentiates the Antitumoral Effect of the dl922-947 Oncolytic Virus in Malignant Mesothelioma Cell Lines" International Journal of Molecular Sciences 21, no. 19: 7333. https://doi.org/10.3390/ijms21197333
APA StyleIannuzzi, C. A., Indovina, P., Forte, I. M., Di Somma, S., Malfitano, A. M., Bruno, M., Portella, G., Pentimalli, F., & Giordano, A. (2020). Pharmacological Inhibition of WEE1 Potentiates the Antitumoral Effect of the dl922-947 Oncolytic Virus in Malignant Mesothelioma Cell Lines. International Journal of Molecular Sciences, 21(19), 7333. https://doi.org/10.3390/ijms21197333