N-Acetyl Cysteine Modulates the Inflammatory and Oxidative Stress Responses of Rescued Growth-Arrested Dental Pulp Microtissues Exposed to TEGDMA in ECM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. NAC Supports the Proliferation and Viability of Human Dental Pulp Cells Affected by TEGDMA
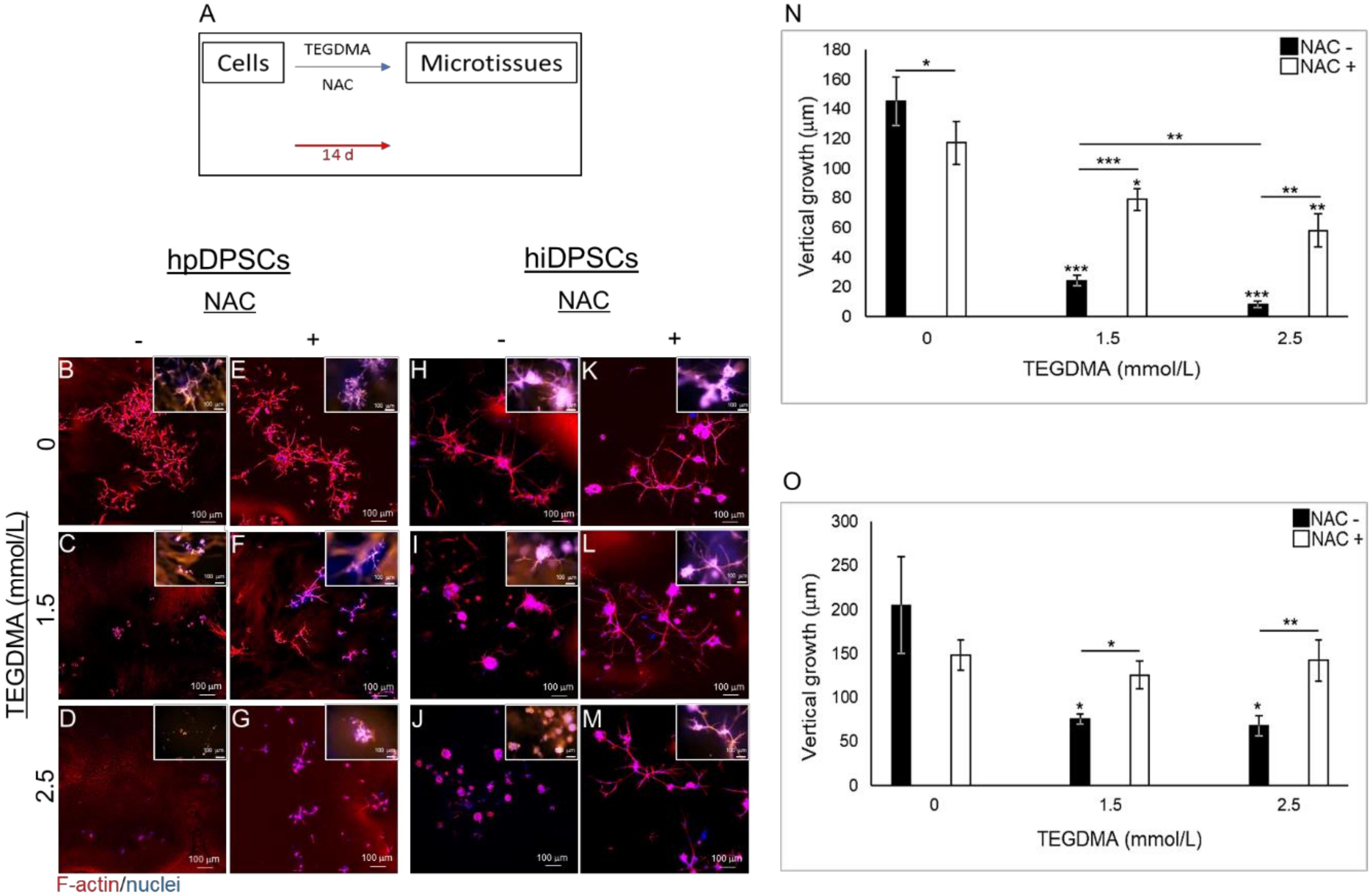
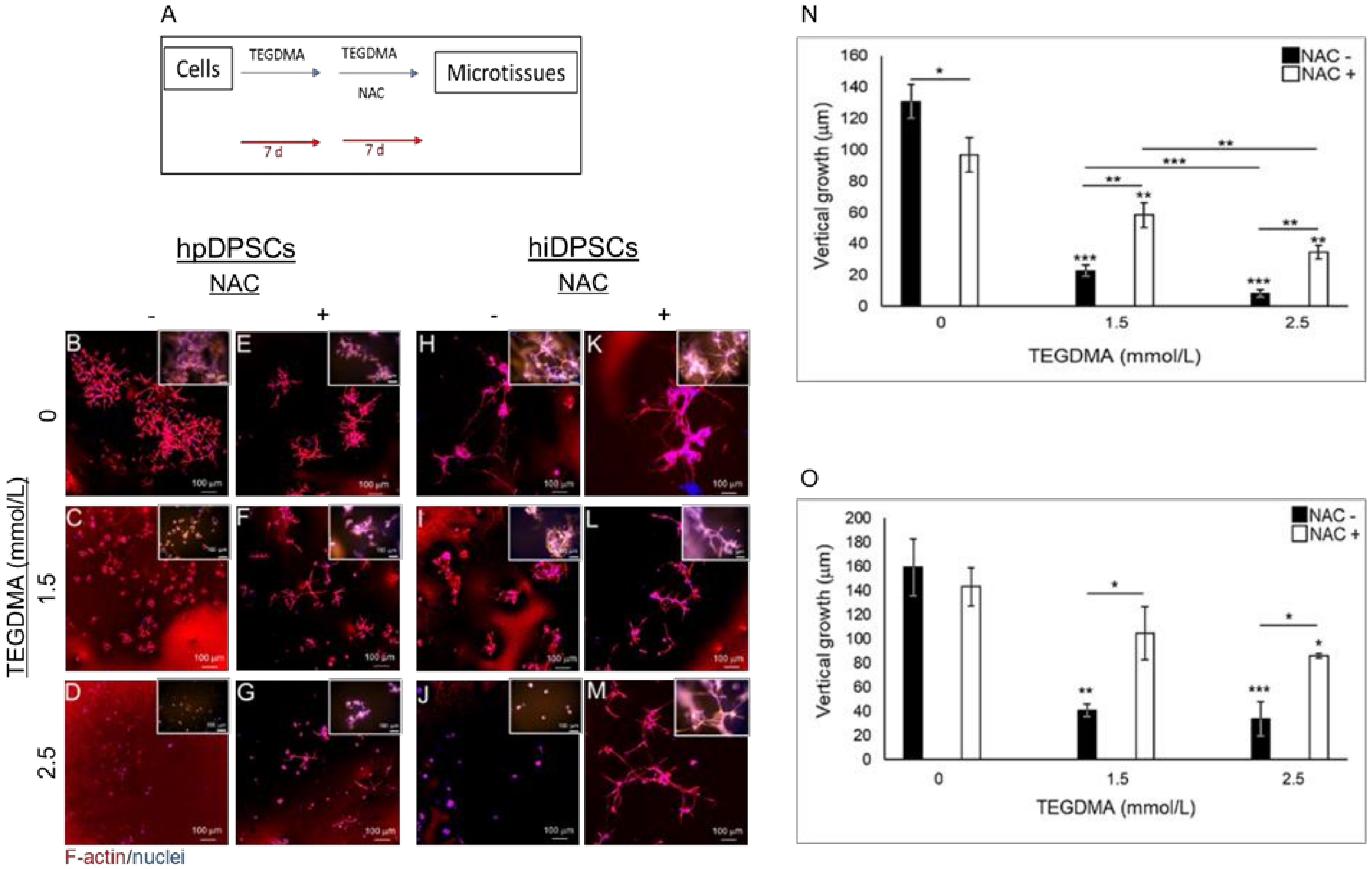
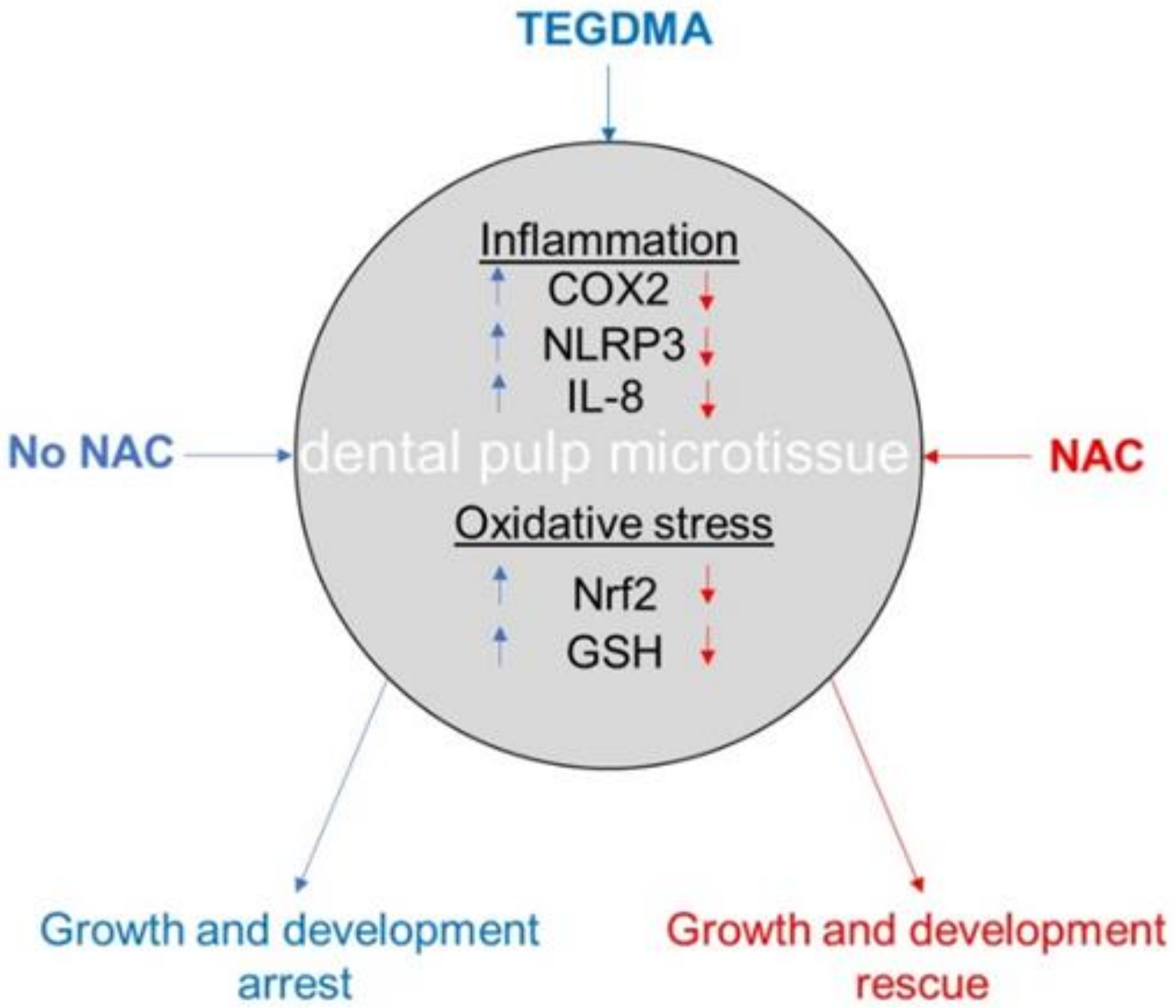
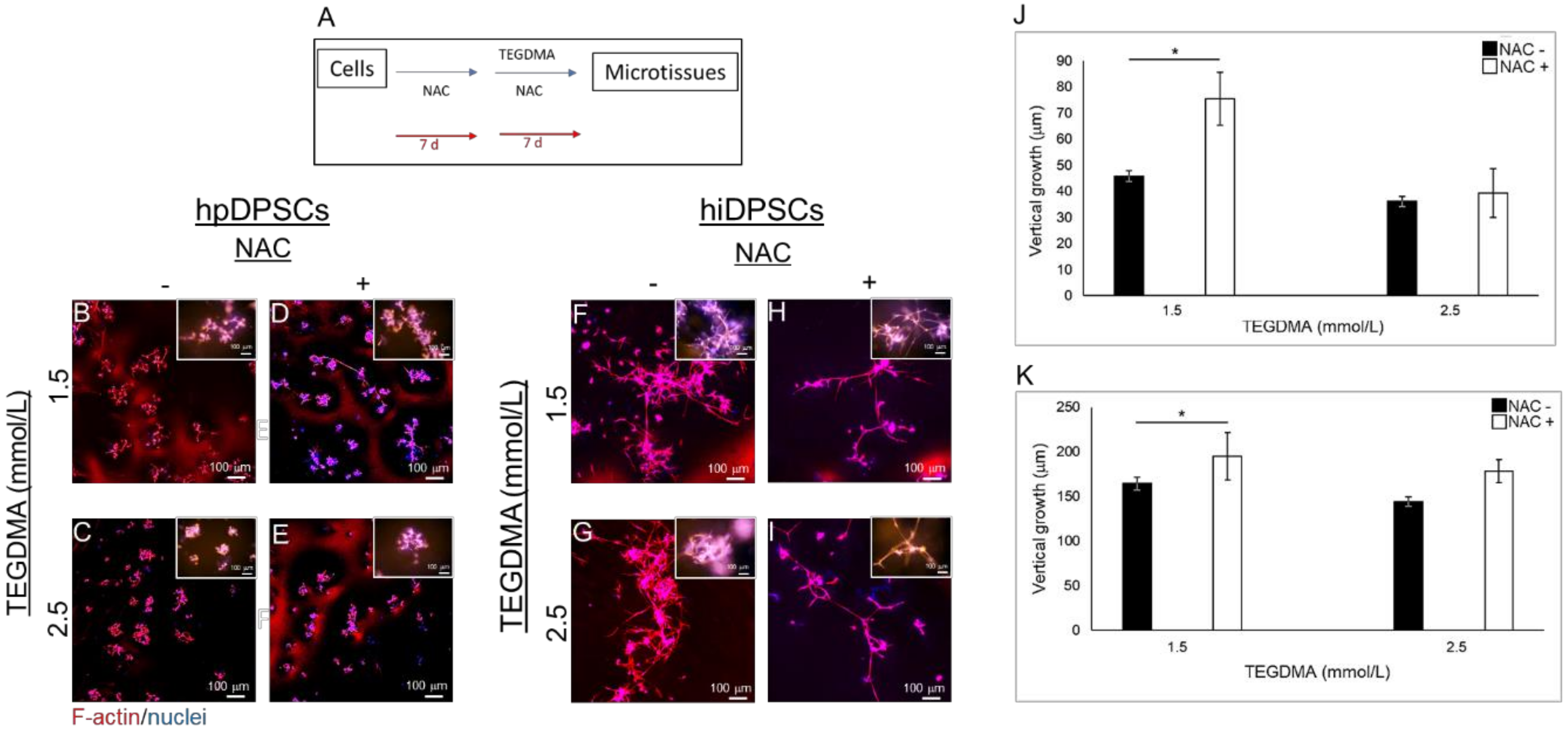
2.2. NAC Resumes the Development of Growth-Arrested Microtissues Exposed and Pre-Exposed to TEGDMA
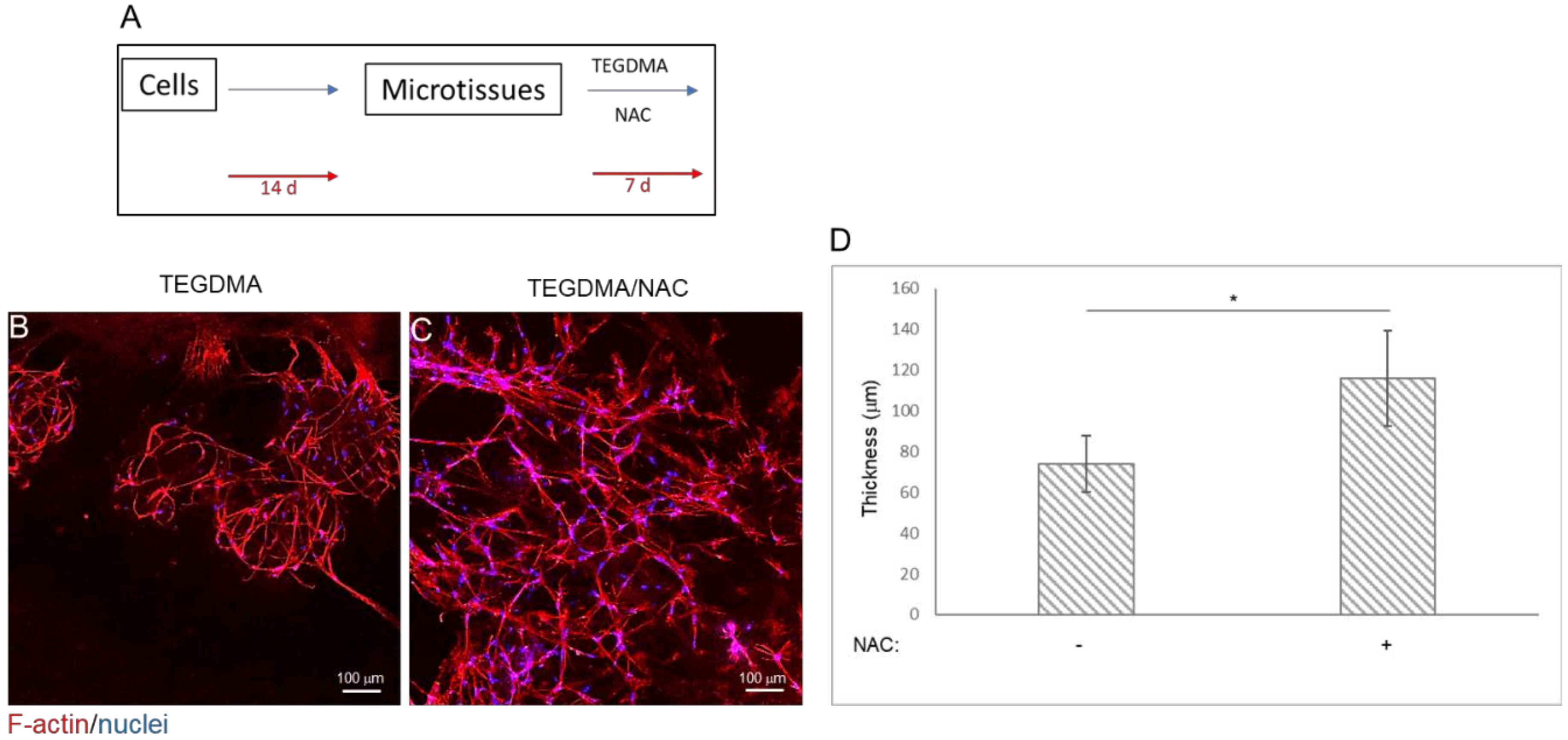
2.3. NAC Prevents Structural Deterioration of Developed and Matured Microtissues Exposed to TEGDMA
2.4. Modulation of the Inflammatory Response Correlates to the Regrowth and Development of TEGDMA-Affected Microtissues Treated by NAC
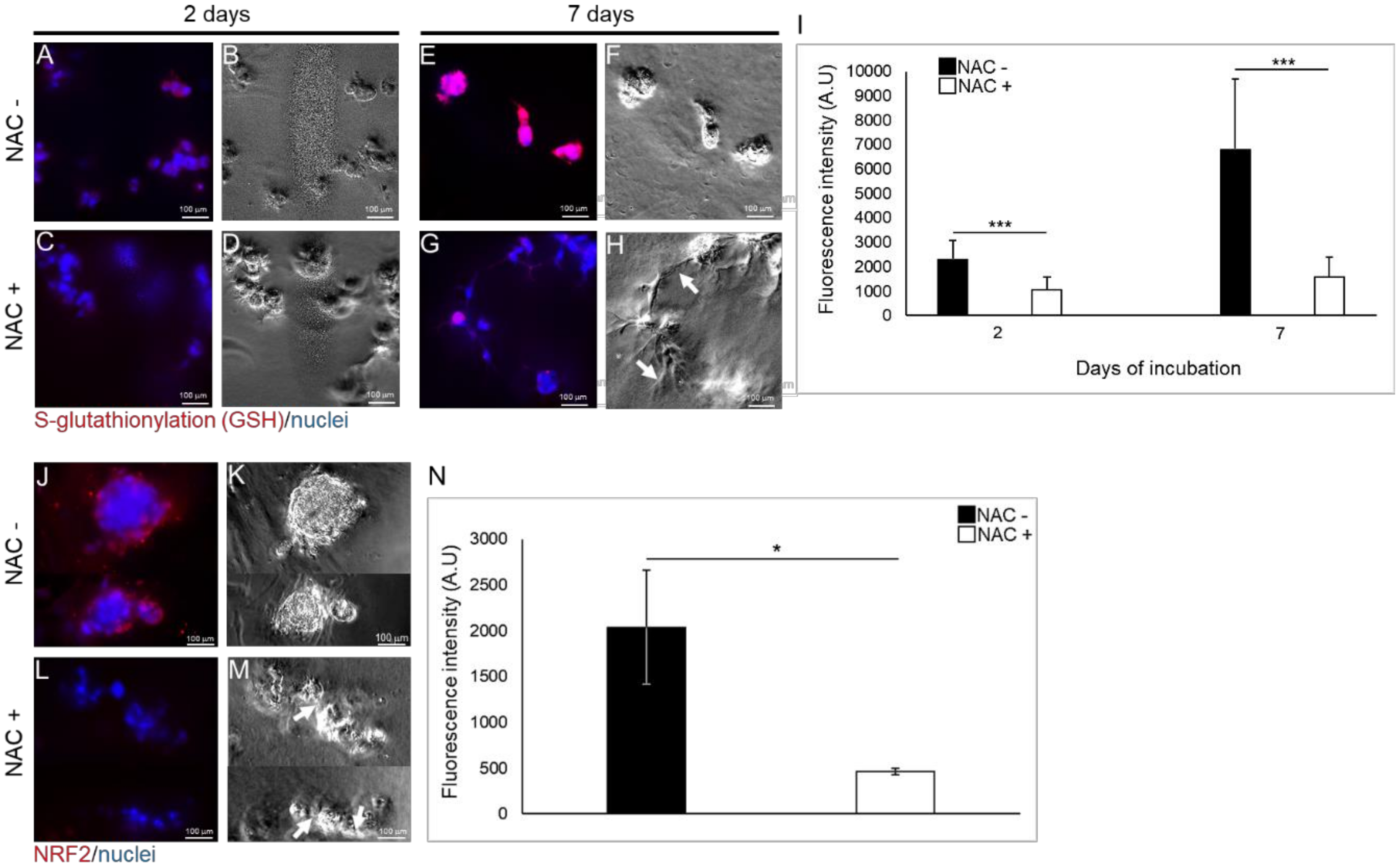
2.5. NAC Moderates the Expression of the Anti-Oxidative Response Elements in TEGDMA-Affected Microtissues
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. 3D ECM Cultures
4.3. Cell Proliferation and Viability
4.4. Cell Staining
4.5. Microscopy and Image Analysis
4.6. 3D Cell Culture ELISA
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADA | American Dental Association |
| ANOVA | Analysis of Variance |
| AU | Absorbance Units |
| COX2 | Cyclooxygenase 2 |
| d | Days |
| DAPI | 6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DPBS | Dulbecco’s Phosphate-Buffered Saline |
| ECM | Extracellular Matrix |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| FDA | Food and Drug Administration |
| GSH | Glutathione |
| H | Hours |
| HEMA | 2-hydroxyethyl methacrylate |
| hiDPSC | Human Immortalized Dental Pulp Stem Cell |
| hpDPSC | Human Primary Dental Pulp Stem Cell |
| H2O2 | Hydrogen Peroxide |
| IgG | Immunoglobulin G |
| IL-1β | Interleukin 1β |
| IL-8 | Interleukin 8 |
| LCSM | Laser-Scanning Confocal Microscopy |
| min | Minutes |
| MTT | (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NAC | N-Acetyl Cysteine |
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| Nrf2 | Nuclear Factor Erythroid-2-Related Factor 2 |
| OCT4 | Octamer-Binding Transcription Factor 4 |
| ROS | Reactive Oxygen Species |
| SOX2 | Sex Determined Region y-Box 2 |
| TEGDMA | Triethyleneglycol Dimethacrylate |
| UTHSC | University of Tennessee Health Science Center |
Appendix A

References
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in Chronic Wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in Wound Repair: Molecular and Cellular Mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 1–23. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Hansel, C.; Leyhausen, G.; Mai, U.; Geurtsen, W. Effects of various resin composite (co)monomers and extracts on two caries-associated micro-organisms in vitro. J. Dent. Res. 1998, 77, 60–67. [Google Scholar] [CrossRef]
- Bakopoulou, A.; Papadopoulos, T.; Garefis, P. Molecular Toxicology of Substances Released from Resin–Based Dental Restorative Materials. Int. J. Mol. Sci. 2009, 10, 3861–3899. [Google Scholar] [CrossRef]
- Ferracane, J.L. Elution of leachable components from composites. J. Oral Rehabilit. 1994, 21, 441–452. [Google Scholar] [CrossRef]
- Santerre, J.P.; Shajii, L.; Leung, B. Relation of dental composite formulations to their degradation and the release of hydrolyzed polymeric-resin-derived products. Crit. Rev. Oral Biol. Med. 2001, 12, 136–151. [Google Scholar] [CrossRef]
- Hume, W.; Gerzina, T. Bioavailability of Components of Resin-Based Materials Which Are Applied To Teeth. Crit. Rev. Oral Biol. Med. 1996, 7, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Bouillaguet, S. Biological Risks of Resin-based Materials to the Dentin-Pulp Complex. Crit. Rev. Oral Biol. Med. 2004, 15, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Wataha, J.; Kaga, M.; Lockwood, P.; Volkmann, K.; Sano, H. Components of Dentinal Adhesives Modulate Heat Shock Protein 72 Expression in Heat-stressed THP-1 Human Monocytes at Sublethal Concentrations. J. Dent. Res. 2002, 81, 265–269. [Google Scholar] [CrossRef]
- Modena, K.C.D.S.; Casas-Apayco, L.C.; Atta, M.T.; Costa, C.A.D.S.; Hebling, J.; Sipert, C.R.; Navarro, M.F.D.L.; Santos, C.F. Cytotoxicity and biocompatibility of direct and indirect pulp capping materials. J. Appl. Oral Sci. 2009, 17, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.C.; Segala, A.D.; Costa, C.A. Human pulpal response to direct pulp capping with an adhesive system. Am. J. Dent. 2000, 13, 139–147. [Google Scholar]
- Geurtsen, W.; Leyhausen, G. Chemical-Biological Interactions of the resin monomer triethyleneglycol-dimethacrylate (TEGDMA). J. Dent. Res. 2001, 80, 2046–2050. [Google Scholar] [CrossRef]
- Stanislawski, L.; Lefeuvre, M.; Bourd, K.; Soheili-Majd, E.; Goldberg, M.; Périanin, A. TEGDMA-induced toxicity in human fibroblasts is associated with early and drastic glutathione depletion with subsequent production of oxygen reactive species. J. Biomed. Mater. Res. Part A 2003, 66, 476–482. [Google Scholar] [CrossRef]
- Chang, H.-H.; Chang, M.-C.; Huang, G.-F.; Wang, Y.-L.; Chan, C.-P.; Wang, T.-M.; Lin, P.-S.; Jeng, J.-H. Effect of triethylene glycol dimethacrylate on the cytotoxicity, cyclooxygenase-2 expression and prostanoids production in human dental pulp cells. Int. Endod. J. 2012, 45, 848–858. [Google Scholar] [CrossRef]
- Schweikl, H.; Altmannberger, I.; Hanser, N.; Hiller, K.-A.; Bolay, C.; Brockhoff, G.; Spagnuolo, G.; Galler, K.; Schmalz, G. The effect of triethylene glycol dimethacrylate on the cell cycle of mammalian cells. Biomaterials 2005, 26, 4111–4118. [Google Scholar] [CrossRef]
- Tigani, E.K.; Skrtic, A.; Valerio, M.S.; Kaufman, G. Assessing the effect of triethyleneglycol dimethacrylate on tissue repair in 3D organotypic cultures. J. Appl. Toxicol. 2018, 39, 247–259. [Google Scholar] [CrossRef]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative Stress, Prooxidants, and Antioxidants: The Interplay. Bio. Med. Res. Int. 2014, 2014, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Pojšak, B.; Šuput, D.; Milisav, I. Achieving the Balance between ROS and Antioxidants: When to Use the Synthetic Antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Bodnár, E.; Bakondi, E.; Kovács, K.; Hegedűs, C.; Lakatos, P.; Robaszkiewicz, A.; Regdon, Z.; Virág, L.; Szabó, É. Redox Profiling Reveals Clear Differences between Molecular Patterns of Wound Fluids from Acute and Chronic Wounds. Oxid. Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Bryan, N.; Ashwin, H.; Smart, N.A.; Bayon, Y.; Wohlert, S.; Hunt, J.A. Reactive oxygen species (ROS)—A family of fate deciding molecules pivotal in constructive inflammation and wound healing. Eur. Cell Mater. 2012, 24, 249–265. [Google Scholar] [CrossRef]
- Cano-Sanchez, M.; Lancel, S.; Boulanger, É.; Neviere, R. Targeting Oxidative Stress and Mitochondrial Dysfunction in the Treatment of Impaired Wound Healing: A Systematic Review. Antioxidants 2018, 7, 98. [Google Scholar] [CrossRef]
- Jiao, Y.; Ma, S.; Wang, Y.; Li, J.; Shan, L.; Liu, Q.; Liu, Y.; Song, Q.; Yu, F.; Yu, H.; et al. N-Acetyl Cysteine Depletes Reactive Oxygen Species and Prevents Dental Monomer-Induced Intrinsic Mitochondrial Apoptosis In Vitro in Human Dental Pulp Cells. PLoS ONE 2016, 11, e0147858. [Google Scholar] [CrossRef]
- Yeh, C.-C.; Chang, J.Z.-C.; Yang, W.-H.; Chang, H.-H.; Lai, E.H.-H.; Kuo, M.Y.-P. NADPH oxidase 4 is involved in the triethylene glycol dimethacrylate-induced reactive oxygen species and apoptosis in human embryonic palatal mesenchymal and dental pulp cells. Clin. Oral Investig. 2014, 19, 1463–1471. [Google Scholar] [CrossRef]
- Chang, H.-H. Stimulation of glutathione depletion, ROS production and cell cycle arrest of dental pulp cells and gingival epithelial cells by HEMA. Biomaterials 2005, 26, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Schweikl, H.; Spagnuolo, G.; Schmalz, G. Genetic and cellular toxicology of dental resin monomers. J. Dent. Res. 2006, 85, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Liu, H.; Yang, Y.; Yang, Y.; Jiao, Y.; Tay, F.R.; Chen, J. Biological Activities and Potential Oral Applications of N-Acetylcysteine: Progress and Prospects. Oxidative Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free. Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Šalamon, Š.; Kramar, B.; Marolt, T.P.; Pojšak, B.; Milisav, I. Medical and Dietary Uses of N-Acetylcysteine. Antioxidants 2019, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Shahripour, R.B.; Harrigan, M.R.; Alexandrov, A.V. N-acetylcysteine (NAC) in neurological disorders: Mechanisms of action and therapeutic opportunities. Brain Behav. 2014, 4, 108–122. [Google Scholar] [CrossRef]
- Palacio, J.R.; Markert, U.R.; Martinez, P. Anti-inflammatory properties of N-acetylcysteine on lipopolysaccharide-activated macrophages. Inflamm. Res. 2011, 60, 695–704. [Google Scholar] [CrossRef]
- Cazzola, M.; Calzetta, L.; Facciolo, F.; Rogliani, P.; Matera, M.G. Pharmacological investigation on the anti-oxidant and anti-inflammatory activity of N-acetylcysteine in an ex vivo model of COPD exacerbation. Respir. Res. 2017, 18, 26. [Google Scholar] [CrossRef]
- Calzetta, L.; Rogliani, P.; Facciolo, F.; Rinaldi, B.; Cazzola, M.; Matera, M.G. N-Acetylcysteine protects human bronchi by modulating the release of neurokinin A in an ex vivo model of COPD exacerbation. Biomed. Pharmacother. 2018, 103, 1–8. [Google Scholar] [CrossRef]
- Chen, G.; Shi, J.; Hu, Z.; Hang, C. Inhibitory Effect on Cerebral Inflammatory Response following Traumatic Brain Injury in Rats: A Potential Neuroprotective Mechanism of N-Acetylcysteine. Mediat. Inflamm. 2008, 2008, 1–8. [Google Scholar] [CrossRef]
- Csontos, C.; Rezman, B.; Foldi, V.; Bogar, L.; Drenkovics, L.; Roth, E.; Weber, G.; Lantos, J. Effect of N-acetylcysteine treatment on oxidative stress and inflammation after severe burn. Burns 2012, 38, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, M.; Zhang, W.; Gu, A.; Dong, J.; Li, J.; Shan, A. Protective Effect of N-Acetylcysteine against Oxidative Stress Induced by Zearalenone via Mitochondrial Apoptosis Pathway in SIEC02 Cells. Toxins 2018, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Kerksick, C.; Willoughby, D. The Antioxidant Role of Glutathione and N-Acetyl-Cysteine Supplements and Exercise-Induced Oxidative Stress. J. Int. Soc. Sports Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Yamada, M.; Sugita, Y.; Ogawa, T. N-Acetyl Cysteine Protects TMJ Chondrocytes from Oxidative Stress. J. Dent. Res. 2010, 90, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-H.; Choi, Y.-S.; Lee, H.-W.; Heo, J.S.; Chang, S.W.; Lee, J.-Y. Antibacterial effects of N-acetylcysteine against endodontic pathogens. J. Microbiol. 2016, 54, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-H.; Jang, E.-Y.; Shim, K.S.; Lee, J.-Y. In vitro effects of N-acetyl cysteine alone and in combination with antibiotics on Prevotella intermedia. J. Microbiol. 2015, 53, 321–329. [Google Scholar] [CrossRef]
- Rasmussen, K.; Nikrad, J.; Reilly, C.; Li, Y.; Jones, R. N-Acetyl-l-cysteine effects on multi-species oral biofilm formation and bacterial ecology. Lett. Appl. Microbiol. 2015, 62, 30–38. [Google Scholar] [CrossRef]
- Blasi, F.; Page, C.P.; Rossolini, G.M.; Pallecchi, L.; Matera, M.G.; Rogliani, P.; Cazzola, M. The effect of N -acetylcysteine on biofilms: Implications for the treatment of respiratory tract infections. Respir. Med. 2016, 117, 190–197. [Google Scholar] [CrossRef]
- Kumar, J.; Teoh, S.L.; Das, S.; Mahakknaukrauh, P. Oxidative Stress in Oral Diseases: Understanding Its Relation with Other Systemic Diseases. Front. Physiol. 2017, 8, 693. [Google Scholar] [CrossRef]
- Żukowski, P.; Maciejczyk, M.; Waszkiel, D. Sources of free radicals and oxidative stress in the oral cavity. Arch. Oral Biol. 2018, 92, 8–17. [Google Scholar] [CrossRef]
- Żukowski, P.; Maciejczyk, M.; Matczuk, J.; Kurek, K.; Waszkiel, D.; Żendzian-Piotrowska, M.; Zalewska, A. Effect of N-Acetylcysteine on Antioxidant Defense, Oxidative Modification, and Salivary Gland Function in a Rat Model of Insulin Resistance. Oxid. Med. Cell. Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.; Magli, S.; Rabbachin, L.; Sampaolesi, S.; Nicotra, F.; Russo, L. 3D Extracellular Matrix Mimics: Fundamental Concepts and Role of Materials Chemistry to Influence Stem Cell Fate. Biomacromolecules 2020, 21, 1968–1994. [Google Scholar] [CrossRef] [PubMed]
- Millea, P.J. N-acetylcysteine: Multiple clinical applications. Am. Fam. Physician 2009, 80, 265–269. [Google Scholar]
- Chang, M.C.; Lin, L.-D.; Wu, M.-T.; Chan, C.-P.; Chang, H.-H.; Lee, M.-S.; Sun, T.-Y.; Jeng, P.-Y.; Yeung, S.-Y.; Lin, H.-J.; et al. Effects of Camphorquinone on Cytotoxicity, Cell Cycle Regulation and Prostaglandin E2 Production of Dental Pulp Cells: Role of ROS, ATM/Chk2, MEK/ERK and Hemeoxygenase-1. PLoS ONE 2015, 10, e0143663. [Google Scholar] [CrossRef] [PubMed]
- Parasassi, T.; Brunelli, R.; Bracci-Laudiero, L.; Greco, G.; Gustafsson, A.C.; Krasnowska, E.K.; Lundeberg, J.; Lundeberg, T.; Pittaluga, E.; Romano, M.C.; et al. Differentiation of normal and cancer cells induced by sulfhydryl reduction: Biochemical and molecular mechanisms. Cell Death Differ. 2005, 12, 1285–1296. [Google Scholar] [CrossRef]
- Rivabene, R.; Viora, M.; Matarrese, P.; Rainaldi, G.; D’Ambrosio, A.; Malorni, W. N-acetyl-cysteine enhances cell adhesion properties of epithelial and lymphoid cells. Cell Biol. Int. 1995, 19, 681–686. [Google Scholar] [CrossRef]
- Park, W.H.; You, B.R. Antimycin A induces death of the human pulmonary fibroblast cells via ROS increase and GSH depletion. Int. J. Oncol. 2016, 48, 813–820. [Google Scholar] [CrossRef]
- Park, W.H. The effect of MAPK inhibitors and ROS modulators on cell growth and death of H(2)O(2)-treated HeLa cells. Mol. Med. Rep. 2013, 8, 557–564. [Google Scholar] [CrossRef]
- Kaneko, Y.; Tanigawa, N.; Sato, Y.; Kobayashi, T.; Nakamura, S.; Ito, E.; Soma, T.; Miyamoto, K.; Kobayashi, S.; Harato, K.; et al. Oral administration of N-acetyl cysteine prevents osteoarthritis development and progression in a rat model. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Joshi, D.; Mittal, D.K.; Shukla, S.; Srivastav, A.K.; Srivastav, S.K. N-acetyl cysteine and selenium protects mercuric chloride-induced oxidative stress and antioxidant defense system in liver and kidney of rats: A histopathological approach. J. Trace Elements Med. Biol. 2014, 28, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.; Njeh, A.; Uzunoglu, E. Is Pulp Inflammation a Prerequisite for Pulp Healing and Regeneration? Mediat. Inflamm. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Ma, S.; Li, J.; Shan, L.; Wang, Y.; Tian, M.; Yang, Y.; Sun, J.; Ban, J.; Chen, J. N-Acetyl Cysteine (NAC)-Directed Detoxification of Methacryloxylethyl Cetyl Ammonium Chloride (DMAE-CB). PLoS ONE 2015, 10, e0135815. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-C.; Chang, H.-H.; Lee, M.-Y.; Lin, C.-C.; Yeh, H.-W.; Yang, T.-T.; Lin, P.-S.; Tseng, W.-Y.; Jeng, J.-H. Prostaglandin F2α-Induced Interleukin-8 Production in Human Dental Pulp Cells Is Associated With MEK/ERK Signaling. J. Endod. 2009, 35, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Xia, Z.; Jiang, J.; McNeill, J.H. Downregulation of NADPH oxidase, antioxidant enzymes, and inflammatory markers in the heart of streptozotocin-induced diabetic rats by N-acetyl-l-cysteine. Am. J. Physiol. Circ. Physiol. 2007, 292, H1728–H1736. [Google Scholar] [CrossRef] [PubMed]
- Burton, L.; Paget, D.; Binder, N.B.; Bohnert, K.; Nestor, B.J.; Sculco, T.P.; Santambrogio, L.; Ross, F.P.; Goldring, S.R.; Purdue, P.E. Orthopedic wear debris mediated inflammatory osteolysis is mediated in part by NALP3 inflammasome activation. J. Orthop. Res. 2012, 31, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Alizadehgharib, S.; Östberg, A.-K.; Dahlgren, U. Triethylene glycol dimethacrylate: Adjuvant properties and effect on cytokine production. Acta Biomater. Odontol. Scand. 2017, 4, 1–9. [Google Scholar] [CrossRef]
- Hensten-Pettersen, A. Skin and mucosal reactions associated with dental materials. Eur. J. Oral Sci. 1998, 106, 707–712. [Google Scholar]
- Cho, K.A.; Suh, J.W.; Lee, K.H.; Kang, J.L.; Woo, S.-Y. IL-17 and IL-22 enhance skin inflammation by stimulating the secretion of IL-1beta by keratinocytes via the ROS-NLRP3-caspase-1 pathway. Int. Immunol. 2012, 24, 147–158. [Google Scholar] [CrossRef]
- Alizadehgharib, S.; Östberg, A.-K.; Larsson, L.; Dahlgren, U. The Immunomodulatory Properties of 2-Hydroxyethyl Methacrylate are Mediated by the NLRP3 Inflammasome. J. Adhes. Dent. 2018, 20, 213–220. [Google Scholar]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, P.; Ma, X.; Yin, X.; Li, J.; Wang, H.; Jiang, W.; Jia, Q.; Ni, L. Mechanisms that lead to the regulation of NLRP3 inflammasome expression and activation in human dental pulp fibroblasts. Mol. Immunol. 2015, 66, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Lv, H.; Wang, H.; Wang, D.; Sun, S.; Jia, Q.; Wang, P.; Song, B.; Ni, L. Activation of the NLRP3/caspase-1 inflammasome in human dental pulp tissue and human dental pulp fibroblasts. Cell Tissue Res. 2015, 361, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Su, S.-H.; Wu, Y.-F.; Lin, Q.; Wang, D.-P.; Hai, J. URB597 protects against NLRP3 inflammasome activation by inhibiting autophagy dysfunction in a rat model of chronic cerebral hypoperfusion. J. Neuroinflamm. 2019, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat. Immunol. 2008, 9, 866–872. [Google Scholar] [CrossRef]
- Rubartelli, A. Redox control of NLRP3 inflammasome activation in health and disease. J. Leukoc. Biol. 2012, 92, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Jun, J.-H.; Lee, H.-L.; Woo, K.M.; Ryoo, H.-M.; Kim, G.-S.; Baek, J.-H.; Han, S.-B. N-acetyicysteine prevents lps-lnduced pro-inflammatory cytokines and mmp2 production in gingival fibroblasts. Arch. Pharmacal Res. 2007, 30, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Krifka, S.; Seidenader, C.; Hiller, K.-A.; Schmalz, G.; Schweikl, H. Oxidative stress and cytotoxicity generated by dental composites in human pulp cells. Clin. Oral Investig. 2011, 16, 215–224. [Google Scholar] [CrossRef]
- Chang, M.-C.; Chen, L.-I.; Chan, C.-P.; Lee, J.-J.; Wang, T.-M.; Yang, T.-T.; Lin, P.-S.; Lin, H.-J.; Chang, H.-H.; Jeng, J.-H. The role of reactive oxygen species and hemeoxygenase-1 expression in the cytotoxicity, cell cycle alteration and apoptosis of dental pulp cells induced by BisGMA. Biomaterials 2010, 31, 8164–8171. [Google Scholar] [CrossRef]
- Eckhardt, A.; Müller, P.; Hiller, K.-A.; Krifka, S.; Bolay, C.; Spagnuolo, G.; Schmalz, G.; Schweikl, H. Influence of TEGDMA on the mammalian cell cycle in comparison with chemotherapeutic agents. Dent. Mater. 2010, 26, 232–241. [Google Scholar] [CrossRef]
- Schweikl, H.; Hartmann, A.; Hiller, K.-A.; Spagnuolo, G.; Bolay, C.; Brockhoff, G.; Schmalz, G. Inhibition of TEGDMA and HEMA-induced genotoxicity and cell cycle arrest by N-acetylcysteine. Dent. Mater. 2007, 23, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, A.; Cacalano, N.A.; Hume, W.R.; Jewett, A. Mechanisms of N-acetyl Cysteine–mediated Protection From 2-Hydroxyethyl Methacrylate–induced Apoptosis. J. Endod. 2008, 34, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.R.; Lim, B.-S.; Park, H.C.; Son, K.M.; Yang, H.-C. Effects of N-acetylcysteine on TEGDMA- and HEMA-induced suppression of osteogenic differentiation of human osteosarcoma MG63 cells. J. Biomed. Mater. Res. Part B Appl. Biomater. 2011, 98, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, J.; Leyhausen, G.; Leibfritz, D.; Geurtsen, W. Effect of TEGDMA on the intracellular glutathione concentration of human gingival fibroblasts. J. Biomed. Mater. Res. 2002, 63, 746–751. [Google Scholar] [CrossRef]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-Glutathionylation: From Molecular Mechanisms to Health Outcomes. Antioxidants Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef]
- Ghezzi, P.; Romines, B.; Fratelli, M.; Eberini, I.; Gianazza, E.; Casagrande, S.; Laragione, T.; Mengozzi, M.; Herzenberg, L.A. Protein glutathionylation: Coupling and uncoupling of glutathione to protein thiol groups in lymphocytes under oxidative stress and HIV infection. Mol. Immunol. 2002, 38, 773–780. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Li, N.; Alam, J.; Venkatesan, M.I.; Eiguren-Fernandez, A.; Schmitz, D.; Di Stefano, E.; Slaughter, N.; Killeen, E.; Wang, X.; Huang, A.; et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: Protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J. Immunol. 2004, 173, 3467–3481. [Google Scholar] [CrossRef]
- Lee, D.; Kook, S.-H.; Ji, H.; Lee, S.-A.; Choi, K.-C.; Lee, K.-Y.; Lee, J.-C. N-acetyl cysteine inhibits H2O2-mediated reduction in the mineralization of MC3T3-E1 cells by down-regulating Nrf2/HO-1 pathway. BMB Rep. 2015, 48, 636–641. [Google Scholar] [CrossRef]
- Gallorini, M.; Petzel, C.; Bolay, C.; Hiller, K.-A.; Cataldi, A.; Buchalla, W.; Krifka, S.; Schweikl, H. Activation of the Nrf2-regulated antioxidant cell response inhibits HEMA-induced oxidative stress and supports cell viability. Biomaterials 2015, 56, 114–128. [Google Scholar] [CrossRef]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.S.; Goltsov, A.; Langdon, S.P.; Harrison, D.J.; Bown, J.L.; Deeni, Y.Y. Quantitative analysis of NRF2 pathway reveals key elements of the regulatory circuits underlying antioxidant response and proliferation of ovarian cancer cells. J. Biotechnol. 2015, 202, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Diomede, F.; Marconi, G.D.; Guarnieri, S.; D’Attilio, M.; Cavalcanti, M.F.X.B.; Mariggiò, M.A.; Pizzicannella, J.; Trubiani, O. A Novel Role of Ascorbic Acid in Anti-Inflammatory Pathway and ROS Generation in HEMA Treated Dental Pulp Stem Cells. Materials 2019, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Rapino, M.; Di Valerio, V.; Zara, S.; Gallorini, M.; Marconi, G.D.; Sancilio, S.; Marsich, E.; Ghinassi, B.; Di Giacomo, V.; Cataldi, A.; et al. Chitlac-coated Thermosets Enhance Osteogenesis and Angiogenesis in a Co-culture of Dental Pulp Stem Cells and Endothelial Cells. Nanomaterials 2019, 9, 928. [Google Scholar] [CrossRef]
- Hilkens, P.; Gervois, P.; Fanton, Y.; Vanormelingen, J.; Martens, W.; Struys, T.; Politis, C.; Lambrichts, I.; Bronckaers, A. Effect of isolation methodology on stem cell properties and multilineage differentiation potential of human dental pulp stem cells. Cell Tissue Res. 2013, 353, 65–78. [Google Scholar] [CrossRef]
- Kaufman, G.; Kiburi, N.M.; Skrtic, D. The self-renewal dental pulp stem cell microtissues challenged by a toxic dental monomer. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Urraca, N.; Memon, R.; El-Iyachi, I.; Goorha, S.; Valdez, C.; Tran, Q.T.; Scroggs, R.; Miranda-Carboni, G.A.; Donaldson, M.; Weisman, L.; et al. Characterization of neurons from immortalized dental pulp stem cells for the study of neurogenetic disorders. Stem Cell Res. 2015, 15, 722–730. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaufman, G.; Skrtic, D. N-Acetyl Cysteine Modulates the Inflammatory and Oxidative Stress Responses of Rescued Growth-Arrested Dental Pulp Microtissues Exposed to TEGDMA in ECM. Int. J. Mol. Sci. 2020, 21, 7318. https://doi.org/10.3390/ijms21197318
Kaufman G, Skrtic D. N-Acetyl Cysteine Modulates the Inflammatory and Oxidative Stress Responses of Rescued Growth-Arrested Dental Pulp Microtissues Exposed to TEGDMA in ECM. International Journal of Molecular Sciences. 2020; 21(19):7318. https://doi.org/10.3390/ijms21197318
Chicago/Turabian StyleKaufman, Gili, and Drago Skrtic. 2020. "N-Acetyl Cysteine Modulates the Inflammatory and Oxidative Stress Responses of Rescued Growth-Arrested Dental Pulp Microtissues Exposed to TEGDMA in ECM" International Journal of Molecular Sciences 21, no. 19: 7318. https://doi.org/10.3390/ijms21197318
APA StyleKaufman, G., & Skrtic, D. (2020). N-Acetyl Cysteine Modulates the Inflammatory and Oxidative Stress Responses of Rescued Growth-Arrested Dental Pulp Microtissues Exposed to TEGDMA in ECM. International Journal of Molecular Sciences, 21(19), 7318. https://doi.org/10.3390/ijms21197318