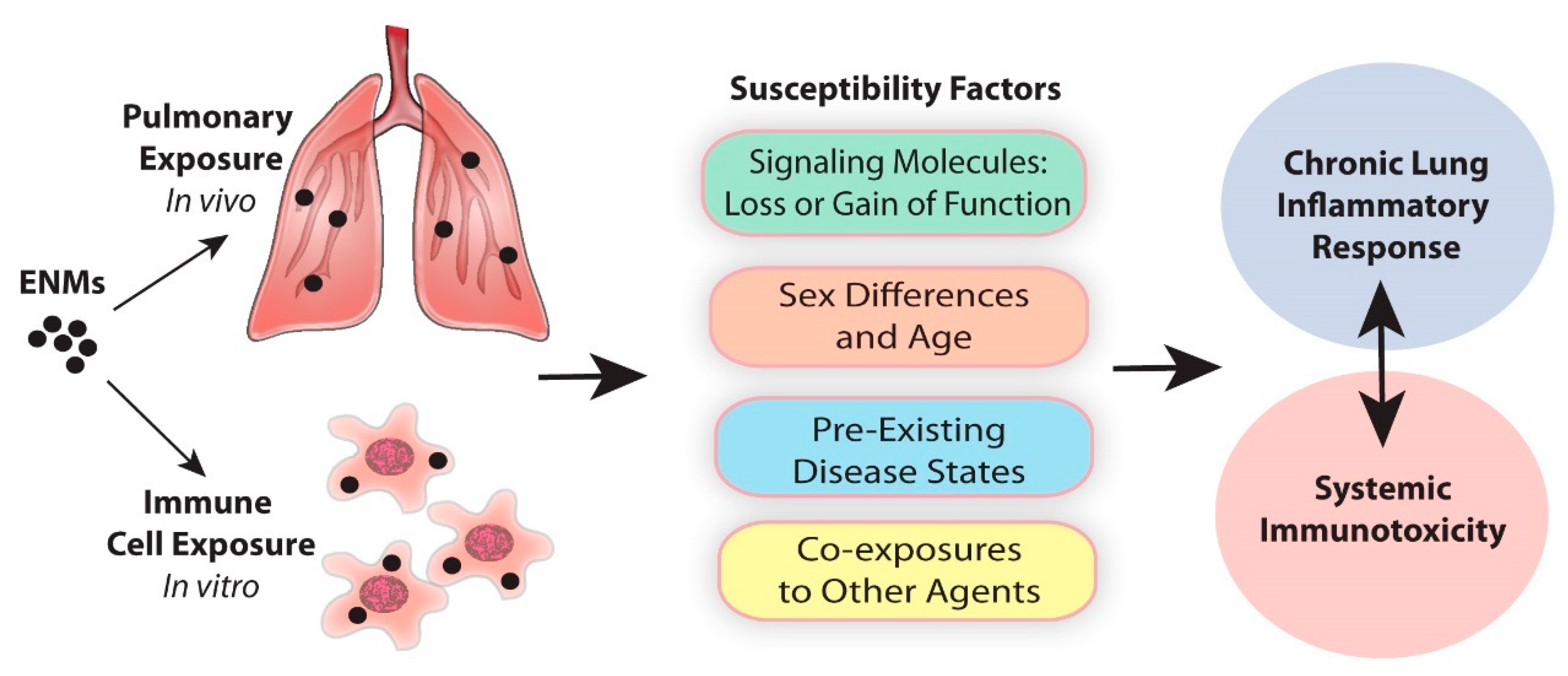
Susceptibility Factors in Chronic Lung Inflammatory Responses to Engineered Nanomaterials

Abstract
1. Introduction
2. Deficiency in Cell Signaling Molecules as Determinants of Susceptibility to ENM-Induced Chronic Lung Inflammation
2.1. Transcription Factors
2.1.1. STAT1
2.1.2. T-bet
2.1.3. Nrf2
2.1.4. P53
2.1.5. BMAL1
2.2. Enzymes/Proteins
2.2.1. NADPH Oxidase
2.2.2. COX-2
2.2.3. TIMP1
2.2.4. MPO
2.2.5. ApoE
2.3. Receptors
2.3.1. AhR
2.3.2. CCR5
2.4. Cytokines/Chemokines
2.4.1. IL-1/Inflammasome
2.4.2. OPN
2.4.3. IL-6
3. Sex
4. Susceptible Organ Systems and Pre-Existing Disease States
4.1. Lung
4.2. Cardiovascular
4.3. Liver
4.4. Spleen
4.5. Brain
5. Co-Exposures to ENMs and Other Agents
6. Challenges and Alternative Approaches
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Magun, B.E.; Wood, L.J. Lung inflammation caused by inhaled toxicants: A review. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of in flammation: State of the art, definitions and terms. FASEB J. 2007, 21, 325–332. [Google Scholar] [CrossRef]
- Schett, G.A.; Neurath, M.F. Resolution of chronic inflammatory disease: Universal and tissue-specific concepts. Nat. Commun. 2018, 9, 3261. [Google Scholar] [CrossRef]
- Barroso, L.C.; Magalhaes, G.S.; Galvão, I.; Reis, A.C.; Souza, D.G.; Sousa, L.P.; Santos, R.A.S.; Campagnole-Santos, M.J.; Pinho, V.; Teixeira, M.M. Angiotensin-(1-7) promotes resolution of neutrophilic inflammation in a model of antigen-induced arthritis in mice. Front. Immunol. 2017, 8, 1596. [Google Scholar] [CrossRef]
- Galvão, I.; Athayde, R.M.; Perez, D.A.; Reis, A.C.; Rezende, L.; De Oliveira, V.L.S.; Rezende, B.M.; Gonçalves, W.A.; Sousa, L.P.; Teixeira, M.M.; et al. ROCK inhibition drives resolution of acute inflammation by enhancing neutrophil apoptosis. Cells 2019, 8, 964. [Google Scholar] [CrossRef]
- Lim, C.S.; Porter, D.W.; Orandle, M.S.; Green, B.J.; Barnes, M.A.; Croston, T.L.; Wolfarth, M.G.; Battelli, L.A.; Andrew, M.E.; Beezhold, D.H.; et al. Resolution of pulmonary inflammation induced by carbon nanotubes and fullerenes in mice: Role of macrophage polarization. Front. Immunol. 2020, 11, 1186. [Google Scholar] [CrossRef]
- Sansbury, B.E.; Spite, M. Resolution of acute inflammation and the role of Resolvins in immunity, thrombosis, and vascular biology. Circ. Res. 2016, 119, 113–130. [Google Scholar] [CrossRef]
- Varela, M.L.; Mogildea, M.; Moreno, I.; Lopes, A. Acute inflammation and metabolism. Inflammation 2018, 41, 1115–1127. [Google Scholar] [CrossRef]
- Robb, C.T.; Regan, K.H.; Dorward, D.A.; Rossi, A.G. Key mechanisms governing resolution of lung inflammation. Semin. Immunopathol. 2016, 38, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Serhan, C.N. Resolution of acute inflammation in the lung. Annu. Rev. Physiol. 2013, 76, 467–492. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Bioactive lipids and chronic inflammation: Managing the fire within. Front. Immunol. 2018, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.R.; Lloyd, C.M. Chronic inflammation and asthma. Mutat. Res. Mol. Mech. Mutagen. 2010, 690, 24–39. [Google Scholar] [CrossRef]
- Jain, A.; Pasare, C. Innate control of adaptive immunity: Beyond the three-signal paradigm. J. Immunol. 2017, 198, 3791–3800. [Google Scholar] [CrossRef]
- Pesic, M.; Greten, F.R. Inflammation and cancer: Tissue regeneration gone awry. Curr. Opin. Cell Biol. 2016, 43, 55–61. [Google Scholar] [CrossRef]
- Barnes, P.J. The cytokine network in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Nasef, N.A.; Mehta, S.; Ferguson, L.R. Susceptibility to chronic inflammation: An update. Arch. Toxicol. 2017, 91, 1131–1141. [Google Scholar] [CrossRef]
- Chapman, D.G.; Irvin, C.G. Mechanisms of airway hyper-responsiveness in asthma: The past, present and yet to come. Clin. Exp. Allergy 2015, 45, 706–719. [Google Scholar] [CrossRef]
- Global Network. The Global Asthma Report. Available online: http://www.globalasthmareport.org/ (accessed on 7 June 2020).
- CDC. Most Recent National Asthma Data. Available online: https://www.cdc.gov/asthma/most_recent_national_asthma_data.htm (accessed on 7 June 2020).
- Hendren, C.O.; Mesnard, X.; Dröge, J.; Wiesner, M.R. Estimating production data for five engineered nanomaterials as a basis for exposure assessment. Environ. Sci. Technol. 2011, 45, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.C. Mesenchymal cell survival in airway and interstitial pulmonary fibrosis. Fibrogenesis Tissue Repair. 2010, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Podila, R.; Brown, J.M. Toxicity of engineered nanomaterials: A physicochemical perspective. J. Biochem. Mol. Toxicol. 2012, 27, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Oberdörster, G. Safety assessment for nanotechnology and nanomedicine: Concepts of nanotoxicology. J. Intern. Med. 2010, 267, 89–105. [Google Scholar] [CrossRef]
- Hochella, M.F.; Mogk, D.W.; Ranville, J.F.; Allen, I.C.; Luther, G.W.I.; Marr, L.C.; McGrail, B.P.; Murayama, M.; Qafoku, N.P.; Rosso, K.M.; et al. Natural, incidental, and engineered nanomaterials and their impacts on the Earth system. Science 2019, 363, eaau8299. [Google Scholar] [CrossRef]
- Bandala, E.R.; Berli, M. Engineered nanomaterials (ENMs) and their role at the nexus of Food, Energy, and Water. Mater. Sci. Energy Technol. 2019, 2, 29–40. [Google Scholar] [CrossRef]
- Kermanizadeh, A.; Gosens, I.; MacCalman, L.; Johnston, H.; Danielsen, P.H.; Jacobsen, N.R.; Lenz, A.G.; Fernandes, T.; Schins, R.P.F.; Cassee, F.R.; et al. A Multilaboratory toxicological assessment of a panel of 10 engineered nanomaterials to human health—ENPRA project—The highlights, limitations, and current and future challenges. J. Toxicol. Environ. Health Part B Crit. Rev. 2016, 19, 1–28. [Google Scholar] [CrossRef]
- Good, K.D.; Bergman, L.E.; Klara, S.S.; Leitch, M.E.; VanBriesen, J.M.; Klara, S.S. Implications of engineered nanomaterials in drinking water sources. J. Am. Water Work. Assoc. 2016, 108, E1–E17. [Google Scholar] [CrossRef]
- Debia, M.; Bakhiyi, B.; Ostiguy, C.; Verbeek, J.H.; Brouwer, D.H.; Murashov, V. A systematic review of reported exposure to engineered nanomaterials. Ann. Occup. Hyg. 2016, 60, 916–935. [Google Scholar] [CrossRef]
- Bonner, J.C. Nanoparticles as a potential cause of pleural and interstitial lung disease. In Proceedings of the American Thoracic Society, New Orleans, LA, USA, 14–19 May 2010; Volume 7, pp. 138–141. [Google Scholar]
- Giese, B.; Klaessig, F.; Park, B.; Kaegi, R.; Steinfeldt, M.; Wigger, H.; Von Gleich, A.; Gottschalk, F. Risks, release and concentrations of engineered nanomaterial in the environment. Sci. Rep. 2018, 8, 1565. [Google Scholar] [CrossRef]
- Ogunsona, E.O.; Muthuraj, R.; Ojogbo, E.; Valerio, O.; Mekonnen, T.H.; Valero, O. Engineered nanomaterials for antimicrobial applications: A review. Appl. Mater. Today 2020, 18, 100473. [Google Scholar] [CrossRef]
- Piccinno, F.; Gottschalk, F.; Seeger, S.; Nowack, B. Industrial production quantities and uses of ten engineered nanomaterials in Europe and the world. J. Nanoparticle Res. 2012, 14, 1109. [Google Scholar] [CrossRef]
- Osman, N.M.; Sexton, D.W.; Saleem, I.Y. Toxicological assessment of nanoparticle interactions with the pulmonary system. Nanotoxicology 2019, 14, 21–58. [Google Scholar] [CrossRef]
- Yetisen, A.K.; Qu, H.; Manbachi, A.; Butt, H.; Dokmeci, M.R.; Hinestroza, J.P.; Skorobogatiy, M.; Khademhosseini, A.; Yun, S.-H. Nanotechnology in textiles. ACS Nano 2016, 10, 3042–3068. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Wu, C.; Li, J.; Guo, A.; Li, Q.; Jiang, H.; Chen, B.; Wang, X. Synergistic effect of functionalized nickel nanoparticles and Quercetin on inhibition of the SMMC-7721 cells proliferation. Nanoscale Res. Lett. 2009, 4, 1395–1402. [Google Scholar] [CrossRef]
- Liang, X.-J.; Chen, C.; Zhao, Y.; Jia, L.; Wang, P.C. Biopharmaceutics and therapeutic potential of engineered nanomaterials. Curr. Drug Metab. 2008, 9, 697–709. [Google Scholar] [CrossRef]
- Karmakar, S.; Saxena, V.; Chandra, P.; Pandey, L. Novel therapeutics and diagnostics strategies based on engineered nanobiomaterials. In Nanotechnology in Modern Animal Biotechnology: Recent Trends and Future Perspectives; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2019; pp. 1–27. [Google Scholar]
- Elkodous, M.A.; El-Sayyad, G.S.; Nasser, H.A.; ElShamy, A.A.; Morsi, M.; Abdelrahman, I.Y.; Kodous, A.S.; Mosallam, F.M.; Gobara, M.; El-Batal, A.I. Engineered nanomaterials as potential candidates for HIV treatment: Between opportunities and challenges. J. Clust. Sci. 2019, 30, 531–540. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Liang, Y.; Li, J.; Liu, Y.; Zhang, J.; Zhang, A.; Fu, J.; Jiang, G. PFOS induced lipid metabolism disturbances in BALB/c mice through inhibition of low density lipoproteins excretion. Sci. Rep. 2014, 4, 1–8. [Google Scholar] [CrossRef]
- Research and Markets Carbon Nanotubes (CNT) Market by Type, Method, Application—Global Forecast to 2023. Available online: https://www.marketsandmarkets.com/PressReleases/carbon-nanotubes.asp (accessed on 7 June 2020).
- Vetrivel, R.; Navinselvakumar, C.; Samuel Ratna Kumar, P.S. Carbon nanotubes and its applications—A review. Int. J. Mech. Prod. Eng. Res. Dev. 2018, 2, 4. [Google Scholar]
- Xu, J.; Cao, Z.; Zhang, Y.; Yuan, Z.; Lou, Z.; Xu, X.; Wang, X. A review of functionalized carbon nanotubes and graphene for heavy metal adsorption from water: Preparation, application, and mechanism. Chemosphere 2018, 195, 351–364. [Google Scholar] [CrossRef]
- Francis, A.P.; Thiyagarajan, D. Toxicity of carbon nanotubes: A review. Toxicol. Ind. Health 2018, 34, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, D.; Patnaik, S.; Sood, S.; Das, N. Carbon nanotubes: Evaluation of toxicity at biointerfaces. J. Pharm. Anal. 2019, 9, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Chen, K.; Yin, L.; Sun, Z.; Li, F.; Cheng, H.-M. The regulating role of carbon nanotubes and Graphene in lithium-ion and lithium-sulfur batteries. Adv. Mater. 2018, 31, e1800863. [Google Scholar] [CrossRef] [PubMed]
- Saleh, H.; Alali, E.; Ebaid, A. Medical applications for the flow of carbon-nanotubes suspended nanofluids in the presence of convective condition using Laplace transform. J. Assoc. Arab. Univ. Basic Appl. Sci. 2017, 24, 206–212. [Google Scholar] [CrossRef]
- Shaki, H.; Raissi, H.; Mollania, F.; Hashemzadeh, H. Modeling the interaction between anti-cancer drug penicillamine and pristine and functionalized carbon nanotubes for medical applications: Density functional theory investigation and a molecular dynamics simulation. J. Biomol. Struct. Dyn. 2019, 38, 1322–1334. [Google Scholar] [CrossRef]
- Glass, D.C.; Mazhar, M.; Xiang, S.D.; Dean, P.; Simpson, P.; Priestly, B.; Plebanski, M.; Abramson, M.J.; Sim, M.R.; Dennekamp, M. Immunological effects among workers who handle engineered nanoparticles. Occup. Environ. Med. 2017, 74, 868–876. [Google Scholar] [CrossRef]
- Poh, T.Y.; Ali, N.A.B.M.; Mac Aogáin, M.; Kathawala, M.H.; Setyawati, M.I.; Ng, K.W.; Chotirmall, S.H. Inhaled nanomaterials and the respiratory microbiome: Clinical, immunological and toxicological perspectives. Part. Fibre Toxicol. 2018, 15, 46. [Google Scholar] [CrossRef]
- Zhou, L.; Forman, H.J.; Ge, Y.; Lunec, J. Multi-walled carbon nanotubes: A cytotoxicity study in relation to functionalization, dose and dispersion. Toxicol. Vitr. 2017, 42, 292–298. [Google Scholar] [CrossRef]
- Chatterjee, N.; Yang, J.; Yoon, D.; Kim, S.; Joo, S.-W.; Choi, J. Differential crosstalk between global DNA methylation and metabolomics associated with cell type specific stress response by pristine and functionalized MWCNT. Biomaterials 2017, 115, 167–180. [Google Scholar] [CrossRef]
- Poulsen, S.S.; Jackson, P.; Kling, K.I.; Knudsen, K.B.; Skaug, V.; Kyjovska, Z.O.; Thomsen, B.L.; Clausen, P.A.; Atluri, R.; Berthing, T.; et al. Multi-walled carbon nanotube physicochemical properties predict pulmonary inflammation and genotoxicity. Nanotoxicology 2016, 10, 1263–1275. [Google Scholar] [CrossRef]
- Poulsen, S.S.; Saber, A.T.; Williams, A.; Andersen, O.; Købler, C.; Atluri, R.; Pozzebon, M.E.; Mucelli, S.P.; Simion, M.; Rickerby, D.G.; et al. MWCNTs of different physicochemical properties cause similar inflammatory responses, but differences in transcriptional and histological markers of fibrosis in mouse lungs. Toxicol. Appl. Pharmacol. 2015, 284, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.B.; Berthing, T.; Jackson, P.; Poulsen, S.S.; Mortensen, A.; Jacobsen, N.R.; Skaug, V.; Szarek, J.; Hougaard, K.S.; Wolff, H.; et al. Physicochemical predictors of Multi-Walled Carbon Nanotube-induced pulmonary histopathology and toxicity one year after pulmonary deposition of 11 different Multi-Walled Carbon Nanotubes in mice. Basic Clin. Pharmacol. Toxicol. 2018, 124, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Sukwong, P.; Somkid, K.; Kongseng, S.; Pissuwan, D.; Yoovathaworn, K. Respiratory tract toxicity of Titanium dioxide nanoparticles and multi-walled carbon nanotubes on mice after intranasal exposure. Micro Nano Lett. 2016, 11, 4. [Google Scholar] [CrossRef]
- Bierkandt, F.; Leibrock, L.; Wagener, S.; Laux, P.; Luch, A. The impact of nanomaterial characteristics on inhalation toxicity. Toxicol. Res. 2018, 7, 321–346. [Google Scholar] [CrossRef]
- Kodali, V.; Littke, M.H.; Tilton, S.C.; Teeguarden, J.; Shi, L.; Frevert, C.W.; Wang, W.; Pounds, J.G.; Thrall, B. Dysregulation of macrophage activation profiles by engineered nanoparticles. ACS Nano 2013, 7, 6997–7010. [Google Scholar] [CrossRef]
- Kämpfer, A.A.; Urbán, P.; La Spina, R.; Jiménez, I.O.; Kanase, N.; Stone, V.; Kinsner-Ovaskainen, A. Ongoing inflammation enhances the toxicity of engineered nanomaterials: Application of an in vitro co-culture model of the healthy and inflamed intestine. Toxicol. Vitr. 2020, 63, 104738. [Google Scholar] [CrossRef]
- Boraschi, D.; Costantino, L.; Italiani, P. Interaction of nanoparticles with immunocompetent cells: Nanosafety considerations. Nanomedicine 2012, 7, 121–131. [Google Scholar] [CrossRef]
- Pietroiusti, A. Health implications of engineered nanomaterials. Nanoscale 2012, 4, 1231. [Google Scholar] [CrossRef]
- Mizutani, N.; Nabe, T.; Yoshino, S. Exposure to multiwalled carbon nanotubes and allergen promotes early- and late-phase increases in airway resistance in mice. Biol. Pharm. Bull. 2012, 35, 2133–2140. [Google Scholar] [CrossRef]
- Chortarea, S.; Zerimariam, F.; Barosova, H.; Septiadi, D.; Clift, M.J.; Petri-Fink, A.; Rothen-Rutishauser, B. Profibrotic activity of Multiwalled carbon nanotubes upon prolonged exposures in different human lung cell types. Appl. Vitr. Toxicol. 2019, 5, 47–61. [Google Scholar] [CrossRef]
- Rahman, L.; Jacobsen, N.R.; Aziz, S.A.; Wu, N.; Williams, A.; Yauk, C.L.; White, P.; Wallin, H.; Vogel, U.B.; Halappanavar, S. Multi-walled carbon nanotube-induced genotoxic, inflammatory and pro-fibrotic responses in mice: Investigating the mechanisms of pulmonary carcinogenesis. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2017, 823, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Baugh, J.A. The significance of nanoparticles in particle-induced pulmonary fibrosis. McGill J. Med. 2008, 11, 43–50. [Google Scholar] [PubMed]
- Ryman-Rasmussen, J.P.; Tewksbury, E.W.; Moss, O.R.; Cesta, M.F.; Wong, B.A.; Bonner, J.C. Inhaled Multiwalled carbon nanotubes potentiate airway fibrosis in murine allergic asthma. Am. J. Respir. Cell Mol. Biol. 2008, 40, 349–358. [Google Scholar] [CrossRef]
- Bonner, J.C. Carbon nanotubes as delivery systems for respiratory disease: Do the dangers outweigh the potential benefits? Expert Rev. Respir. Med. 2011, 5, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.A.; Sayers, B.C.; Glista-Baker, E.E.; Shipkowski, K.A.; Ihrie, M.D.; Duke, K.S.; Taylor, A.J.; Bonner, J.C. STAT1 attenuates murine allergen-induced airway remodeling and exacerbation by carbon nanotubes. Am. J. Respir. Cell Mol. Biol. 2015, 53, 625–636. [Google Scholar] [CrossRef]
- Wang, L.; Feng, M.; Li, Q.; Qiu, C.; Chen, R. Advances in nanotechnology and asthma. Ann. Transl. Med. 2019, 7, 180. [Google Scholar] [CrossRef]
- Lanone, S.; Boczkowski, J. Titanium and gold nanoparticles in asthma: The bad and the ugly. Eur. Respir. J. 2011, 37, 225–227. [Google Scholar] [CrossRef]
- Hussain, S.; Vanoirbeek, J.A.; Luyts, K.; De Vooght, V.; Verbeken, E.; Thomassen, L.; Martens, J.; Dinsdale, D.; Boland, S.; Marano, F.; et al. Lung exposure to nanoparticles modulates an asthmatic response in a mouse model. Eur. Respir. J. 2010, 37, 299–309. [Google Scholar] [CrossRef]
- Fatkhutdinova, L.M.; Khaliullin, T.O.; Vasil’Yeva, O.L.; Zalyalov, R.R.; Mustafin, I.G.; Kisin, E.R.; Birch, M.E.; Yanamala, N.; Shvedova, A.A. Fibrosis biomarkers in workers exposed to MWCNTs. Toxicol. Appl. Pharmacol. 2016, 299, 125–131. [Google Scholar] [CrossRef]
- Dong, J.; Porter, D.W.; Batteli, L.A.; Wolfarth, M.G.; Richardson, D.L.; Ma, Q. Pathologic and molecular profiling of rapid-onset fibrosis and inflammation induced by multi-walled carbon nanotubes. Arch. Toxicol. 2014, 89, 621–633. [Google Scholar] [CrossRef]
- He, X.; Young, S.-H.; Schwegler-Berry, D.; Chisholm, W.P.; Fernback, J.E.; Ma, Q. Multiwalled carbon nanotubes induce a Fibrogenic response by stimulating reactive oxygen species production, activating NF-κB signaling, and promoting fibroblast-to-Myofibroblast transformation. Chem. Res. Toxicol. 2011, 24, 2237–2248. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Kiliç, G.; Costa, P.M.; Fadeel, B. Cytotoxicity screening and cytokine profiling of nineteen nanomaterials enables hazard ranking and grouping based on inflammogenic potential. Nanotoxicology 2017, 11, 809–826. [Google Scholar] [PubMed]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.R.; Takahashi, S.; Severgnini, M.; Fanburg, B.L.; Cochran, B.H. Role of the JAK-STAT pathway in PDGF-stimulated proliferation of human airway smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2002, 282, L1296–L1304. [Google Scholar] [CrossRef] [PubMed]
- Boisson-Dupuis, S.; Kong, X.-F.; Okada, S.; Cypowyj, S.; Puel, A.; Abel, L.; Casanova, J.-L. Inborn errors of human STAT1: Allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 2012, 24, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Antao-Menezes, A.; Ingram, J.L.; Rice, A.B.; Nyska, A.; Tani, Y.; Kleeberger, S.R.; Bonner, J.C. Susceptibility of signal transducer and activator of transcription-1-deficient mice to pulmonary Fibrogenesis. Am. J. Pathol. 2005, 167, 1221–1229. [Google Scholar] [CrossRef]
- Duke, K.S.; Taylor-Just, A.J.; Ihrie, M.; Shipkowski, K.A.; Thompson, E.A.; Dandley, E.C.; Parsons, G.N.; Bonner, J.C. STAT1-dependent and -independent pulmonary allergic and fibrogenic responses in mice after exposure to tangled versus rod-like multi-walled carbon nanotubes. Part. Fibre Toxicol. 2017, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.A.; Sayers, B.C.; Glista-Baker, E.E.; Shipkowski, K.A.; Ihrie, M.; Duke, K.S.; Taylor, A.J.; Bonner, J.C. Role of signal transducer and activator of transcription 1 in murine allergen–induced airway remodeling and exacerbation by carbon nanotubes. Am. J. Respir. Cell Mol. Biol. 2015, 53, 625–636. [Google Scholar] [CrossRef]
- Lindahl, G.E.; Stock, C.J.; Shiwen, X.; Leoni, P.; Sestini, P.; Howat, S.; Bou-Gharios, G.; Nicholson, A.G.; Denton, C.P.; Grutters, J.C.; et al. Microarray profiling reveals suppressed interferon stimulated gene program in fibroblasts from scleroderma-associated interstitial lung disease. Respir. Res. 2013, 14, 80. [Google Scholar] [CrossRef]
- Lazarevic, V.; Glimcher, L.H.; Lord, G.M. T-bet: A bridge between innate and adaptive immunity. Nat. Rev. Immunol. 2013, 13, 777–789. [Google Scholar] [CrossRef]
- Finotto, S.; Neurath, M.F.; Glickman, J.N.; Qin, S.; Lehr, H.A.; Green, F.H.Y.; Ackerman, K.; Haley, K.; Galle, P.R.; Szabo, S.J.; et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science 2002, 295, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Glista-Baker, E.E.; Taylor-Just, A.J.; Sayers, B.C.; Thompson, E.A.; Bonner, J.C. Nickel nanoparticles cause exaggerated lung and airway remodeling in mice lacking the T-box transcription factor, TBX21 (T-bet). Part. Fibre Toxicol. 2014, 11, 7. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and its activators in respiratory diseases. Oxidative Med. Cell. Longev. 2019, 2019, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Ma, Q. Suppression of basal and carbon nanotube-induced oxidative stress, inflammation and fibrosis in mouse lungs by Nrf2. Nanotoxicology 2015, 10, 699–709. [Google Scholar] [CrossRef]
- Liu, W.; Hu, T.; Zhou, L.; Wu, D.; Huang, X.; Ren, X.; Lv, Y.; Hong, W.; Huang, G.; Lin, Z.; et al. Nrf2 protects against oxidative stress induced by SiO2 nanoparticles. Nanomedicine 2017, 12, 2303–2318. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef]
- Takahashi, T.; Munakata, M.; Ohtsuka, Y.; Nisihara, H.; Nasuhara, Y.; Kamachi-Satoh, A.; Dosaka-Akita, H.; Homma, Y.; Kawakami, Y. Expression and alteration of ras and p53 proteins in patients with lung carcinoma accompanied by idiopathic pulmonary fibrosis. Cancer 2002, 95, 624–633. [Google Scholar] [CrossRef]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic pulmonary fibrosis: A disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef]
- Wang, L.; Luanpitpong, S.; Castranova, V.; Tse, W.; Lu, Y.; Pongrakhananon, V.; Rojanasakul, Y. Carbon nanotubes induce malignant transformation and Tumorigenesis of human lung epithelial cells. Nano Lett. 2011, 11, 2796–2803. [Google Scholar] [CrossRef]
- Takagi, A.; Hirose, A.; Nishimura, T.; Fukumori, N.; Ogata, A.; Ohashi, N.; Kitajima, S.; Kanno, J. Induction of mesothelioma in p53+/- mouse by intraperitoneal application of multi-wall carbon nanotube. J. Toxicol. Sci. 2008, 33, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Duke, K.S.; Thompson, E.A.; Ihrie, M.D.; Taylor-Just, A.J.; Ash, E.A.; Shipkowski, K.A.; Hall, J.R.; Tokarz, D.A.; Cesta, M.F.; Hubbs, A.F.; et al. Role of p53 in the chronic pulmonary immune response to tangled or rod-like multi-walled carbon nanotubes. Nanotoxicology 2018, 12, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Luyts, K.; Smulders, S.; Napierska, D.; Van kerckhoven, S.; Poels, K.; Scheers, H.; Hemmeryckx, B.; Nemery, B.; Hoylaerts, M.F.; Hoet, P.H.M. Pulmonary and hemostatic toxicity of multi-walled carbon nanotubes and zinc oxide nanoparticles after pulmonary exposure in Bmal1 knockout mice. Part. Fibre Toxicol. 2014, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Somanath, P.R.; Podrez, E.A.; Chen, J.; Ma, Y.; Marchant, K.; Antoch, M.; Byzova, T. Deficiency in core circadian protein Bmal1 is associated with a prothrombotic and vascular phenotype. J. Cell. Physiol. 2010, 226, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2014, 12, 5–23. [Google Scholar] [CrossRef]
- Shvedova, A.A.; Kisin, E.; Murray, A.; Kommineni, C.; Castranova, V.; Fadeel, B.; Kagan, V. Increased accumulation of neutrophils and decreased fibrosis in the lung of NADPH oxidase-deficient C57BL/6 mice exposed to carbon nanotubes. Toxicol. Appl. Pharmacol. 2008, 231, 235–240. [Google Scholar] [CrossRef]
- Sayers, B.C.; Taylor, A.J.; Glista-Baker, E.E.; Shipley-Phillips, J.K.; Dackor, R.T.; Edin, M.L.; Lih, F.B.; Tomer, K.B.; Zeldin, D.C.; Langenbach, R.; et al. Role of Cyclooxygenase-2 in exacerbation of allergen-induced airway remodeling by multiwalled carbon nanotubes. Am. J. Respir. Cell Mol. Biol. 2013, 49, 525–535. [Google Scholar] [CrossRef]
- Arima, K.; Umeshita-Suyama, R.; Sakata, Y.; Akaiwa, M.; Mao, X.-Q.; Enomoto, T.; Dake, Y.; Shimazu, S.-I.; Yamashita, T.; Sugawara, N.; et al. Upregulation of IL-13 concentration in vivo by the IL13 variant associated with bronchial asthma. J. Allergy Clin. Immunol. 2002, 109, 980–987. [Google Scholar] [CrossRef]
- Kuperman, D.A.; Huang, X.; Koth, L.L.; Chang, G.H.; Dolganov, G.M.; Zhu, Z.; Elias, J.A.; Sheppard, D.; Erle, D.J. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 2002, 8, 885–889. [Google Scholar] [CrossRef]
- Pierzchalska, M.; Soja, J.; Woś, M.; Szabó, Z.; Nizankowska-Mogielnicka, E.; Sanak, M.; Szczeklik, A. Deficiency of cyclooxygenases transcripts in cultured primary bronchial epithelial cells of aspirin-sensitive asthmatics. J. Physiol. Pharmacol. 2007, 58, 207–218. [Google Scholar] [PubMed]
- Trudeau, J.; Hu, H.; Chibana, K.; Chu, H.W.; Westcott, J.Y.; Wenzel, S.E. Selective downregulation of prostaglandin E 2-related pathways by the TH2 cytokine IL-13. J. Allergy Clin. Immunol. 2006, 117, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, S.; Zhang, S.; Cai, G.; Jiang, H.; Su, H.; Li, X.; Hong, Q.; Zhang, X.; Chen, X. Tissue inhibitor of Metalloproteinase-1 promotes NIH3T3 fibroblast proliferation by activating p-Akt and cell cycle progression. Mol. Cells 2011, 31, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Yu, X.; Porter, D.W.; Battelli, L.A.; Kashon, M.L.; Ma, Q. Common and distinct mechanisms of induced pulmonary fibrosis by particulate and soluble chemical fibrogenic agents. Arch. Toxicol. 2015, 90, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Stetler-Stevenson, W.G.; Fleming, M.V.; Fishback, N.; Koss, M.N.; Liotta, L.A.; Ferrans, V.J.; Travis, W.D. Immunohistochemical study of metalloproteinases and their tissue inhibitors in the lungs of patients with diffuse alveolar damage and idiopathic pulmonary fibrosis. Am. J. Pathol. 1996, 149, 1241–1256. [Google Scholar] [PubMed]
- Manoury, B.; Caulet-Maugendre, S.; Guénon, I.; Lagente, V.; Boichot, E. Timp-1 is a key factor of fibrogenic response to bleomycin in mouse lung. Int. J. Immunopathol. Pharmacol. 2006, 19, 471–487. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Ma, Q. TIMP1 promotes multi-walled carbon nanotube-induced lung fibrosis by stimulating fibroblast activation and proliferation. Nanotoxicology 2016, 11, 41–51. [Google Scholar] [CrossRef]
- Kotchey, G.P.; Zhao, Y.; Kagan, V.E.; Star, A. Peroxidase-mediated biodegradation of carbon nanotubes in vitro and in vivo. Adv. Drug Deliv. Rev. 2013, 65, 1921–1932. [Google Scholar] [CrossRef]
- Tomonaga, T.; Izumi, H.; Yoshiura, Y.; Myojo, T.; Oyabu, T.; Lee, B.-W.; Okada, T.; Marui, T.; Wang, K.-Y.; Kubo, M.; et al. Usefulness of myeloperoxidase as a biomarker for the ranking of pulmonary toxicity of nanomaterials. Part. Fibre Toxicol. 2018, 15, 41. [Google Scholar] [CrossRef]
- Shvedova, A.A.; Kapralov, A.A.; Feng, W.H.; Kisin, E.R.; Murray, A.R.; Mercer, R.R.; Croix, C.M.S.; Lang, M.A.; Watkins, S.C.; Konduru, N.V.; et al. Impaired clearance and enhanced pulmonary inflammatory/fibrotic response to carbon nanotubes in myeloperoxidase-deficient mice. PLoS ONE 2012, 7, e30923. [Google Scholar] [CrossRef]
- Gao, J.; Katagiri, H.; Ishigaki, Y.; Yamada, T.; Ogihara, T.; Imai, J.; Uno, K.; Hasegawa, Y.; Kanzaki, M.; Yamamoto, T.T.; et al. Involvement of Apolipoprotein E in excess fat accumulation and insulin resistance. Diabetes 2006, 56, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.S.; Sheykhzade, M.; Møller, P.; Folkmann, J.K.; Amtorp, O.; Jonassen, T.; Loft, S. Diesel exhaust particles induce endothelial dysfunction in apoE-/- mice. Toxicol. Appl. Pharmacol. 2007, 219, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Vesterdal, L.K.; Folkmann, J.K.; Jacobsen, N.R.; Sheykhzade, M.; Wallin, H.; Loft, S.; Møller, P. Modest vasomotor dysfunction induced by low doses of C60 fullerenes in apolipoprotein E knockout mice with different degree of atherosclerosis. Part. Fibre Toxicol. 2009, 6, 5. [Google Scholar] [CrossRef]
- Jacobsen, N.R.; Møller, P.; Jensen, K.A.; Vogel, U.; Ladefoged, O.; Loft, S.; Wallin, H. Lung inflammation and genotoxicity following pulmonary exposure to nanoparticles in ApoE-/-mice. Part. Fibre Toxicol. 2009, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Christophersen, D.V.; Jacobsen, N.R.; Andersen, M.H.G.; Connell, S.; Barfod, K.K.; Thomsen, M.B.; Miller, M.R.; Duffin, R.; Lykkesfeldt, J.; Vogel, U.B.; et al. Cardiovascular health effects of oral and pulmonary exposure to multi-walled carbon nanotubes in ApoE-deficient mice. Toxicology 2016, 371, 29–40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suzuki, Y.; Tada-Oikawa, S.; Hayashi, Y.; Izuoka, K.; Kataoka, M.; Ichikawa, S.; Wu, W.; Zong, C.; Ichihara, G.; Ichihara, S. Single- and double-walled carbon nanotubes enhance atherosclerogenesis by promoting monocyte adhesion to endothelial cells and endothelial progenitor cell dysfunction. Part. Fibre Toxicol. 2015, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jacobsen, N.R.; Danielsen, P.H.; Lenz, A.G.; Stoeger, T.; Loft, S.; Wallin, H.; Roursgaard, M.; Mikkelsen, L.; Møller, P. Vascular effects of multiwalled carbon nanotubes in dyslipidemic apoe-/- mice and cultured endothelial cells. Toxicol. Sci. 2014, 138, 104–116. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Beamer, C.A.; Shepherd, D.M. Role of the aryl hydrocarbon receptor (AhR) in lung inflammation. Semin. Immunopathol. 2013, 35, 693–704. [Google Scholar] [CrossRef]
- Ho, C.-C.; Lee, H.-L.; Chen, C.-Y.; Luo, Y.-H.; Tsai, M.-H.; Tsai, H.-T.; Lin, P. Involvement of the cytokine–IDO1–AhR loop in zinc oxide nanoparticle-induced acute pulmonary inflammation. Nanotoxicology 2017, 11, 360–370. [Google Scholar] [CrossRef]
- Trinh, L.; Brignole-Baudouin, F.; Pauly, A.; Liang, H.; Houssier, M.; Baudouin, C. Th1- and Th2-related chemokine and chemokine receptor expression on the ocular surface in endotoxin-induced uveitis. Mol. Vis. 2008, 14, 2428–2434. [Google Scholar] [PubMed]
- De-Oliveira-Pinto, L.M.; Marinho, C.F.; Povoa, T.F.; De Azeredo, E.L.; De Souza, L.A.; Barbosa, L.D.R.; Motta-Castro, A.R.C.; Alves, A.M.B.; Avila, C.A.L.; De Souza, L.J.; et al. Regulation of inflammatory chemokine receptors on blood T cells associated to the circulating versus liver Chemokines in dengue fever. PLoS ONE 2012, 7, e38527. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Roh, J.; Kim, S.N.; Kim, Y.; Han, S.-B.; Hong, J.T. CCR5 plays an important role in resolving an inflammatory response to single-walled carbon nanotubes. J. Appl. Toxicol. 2012, 33, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2017, 281, 8–27. [Google Scholar] [CrossRef]
- Liew, F.Y.; Pitman, N.I.; McInnes, I.B. Disease-associated functions of IL-33: The new kid in the IL-1 family. Nat. Rev. Immunol. 2010, 10, 103–110. [Google Scholar] [CrossRef]
- Yazdi, A.S.; Guarda, G.; Riteau, N.; Drexler, S.K.; Tardivel, A.; Couillin, I.; Tschopp, J. Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1α and IL-1β. Proc. Natl. Acad. Sci. USA 2010, 107, 19449–19454. [Google Scholar] [CrossRef]
- Shipkowski, K.A.; Taylor, A.J.; Thompson, E.A.; Glista-Baker, E.E.; Sayers, B.C.; Messenger, Z.J.; Bauer, R.N.; Jaspers, I.; Bonner, J.C. An allergic lung microenvironment suppresses carbon nanotube-induced Inflammasome activation via STAT6-dependent inhibition of caspase-1. PLoS ONE 2015, 10, e0128888. [Google Scholar] [CrossRef]
- Girtsman, T.A.; Beamer, C.A.; Wu, N.; Buford, M.; Holian, A. IL-1R signalling is critical for regulation of multi-walled carbon nanotubes-induced acute lung inflammation in C57Bl/6 mice. Nanotoxicology 2012, 8, 17–27. [Google Scholar] [CrossRef]
- Nikota, J.; Banville, A.; Goodwin, L.R.; Wu, D.; Williams, A.; Yauk, C.L.; Wallin, H.; Vogel, U.B.; Halappanavar, S. Stat-6 signaling pathway and not Interleukin-1 mediates multi-walled carbon nanotube-induced lung fibrosis in mice: Insights from an adverse outcome pathway framework. Part. Fibre Toxicol. 2017, 14, 37. [Google Scholar] [CrossRef]
- Rydman, E.M.; Ilves, M.; Vanhala, E.; Vippola, M.; Lehto, M.; Kinaret, P.A.S.; Pylkkänen, L.; Happo, M.; Hirvonen, M.-R.; Greco, D.; et al. A single aspiration of rod-like carbon nanotubes induces asbestos-like pulmonary inflammation mediated in part by the IL-1 receptor. Toxicol. Sci. 2015, 147, 140–155. [Google Scholar] [CrossRef]
- Dong, J.; Ma, Q. Osteopontin enhances multi-walled carbon nanotube-triggered lung fibrosis by promoting TGF-β1 activation and myofibroblast differentiation. Part. Fibre Toxicol. 2017, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Mangum, J.B.; Bermudez, E.; Sar, M.; Everitt, J. Osteopontin expression in particle-induced lung disease. Exp. Lung Res. 2004, 30, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G.; Marin, V.; Montero-Julian, F.; Mantovani, A.; Farnarier, C. IL-6: A regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003, 24, 25–29. [Google Scholar] [CrossRef]
- Kurjane, N.; Zvagule, T.; Reste, J.; Martinsone, Z.; Pavlovska, I.; Martinsone, I.; Vanadzins, I. The effect of different workplace nanoparticles on the immune systems of employees. J. Nanoparticle Res. 2017, 19, 320. [Google Scholar] [CrossRef]
- Khan, H.; Abdelhalim, M.A.K.; Alhomida, A.S.; Al-Ayed, M.S. Effects of naked gold nanoparticles on Proinflammatory cytokines mRNA expression in rat liver and kidney. BioMed Res. Int. 2013, 2013, 1–6. [Google Scholar] [CrossRef]
- Thompson, L.C.; Holland, N.A.; Snyder, R.J.; Luo, B.; Becak, D.P.; Odom, J.T.; Harrison, B.S.; Brown, J.M.; Gowdy, K.M.; Wingard, C.J. Pulmonary instillation of MWCNT increases lung permeability, decreases gp130 expression in the lungs, and initiates cardiovascular IL-6 transsignaling. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L142–L154. [Google Scholar] [CrossRef][Green Version]
- Dandley, E.C.; Taylor, A.J.; Duke, K.S.; Ihrie, M.; Shipkowski, K.A.; Parsons, G.N.; Bonner, J.C. Atomic layer deposition coating of carbon nanotubes with zinc oxide causes acute phase immune responses in human monocytes in vitro and in mice after pulmonary exposure. Part. Fibre Toxicol. 2015, 13, 29. [Google Scholar] [CrossRef]
- Liu, J.; Pang, Z.; Wang, G.; Guan, X.; Fang, K.; Wang, Z.; Wang, F. Advanced role of neutrophils in common respiratory diseases. J. Immunol. Res. 2017, 2017, 1–21. [Google Scholar] [CrossRef]
- Fielding, C.A.; McLoughlin, R.M.; McLeod, L.; Colmont, C.S.; Najdovska, M.; Grail, D.; Ernst, M.; Jones, S.A.; Topley, N.; Jenkins, B.J. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J. Immunol. 2008, 181, 2189–2195. [Google Scholar] [CrossRef]
- Wang, Y.; Van Boxel-Dezaire, A.H.H.; Cheon, H.; Yang, J.; Stark, G.R. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 16975–16980. [Google Scholar] [CrossRef]
- Qu, Z.; Sun, F.; Zhou, J.; Li, L.; Shapiro, S.D.; Xiao, G. Interleukin-6 prevents the initiation but enhances the progression of lung cancer. Cancer Res. 2015, 75, 3209–3215. [Google Scholar] [CrossRef] [PubMed]
- You, D.J.; Lee, H.Y.; Taylor-Just, A.J.; Linder, K.E.; Bonner, J.C. Sex differences in the acute and subchronic lung inflammatory responses of mice to nickel nanoparticles. Nanotoxicology 2020, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Ray, J.L.; Holian, A. Sex differences in the inflammatory immune response to multi-walled carbon nanotubes and crystalline silica. Inhal. Toxicol. 2019, 31, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Ray, J.L.; Fletcher, P.; Burmeister, R.; Holian, A. The role of sex in particle-induced inflammation and injury. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 12, e1589. [Google Scholar] [CrossRef]
- Serpooshan, V.; Sheibani, S.; Pushparaj, P.; Wojcik, M.; Jang, A.Y.; Santoso, M.R.; Jang, J.H.; Huang, H.; Safavi-Sohi, R.; Haghjoo, N.; et al. Effect of cell sex on uptake of nanoparticles: The overlooked factor at the Nanobio interface. ACS Nano 2018, 12, 2253–2266. [Google Scholar] [CrossRef]
- NIH Office of Research on Women’s Health (ORWH). NIH Office of Research on Women’s Health (ORWH). Available online: https://orwh.od.nih.gov/ (accessed on 7 June 2020).
- Lee, T.; Park, J.Y.; Lee, H.Y.; Cho, Y.-J.; Yoon, H.I.; Lee, J.H.; Jheon, S.; Lee, C.-T.; Park, J.S. Lung cancer in patients with idiopathic pulmonary fibrosis: Clinical characteristics and impact on survival. Respir. Med. 2014, 108, 1549–1555. [Google Scholar] [CrossRef]
- Yung, J.A.; Fuseini, H.; Newcomb, D.C. Hormones, sex, and asthma. Ann. Allergy Asthma Immunol. 2018, 120, 488–494. [Google Scholar] [CrossRef]
- Barnes, P.J. Sex differences in chronic obstructive pulmonary disease mechanisms. Am. J. Respir. Crit. Care Med. 2016, 193, 813–814. [Google Scholar] [CrossRef]
- Khalifa, A.R.M.; Abdel-Rahman, E.A.; Mahmoud, A.M.; Ali, M.H.; Noureldin, M.; Saber, S.H.; Mohsen, M.; Ali, S.S. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol. Rep. 2017, 5, e13125. [Google Scholar] [CrossRef]
- Voltz, J.W.; Card, J.W.; Carey, M.A.; DeGraff, L.M.; Ferguson, C.D.; Flake, G.P.; Bonner, J.C.; Korach, K.S.; Zeldin, D.C. Male sex hormones exacerbate lung function impairment after Bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 45–52. [Google Scholar] [CrossRef]
- Gleeson, M.; Bishop, N.; Oliveira, M.; McCauley, T.; Tauler, P. Sex differences in immune variables and respiratory infection incidence in an athletic population. Exerc. Immunol. Rev. 2011, 17, 122–135. [Google Scholar] [PubMed]
- Kadioglu, A.; Cuppone, A.M.; Trappetti, C.; List, T.; Spreafico, A.; Pozzi, G.; Andrew, P.W.; Oggioni, M.R. Sex-based differences in susceptibility to respiratory and systemic pneumococcal disease in mice. J. Infect. Dis. 2011, 204, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Mourtzoukou, E.G.; Vardakas, K.Z. Sex differences in the incidence and severity of respiratory tract infections. Respir. Med. 2007, 101, 1845–1863. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- LoMauro, A.; Aliverti, A. Sex differences in respiratory function. Breathe 2018, 14, 131–140. [Google Scholar] [CrossRef]
- Joseph, V.; Uppari, N.; Kouchi, H.; De Bruyn, C.; Boukari, R.; Bairam, A. Respiratory regulation by steroids in newborn rats: A sex-specific balance between allopregnanolone and progesterone receptors. Exp. Physiol. 2018, 103, 276–290. [Google Scholar] [CrossRef]
- Shah, R.; Newcomb, D.C. Sex bias in asthma prevalence and pathogenesis. Front. Immunol. 2018, 9, 2997. [Google Scholar] [CrossRef]
- Casimir, G.J.; Lefèvre, N.; Corazza, F.; Duchateau, J. Sex and inflammation in respiratory diseases: A clinical viewpoint. Biol. Sex Differ. 2013, 4, 16. [Google Scholar] [CrossRef]
- Steeg, L.G.V.; Vermillion, M.S.; Hall, O.J.; Alam, O.; McFarland, R.; Chen, H.; Zirkin, B.; Klein, S.L. Age and testosterone mediate influenza pathogenesis in male mice. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L1234–L1244. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019-Novel Coronavirus (2019-nCoV) pneumonia in Wuhan, China: A descriptive study. SSRN Electron. J. 2020, 395, 10223. [Google Scholar] [CrossRef]
- Almqvist, C.; Worm, M.; Leynaert, B.; Working Group of GA2LEN WP 2.5 Gender. Impact of gender on asthma in childhood and adolescence: A GA2LEN review. Allergy 2007, 63, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Kasai, T.; Umeda, Y.; Ohnishi, M.; Mine, T.; Kondo, H.; Takeuchi, T.; Matsumoto, M.; Fukushima, S. Lung carcinogenicity of inhaled multi-walled carbon nanotube in rats. Part. Fibre Toxicol. 2016, 13, 53. [Google Scholar] [CrossRef] [PubMed]
- Brass, D.M.; McGee, S.P.; Dunkel, M.K.; Reilly, S.M.; Tobolewski, J.M.; Sabo-Attwood, T.; Fattman, C.L. Gender influences the response to experimental silica-induced lung fibrosis in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L664–L671. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Latoche, J.D.; Ufelle, A.C.; Fazzi, F.; Ganguly, K.; Leikauf, G.; Fattman, C.L. Secreted Phosphoprotein 1 and sex-specific differences in silica-induced pulmonary fibrosis in mice. Environ. Health Perspect. 2016, 124, 1199–1207. [Google Scholar] [CrossRef]
- Ihrie, M.; Bonner, J.C. The toxicology of engineered nanomaterials in asthma. Curr. Environ. Health Rep. 2018, 5, 100–109. [Google Scholar] [CrossRef]
- Nakae, S.; Lunderius, C.; Ho, L.H.; Schäfer, B.; Tsai, M.; Galli, S.J. TNF can contribute to multiple features of ovalbumin-induced allergic inflammation of the airways in mice. J. Allergy Clin. Immunol. 2007, 119, 680–686. [Google Scholar] [CrossRef]
- Han, H.; Park, Y.H.; Park, H.J.; Lee, K.; Um, K.; Park, J.-W.; Lee, J.-H. Toxic and adjuvant effects of silica nanoparticles on ovalbumin-induced allergic airway inflammation in mice. Respir. Res. 2016, 17, 60. [Google Scholar] [CrossRef]
- Kim, B.-G.; Lee, P.-H.; Lee, S.-H.; Park, M.-K.; Jang, A.-S. Effect of TiO2Nanoparticles on Inflammasome-Mediated Airway Inflammation and Responsiveness. Allergy Asthma Immunol. Res. 2017, 9, 257–264. [Google Scholar] [CrossRef]
- Huang, K.-L.; Lee, Y.-H.; Chen, H.-I.; Liao, H.-S.; Chiang, B.-L.; Cheng, T.-J. Zinc oxide nanoparticles induce eosinophilic airway inflammation in mice. J. Hazard. Mater. 2015, 297, 304–312. [Google Scholar] [CrossRef]
- Ihrie, M.; Taylor-Just, A.J.; Walker, N.J.; Stout, M.D.; Gupta, A.; Richey, J.S.; Hayden, B.K.; Baker, G.L.; Sparrow, B.R.; Duke, K.S.; et al. Inhalation exposure to multi-walled carbon nanotubes alters the pulmonary allergic response of mice to house dust mite allergen. Inhal. Toxicol. 2019, 31, 192–202. [Google Scholar] [CrossRef]
- Lee, B.-J.; Kim, B.; Lee, K. Air pollution exposure and cardiovascular disease. Toxicol. Res. 2014, 30, 71–75. [Google Scholar] [CrossRef]
- Sjögren, B. Occupational exposure to dust: Inflammation and ischaemic heart disease. Occup. Environ. Med. 1997, 54, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Holland, N.A.; Thompson, L.C.; Vidanapathirana, A.K.; Urankar, R.N.; Lust, R.M.; Fennell, T.R.; Wingard, C.J. Impact of pulmonary exposure to gold core silver nanoparticles of different size and capping agents on cardiovascular injury. Part. Fibre Toxicol. 2015, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Minarchick, V.C.; Stapleton, P.A.; Porter, D.W.; Wolfarth, M.G.; Ciftyürek, E.; Barger, M.; Sabolsky, E.M.; Nurkiewicz, T.R. Pulmonary cerium dioxide nanoparticles exposure differentially impairs coronary and mesenteric arteriolar reactivity. Cardiovasc. Toxicol. 2013, 13, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Abukabda, A.B.; Stapleton, P.A.; McBride, C.R.; Yi, J.; Nurkiewicz, T.R. Heterogeneous vascular bed responses to pulmonary titanium dioxide nanoparticle exposure. Front. Cardiovasc. Med. 2017, 4, 33. [Google Scholar] [CrossRef]
- Kaur, S.; Soto-Pantoja, D.R.; Stein, E.V.; Liu, C.; Elkahloun, A.G.; Pendrak, M.L.; Nicolae, A.; Singh, S.P.; Nie, Z.; Levens, D.; et al. Thrombospondin-1 signaling through CD47 inhibits self-renewal by regulating c-Myc and other stem cell transcription factors. Sci. Rep. 2013, 3, srep01673. [Google Scholar] [CrossRef]
- Bazzazi, H.; Isenberg, J.S.; Popel, A.S. Inhibition of VEGFR2 activation and its downstream signaling to ERK1/2 and calcium by thrombospondin-1 (TSP1): In silico investigation. Front. Physiol. 2017. [Google Scholar] [CrossRef]
- Mandler, W.K.; Nurkiewicz, T.R.; Porter, D.W.; Olfert, I.M. Thrombospondin-1 mediates multi-walled carbon nanotube induced impairment of arteriolar dilation. Nanotoxicology 2017, 11, 112–122. [Google Scholar] [CrossRef]
- Mandler, W.K.; Nurkiewicz, T.R.; Porter, D.W.; Kelley, E.E.; Olfert, I.M. Microvascular dysfunction following multiwalled carbon nanotube exposure is mediated by thrombospondin-1 receptor CD47. Toxicol. Sci. 2018, 165, 90–99. [Google Scholar] [CrossRef]
- Bauer, E.M.; Qin, Y.; Miller, T.W.; Bandle, R.W.; Csanyi, G.; Pagano, P.J.; Bauer, P.M.; Schnermann, J.; Roberts, D.D.; Isenberg, J.S. Thrombospondin-1 supports blood pressure by limiting eNOS activation and endothelial-dependent vasorelaxation. Cardiovasc. Res. 2010, 88, 471–481. [Google Scholar] [CrossRef]
- Rogers, N.M.; Sharifi-Sanjani, M.; Yao, M.; Ghimire, K.; Bienes-Martinez, R.; Mutchler, S.M.; Knupp, H.E.; Baust, J.; Novelli, E.M.; Ross, M.; et al. TSP1–CD47 signaling is upregulated in clinical pulmonary hypertension and contributes to pulmonary arterial vasculopathy and dysfunction. Cardiovasc. Res. 2016, 113, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Bitar, M.S. Diabetes impairs angiogenesis and induces endothelial cell senescence by up-regulating thrombospondin-CD47-dependent signaling. Int. J. Mol. Sci. 2019, 20, 673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, M.; Yin, L.; Fu, G.; Liu, Z. Role of thrombospondin-1 and thrombospondin-2 in cardiovascular diseases (Review). Int. J. Mol. Med. 2020, 45, 1275–1293. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, S.S.; Saber, A.T.; Mortensen, A.; Szarek, J.; Wu, D.; Williams, A.; Andersen, O.; Jacobsen, N.R.; Yauk, C.L.; Wallin, H.; et al. Changes in cholesterol homeostasis and acute phase response link pulmonary exposure to multi-walled carbon nanotubes to risk of cardiovascular disease. Toxicol. Appl. Pharmacol. 2015, 283, 210–222. [Google Scholar] [CrossRef]
- Dong, Z.; Wu, T.; Qin, W.; An, C.; Wang, Z.; Zhang, M.; Zhang, Y.; Zhang, C.; An, F. Serum amyloid A directly accelerates the progression of atherosclerosis in Apolipoprotein E–deficient mice. Mol. Med. 2011, 17, 1357–1364. [Google Scholar] [CrossRef]
- Saber, A.T.; Lamson, J.S.; Jacobsen, N.R.; Ravn-Haren, G.; Hougaard, K.S.; Nyendi, A.N.; Wahlberg, P.; Madsen, A.M.; Jackson, P.; Wallin, H.; et al. Particle-Induced Pulmonary Acute Phase Response Correlates with Neutrophil Influx Linking Inhaled Particles and Cardiovascular Risk. PLoS ONE 2013, 8, 7. [Google Scholar] [CrossRef]
- Lee, D.-K.; Jang, H.S.; Chung, H.; Jeon, S.; Jeong, J.; Choi, J.-H.; Cho, W.-S. Aggravation of atherosclerosis by pulmonary exposure to indium oxide nanoparticles. Nanotoxicology. 2020, 14, 355–371. [Google Scholar] [CrossRef]
- Crauwels, H.M.; Van Hove, C.E.; Holvoet, P.; Herman, A.G.; Bult, H. Plaque-associated endothelial dysfunction in apolipoprotein E-deficient mice on a regular diet. Effect of human apolipoprotein AI. Cardiovasc. Res. 2003, 59, 189–199. [Google Scholar] [CrossRef]
- Chen, T.; Hu, J.; Chen, C.; Pu, J.; Cui, X.; Jia, G. Cardiovascular effects of pulmonary exposure to titanium dioxide nanoparticles in ApoE knockout mice. J. Nanosci. Nanotechnol. 2013, 13, 3214–3222. [Google Scholar] [CrossRef]
- Kang, G.S.; Gillespie, P.A.; Gunnison, A.; Moreira, A.L.; Tchou-Wong, K.-M.; Chen, L.C. Long-term inhalation exposure to nickel nanoparticles exacerbated atherosclerosis in a susceptible mouse model. Environ. Health Perspect. 2010, 119, 176–181. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Chen, R.-L.; Shao, Y.; Wang, H.; Liu, Z.-G. Effects of exposure of adult mice to multi-walled carbon nanotubes on the liver lipid metabolism of their offspring. Toxicol. Res. 2018, 7, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, F.; Zhang, J.; Zhu, A.; Zou, L.; Han, A.; Li, J.; Chang, X.; Sun, Y.; Wang, C. Role of oxidative stress in liver toxicity induced by nickel oxide nanoparticles in rats. Mol. Med. Rep. 2017, 17, 3133–3139. [Google Scholar] [CrossRef] [PubMed]
- Dumková, J.; Smutná, T.; Vrlíková, L.; Le Coustumer, P.; Večeřa, Z.; Dočekal, B.; Mikuška, P.; Čapka, L.; Fictum, P.; Hampl, A.; et al. Sub-chronic inhalation of lead oxide nanoparticles revealed their broad distribution and tissue-specific subcellular localization in target organs. Part. Fibre Toxicol. 2017, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, L.; Babadi, V.Y.; Espanani, H.R. Toxic effects of the Fe2O3 nanoparticles on the liver and lung tissue. Bratisl Lek List. 2015, 116, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Ji, J.H.; Park, J.D.; Yoon, J.U.; Kim, D.S.; Jeon, K.S.; Song, M.Y.; Jeong, J.; Han, B.S.; Han, J.H.; et al. Subchronic inhalation toxicity of silver nanoparticles. Toxicol. Sci. 2008, 108, 452–461. [Google Scholar] [CrossRef]
- Suker, D.K.; Jasim, F.A. Liver histopathological alteration after repeated intra-tracheal instillation of titanium dioxide in male rats. Gastroenterol. Hepatol. Bed Bench 2018, 11, 159–168. [Google Scholar]
- Mitchell, L.A.; Lauer, F.T.; Burchiel, S.W.; McDonald, J.D. Mechanisms for how inhaled multiwalled carbon nanotubes suppress systemic immune function in mice. Nat. Nanotechnol. 2009, 4, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.A.; Gao, J.; Wal, R.V.; Gigliotti, A.; Burchiel, S.W.; McDonald, J.D. Pulmonary and systemic immune response to inhaled multiwalled carbon nanotubes. Toxicol. Sci. 2007, 100, 203–214. [Google Scholar] [CrossRef]
- Kido, T.; Tsunoda, M.; Kasai, T.; Sasaki, T.; Umeda, Y.; Senoh, H.; Yanagisawa, H.; Asakura, M.; Aizawa, Y.; Fukushima, S. The increases in relative mRNA expressions of inflammatory cytokines and chemokines in splenic macrophages from rats exposed to multi-walled carbon nanotubes by whole-body inhalation for 13 weeks. Inhal. Toxicol. 2014, 26, 750–758. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Oppenheim, H.A.; Lucero, J.; Guyot, A.-C.; Herbert, L.M.; McDonald, J.D.; Mabondzo, A.; Lund, A. Exposure to vehicle emissions results in altered blood brain barrier permeability and expression of matrix metalloproteinases and tight junction proteins in mice. Part. Fibre Toxicol. 2013, 10, 62. [Google Scholar] [CrossRef]
- Block, M.L.; Elder, A.; Auten, R.L.; Bilbo, S.D.; Chen, H.; Chen, J.-C.; Cory-Slechta, D.A.; Costa, D.; Diaz-Sanchez, D.; Dorman, D.C.; et al. The outdoor air pollution and brain health workshop. NeuroToxicology 2012, 33, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Elder, A.; Gelein, R.; Silva, V.; Feikert, T.; Opanashuk, L.; Carter, J.; Potter, R.; Maynard, A.D.; Ito, Y.; Finkelstein, J.; et al. Translocation of inhaled ultrafine manganese oxide particles to the central nervous system. Environ. Health Perspect. 2006, 114, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Petitot, F.; Lestaevel, P.; Tourlonias, E.; Mazzucco, C.; Jacquinot, S.; Dhieux, B.; Delissen, O.; Tournier, B.B.; Gensdarmes, F.; Beaunier, P.; et al. Inhalation of uranium nanoparticles: Respiratory tract deposition and translocation to secondary target organs in rats. Toxicol. Lett. 2013, 217, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, C.; Sun, J.; Xue, Y. Neurotoxicity of silica nanoparticles: Brain localization and dopaminergic neurons damage pathways. ACS Nano 2011, 5, 4476–4489. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Du, Q.; Ran, B.; Liu, X.; Wang, X.; Ma, X.; Cheng, F.; Sun, B. The neurotoxicity induced by engineered nanomaterials. Int. J. Nanomed. 2019, 14, 4167–4186. [Google Scholar] [CrossRef]
- Wei, L.; Guo, X.-Y.; Yang, T.; Yu, M.-Z.; Chen, D.-W.; Wang, J.-C. Brain tumor-targeted therapy by systemic delivery of siRNA with Transferrin receptor-mediated core-shell nanoparticles. Int. J. Pharm. 2016, 510, 394–405. [Google Scholar] [CrossRef]
- Agemy, L.; Friedmann-Morvinski, D.; Kotamraju, V.R.; Roth, L.; Sugahara, K.N.; Girard, O.; Mattrey, R.F.; Verma, I.M.; Ruoslahti, E.M. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17450–17455. [Google Scholar] [CrossRef]
- Rajora, M.A.; Ding, L.; Valic, M.; Jiang, W.; Overchuk, M.; Chen, J.; Zheng, G. Correction: Tailored theranostic apolipoprotein E3 porphyrin-lipid nanoparticles target glioblastoma. Chem. Sci. 2017, 8, 5803. [Google Scholar] [CrossRef]
- Aragon, M.J.; Topper, L.; Tyler, C.R.; Sanchez, B.; Zychowski, K.; Young, T.L.; Herbert, G.; Hall, P.R.; Erdely, A.; Eye, T.; et al. Serum-borne bioactivity caused by pulmonary multiwalled carbon nanotubes induces neuroinflammation via blood–brain barrier impairment. Proc. Natl. Acad. Sci. USA 2017, 114, E1968–E1976. [Google Scholar] [CrossRef]
- Samiei, F.; Shirazi, F.H.; Naserzadeh, P.; Dousti, F.; Seydi, E.; Pourahmad, J. Toxicity of multi-wall carbon nanotubes inhalation on the brain of rats. Environ. Sci. Pollut. Res. 2020, 27, 12096–12111. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.-T.; Seo, G.-B.; Jo, E.; Lee, M.; Kim, H.-M.; Shim, I.; Lee, B.-W.; Yoon, B.-I.; Kim, P.; Choi, K. Aluminum nanoparticles induce ERK and p38MAPK activation in rat brain. Toxicol. Res. 2013, 29, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Choi, Y.-J.; Jung, E.-J.; Yin, H.-Q.; Kwon, J.-T.; Kim, J.-E.; Im, H.-T.; Cho, M.-H.; Kim, J.-H.; Kim, H.-Y.; et al. Genomics-based screening of differentially expressed genes in the brains of mice exposed to silver nanoparticles via inhalation. J. Nanoparticle Res. 2009, 12, 1567–1578. [Google Scholar] [CrossRef]
- Cesta, M.F.; Ryman-Rasmussen, J.P.; Wallace, D.G.; Masinde, T.; Hurlburt, G.; Taylor, A.J.; Bonner, J.C. Bacterial lipopolysaccharide enhances PDGF signaling and pulmonary fibrosis in rats exposed to carbon nanotubes. Am. J. Respir. Cell Mol. Biol. 2009, 43, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.I.; Ernst, R.K.; Bader, M.W. LPS, TLR4 and infectious disease diversity. Nat. Rev. Genet. 2005, 3, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Dash, D.; Singh, R. Lipopolysaccharide (LPS) exposure differently affects allergic asthma exacerbations and its amelioration by intranasal curcumin in mice. Cytokine 2015, 76, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Gorbet, M.B.; Sefton, M.V. Endotoxin: The uninvited guest. Biomaterials 2005, 26, 6811–6817. [Google Scholar] [CrossRef]
- Thorn, J. The inflammatory response in humans after inhalation of bacterial endotoxin: A review. Inflamm. Res. 2001, 50, 254–261. [Google Scholar] [CrossRef]
- Sandström, T.; Bjermer, L.; Rylander, R. Lipopolysaccharide (LPS) inhalation in healthy subjects increases neutrophils, lymphocytes and fibronectin levels in bronchoalveolar lavage fluid. Eur. Respir. J. 1992, 5, 992–996. [Google Scholar]
- Bonner, J.C.; Rice, A.B.; Lindroos, P.M.; O’Brien, P.O.; Dreher, K.L.; Rosas, I.; Alfaro-Moreno, E.; Osornio-Vargas, A.R. Induction of the lung Myofibroblast PDGF receptor system by urban ambient particles from Mexico City. Am. J. Respir. Cell Mol. Biol. 1998, 19, 672–680. [Google Scholar] [CrossRef]
- Inoue, K.-I.; Takano, H.; Yanagisawa, R.; Hirano, S.; Sakurai, M.; Shimada, A.; Yoshikawa, T. Effects of airway exposure to nanoparticles on lung inflammation induced by bacterial endotoxin in mice. Environ. Health Perspect. 2006, 114, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Agache, I.; Lau, S.; Akdis, C.A.; Smolinska, S.; Bonini, M.; Cavkaytar, O.; Flood, B.; Gajdanowicz, P.; Izuhara, K.; Kalayci, O.; et al. EAACI Guidelines on Allergen Immunotherapy: House dust mite-driven allergic asthma. Allergy Eur. J. Allergy Clin. Immunol. 2019, 74, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Waldron, R.; McGowan, J.; Gordon, N.; McCarthy, C.; Mitchell, E.B.; Fitzpatrick, D.A. Proteome and allergenome of the European house dust mite Dermatophagoides pteronyssinus. PLoS ONE 2019, 14, e0216171. [Google Scholar] [CrossRef] [PubMed]
- Sears, M.R.; Herbison, P.; Holdaway, M.D.; Hewitt, C.J.; Flannery, E.M.; Silva, P.A. The relative risks of sensitivity to grass pollen, house dust mite and cat dander in the development of childhood asthma. Clin. Exp. Allergy 1989, 19, 419–424. [Google Scholar] [CrossRef]
- Arshad, S.H.; Tariq, S.M.; Matthews, S.; Hakim, E. Sensitization to common allergens and its association with allergic disorders at age 4 years: A whole population birth cohort study. Pediatrics 2001, 108, e33. [Google Scholar] [CrossRef]
- Wilson, J.M.; Platts-Mills, T.A. Home environmental interventions for house dust mite. J. Allergy Clin. Immunol. Pract. 2018, 6, 1–7. [Google Scholar] [CrossRef]


{kind=link}
| Transcription Factor | Type of ENM | Dosing and Exposure Method | Duration of Exposure | Findings in KO mice | References |
|---|---|---|---|---|---|
| STAT1 | Multi-Walled Carbon Nanotubes (MWCNTs) (tangled or rod-like) | 4 mg/kg via oropharyngeal aspiration | Single Exposure | Increased lung fibrosis with higher TGF-β1 in bronchoalveolar lavage fluid (BALF). Increased Smad2/3 phosphorylation in lung tissue. | [82] |
| T-bet | Nickel Nanoparticles (NiNPs or MWCNTs | 4 mg/kg via oropharyngeal aspiration | Single exposure | Enhanced mucous cell metaplasia. Increased MUC5AC and MUC5B mRNAs. Persistent eosinophils and lymphocytes in BALF. Greater interstitial lung fibrosis. | [87] |
| Nrf2 | MWCNTs | 5, 20, and 40 μg via pharyngeal aspiration | Single exposure | Higher level of inflammation and fibrosis. Increased inflammatory cell infiltrates. Increased ROS generation and oxidative damage. | [90] |
| Silica NPs | 10 mg/kg via intranasal instillation | Once a day for 2 weeks | Increased reactive oxygen species. Decreased total antioxidant capacity. | [91] | |
| P53 | MWCNTs (tangled or rod-like) | 1 mg/kg via oropharyngeal aspiration | Once a week for 4 weeks | Increased incidence of larger granuloma formation, lymphoid aggregates, and epithelial cell hyperplasia in the lungs of heterozygous p53(+/−). | [97] |
| BMAL1 | MWCNTs | 6.4 or 25.6 μg via oropharyngeal aspiration | Once a week, for 5 consecutive weeks | Increased inflammatory cytokines, oxidative stress, and procoagulant effect in serum. | [98] |
| ZnONPs | 6.4 or 12.8 μg via oropharyngeal aspiration | Once a week, for 5 consecutive weeks | Decreased inflammatory cytokines, decreased oxidative stress, and increased procoagulant effect. | [98] |
| Enzymes/Proteins | Type of ENMs | Dosing and Exposure Method | Duration of Exposure | Findings in KO Mice | References |
|---|---|---|---|---|---|
| NADPH Oxidase | Single-Walled Carbon Nanotubes (SWCNTs) | 40 μg/mouse via pharyngeal aspiration | Single exposure | Augmented lung inflammation by producing higher numbers of neutrophils, apoptotic cells, pro-inflammatory cytokines including TNF-α, MCP-1 (CCL2), and IL-6, and reduced anti-inflammatory cytokine, TGF-β1. Prolonged increase in neutrophils and pro-inflammatory cytokines. | [102] |
| COX-2 | MWCNTs | 4mg/kg via oropharyngeal aspiration | Single MWCNTs exposure after ovalbumin (OVA) sensitization and challenges | More susceptible to eosinophilic lung inflammation, airway mucous cell metaplasia, and airway fibrosis with ovalbumin allergen sensitization. Significantly higher Th2 cytokines including IL-13, Th1 cytokines such as CXCL10, and the Th17 cytokine IL-17A detected. | [103] |
| TIMP1 | MWCNTs | 40 µg/mouse via pharyngeal aspiration | Single exposure | Induced lung fibrosis through activation of intracellular ERK pathway | [112] |
| MPO | SWCNTs | 40 µg/mouse via pharyngeal aspiration | Single exposure | Less efficient clearance of CNTs causing a profound inflammatory response. However, wildtype mice also showed that increased MPO was also associated with number of total cells and neutrophils. | [115] |
| ApoE | AuNPs C60 SWCNTs Carbon Black NPs (CBNPs) | 0.54 µg AuNPs, 54 µg C60, 54 µg SWCNTs, 18 or 54 µg CBNPs via instillation or inhalation | Single exposure | Increased the DNA damage of inflammatory cells, neutrophil percentage, and higher protein level in BALF. | [119] |
| MWCNTs | 4 or 40 μg/mouse via intratracheal instillation | Once a week for 4 weeks | Increased pulmonary inflammation and oxidative stress/damage to DNA in lung tissue. | [120] | |
| SWCNTs Or Double-Walled CNTs | 10 or 40 μg/mouse via pharyngeal aspiration | Once every other week for 10 weeks | Dysregulation of endothelial progenitor cell (EPC) function contributing to developing atherosclerosis, buildup of cholesterol plaques in the walls of arteries. | [121] | |
| MWCNTs | 6.4 or 25.6 μg/mouse via intratracheal instillation | Once a week for 5 weeks | More susceptible to oxidative damage to DNA in lung tissue. Accelerated progression of atherosclerosis. | [122] |
| Cytokines/Chemokines | Type of ENMs | Dosing and Exposure Method | Duration of Exposure | Findings in KO Mice | References |
|---|---|---|---|---|---|
| IL1/Inflammasome | MWCNTs | 50 µg low nickel or high nickel containing MWCNT via oropharyngeal aspiration | Single exposure | Reduced acute inflammation and airway resistance but increased IL-6 protein production within 1 day. | [133] |
| Induced significantly higher number of pulmonary granulomas formation and significant inflammation post 28 days. | |||||
| 162 μg/mouse Mitsui-7 MWCNTs via intratracheal instillation | Single exposure | Acute inflammation at day 1 was suppressed. Fibrotic lesions still developed in KO mice 28 days post exposure. | [134] | ||
| 10 µg rod-like MWCNTs for 4hr via pharyngeal aspiration 10 and 40 µg rod-like MWCNTs for 28 days via pharyngeal aspiration | Single exposure | Reduced neutrophils in BALF and neutrophil chemoattractant CXCL5 mRNA levels 4hr after the exposure. Neutrophils were still reduced in BALF and TNF-α mRNA level was suppressed after 28 days. No changes were observed in Th2 related signals including IL-13 and TGF-β1 mRNA levels. | [135] | ||
| OPN | MWCNTs | 40 μg via pharyngeal aspiration | Single exposure | Reduced fibrotic formation and myofibroblast accumulation in the lungs. | [136] |
| AhR | ZnONPs | 5, 20, and 80 μg/mice via oropharyngeal aspiration | Single exposure | Reduced pulmonary inflammation, cytokine secretion, CYP1A1, and KYN production. Reduced cell number, total protein, and LDH activity in BALF. | [125] |
| CCR5 | SWCNTs | 100 μg/kg via intratracheal instillation | Single exposure | Dominated by B cells and CD8+ T cells instead of T cells and CD4+ T cells in the lungs. Increased IL-6, IL-13, and IL-17 in BALF. More frequent histopathological lesions were detected. | [128] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, D.J.; Bonner, J.C. Susceptibility Factors in Chronic Lung Inflammatory Responses to Engineered Nanomaterials. Int. J. Mol. Sci. 2020, 21, 7310. https://doi.org/10.3390/ijms21197310
You DJ, Bonner JC. Susceptibility Factors in Chronic Lung Inflammatory Responses to Engineered Nanomaterials. International Journal of Molecular Sciences. 2020; 21(19):7310. https://doi.org/10.3390/ijms21197310
Chicago/Turabian StyleYou, Dorothy J., and James C. Bonner. 2020. "Susceptibility Factors in Chronic Lung Inflammatory Responses to Engineered Nanomaterials" International Journal of Molecular Sciences 21, no. 19: 7310. https://doi.org/10.3390/ijms21197310
APA StyleYou, D. J., & Bonner, J. C. (2020). Susceptibility Factors in Chronic Lung Inflammatory Responses to Engineered Nanomaterials. International Journal of Molecular Sciences, 21(19), 7310. https://doi.org/10.3390/ijms21197310