Functions of Cytochrome c Oxidase Assembly Factors



Abstract
1. Introduction
2. Cytochrome c Oxidase Structure and Function
3. Cytochrome c Oxidase Assembly Factors
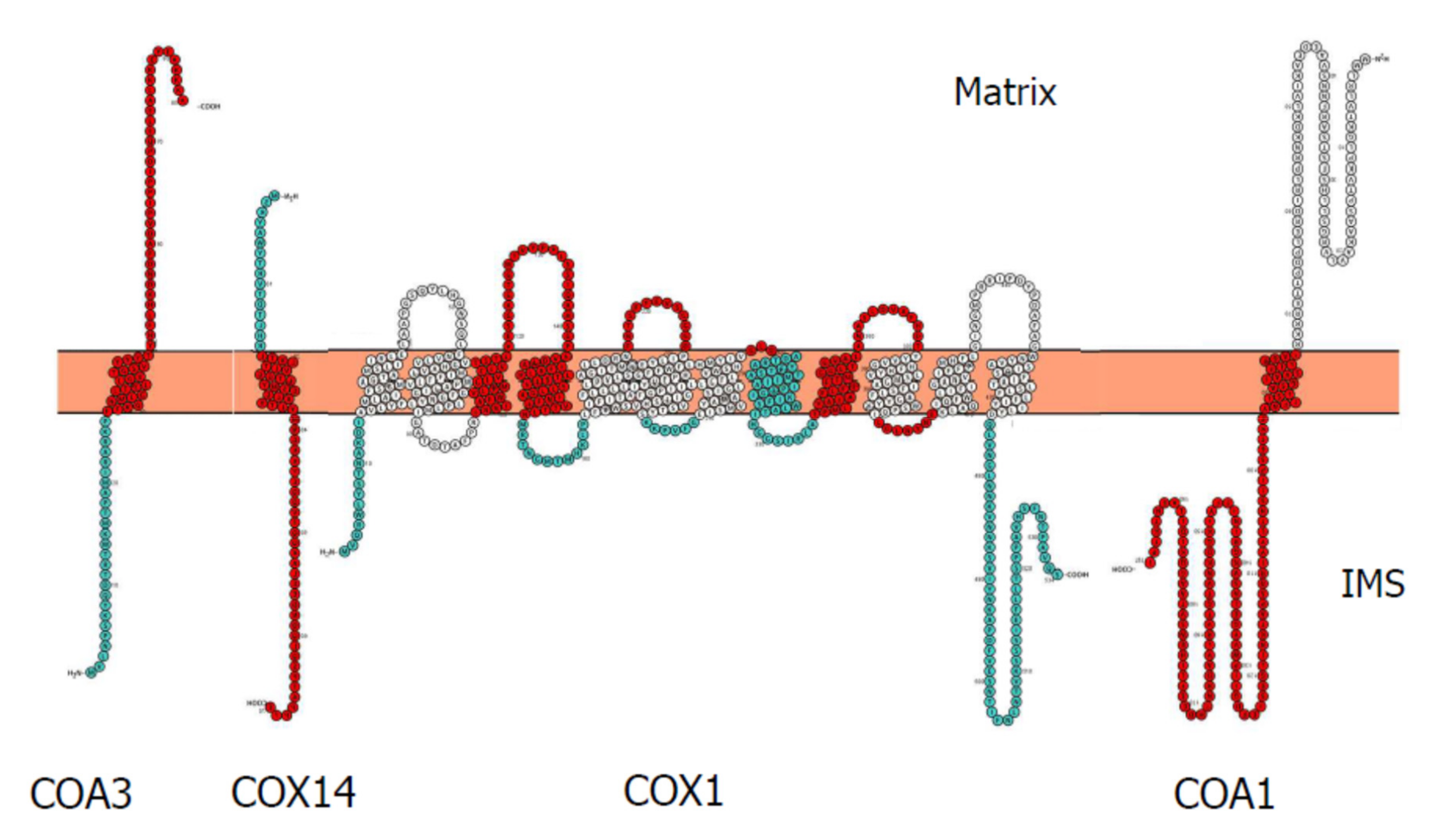
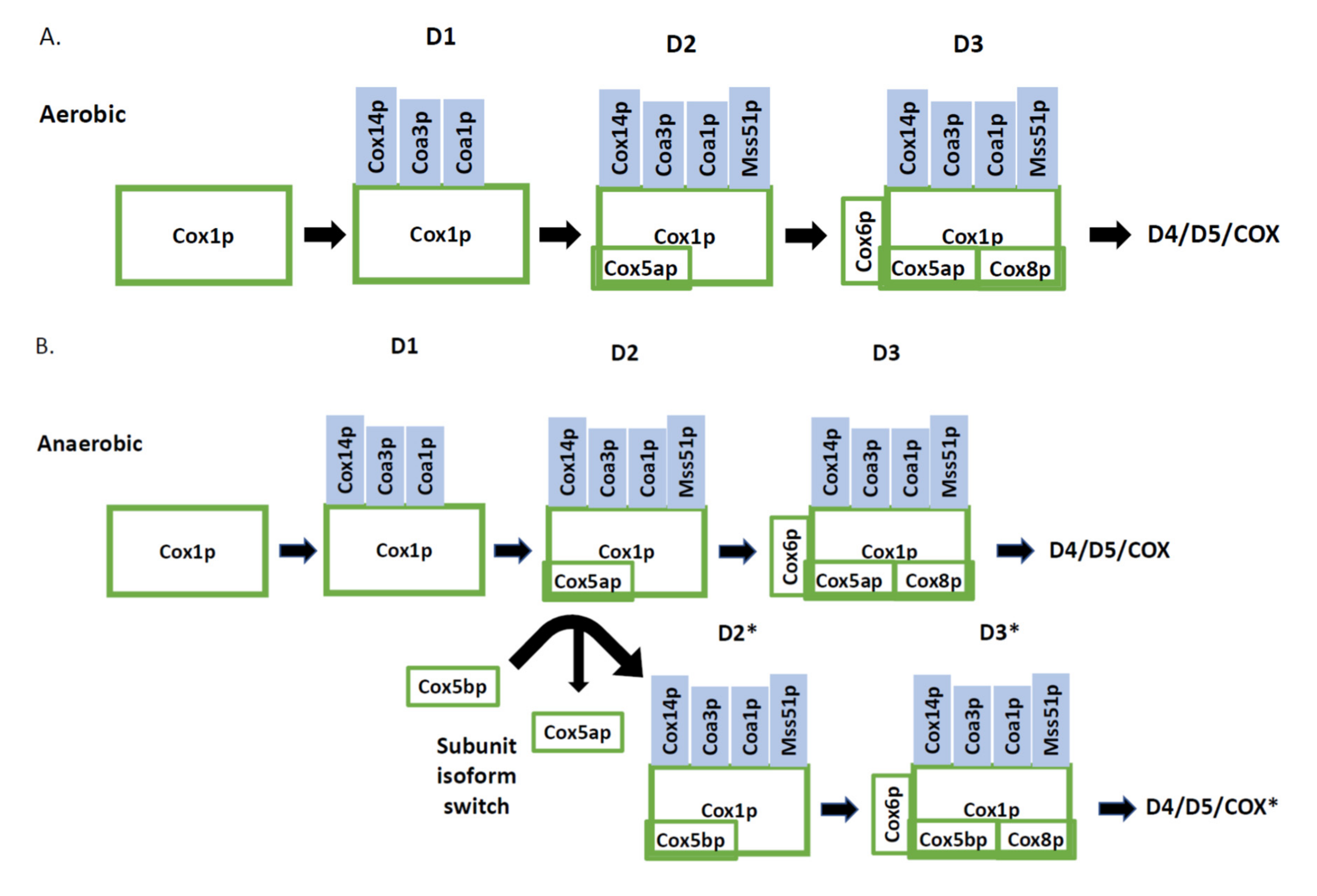
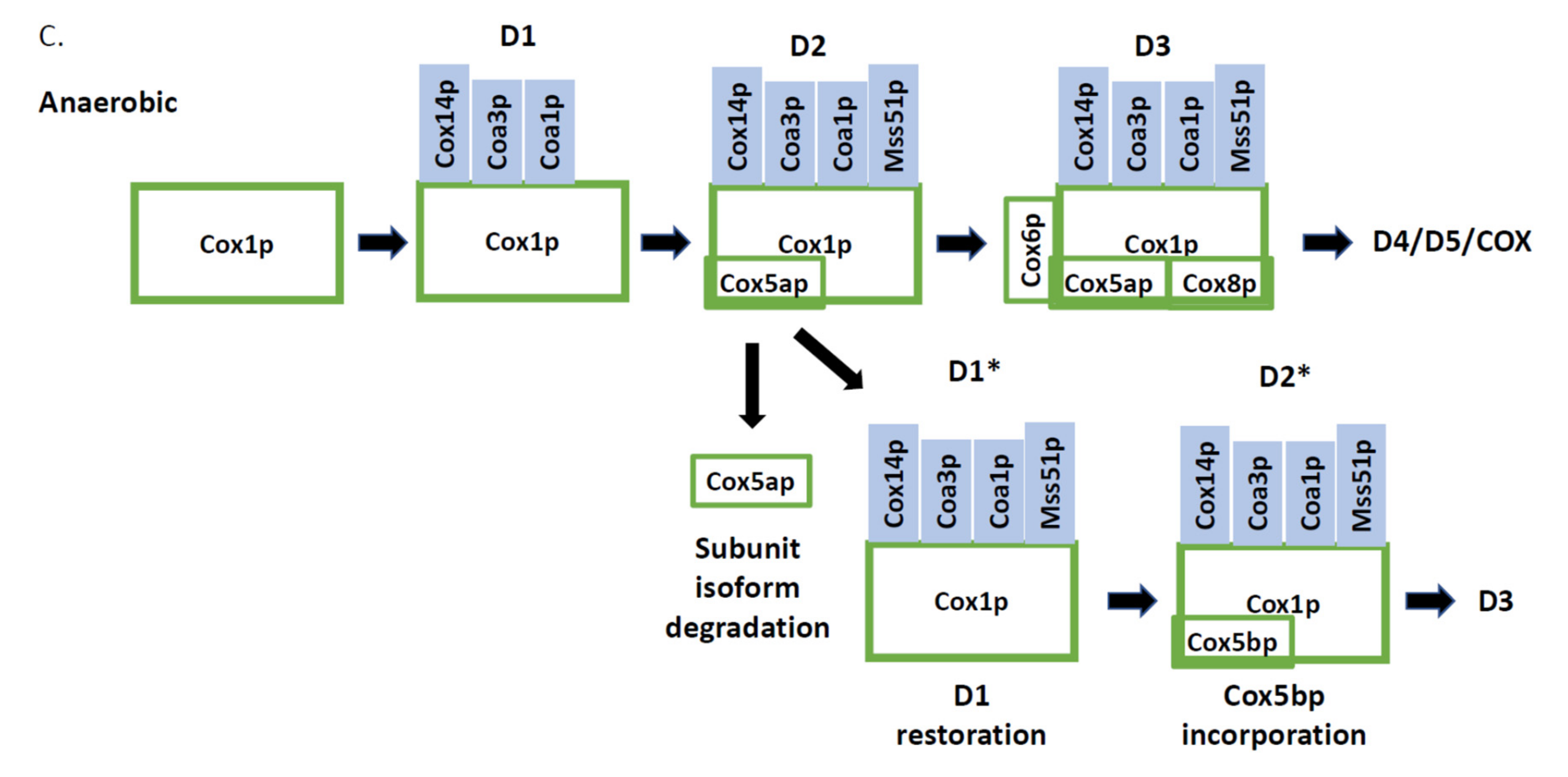
3.1. Cytochrome c Oxidase Subunit 1 Assembly
3.2. Cytochrome c Oxidase Subunit 2 Assembly
3.3. Cytochrome c Oxidase Subunit 3 Assembly
4. Mutations in Cytochrome c Oxidase Assembly Factors as Cause of Human Disease
5. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [PubMed]
- Tzagoloff, A.; Dieckmann, C.L. PET genes of Saccharomyces cerevisiae. Microbiol. Rev. 1990, 54, 211–225. [Google Scholar] [CrossRef]
- Barros, M.H.; McStay, G.P. Modular biogenesis of mitochondrial respiratory complexes. Mitochondrion 2020, 50, 94–114. [Google Scholar] [CrossRef] [PubMed]
- Mick, D.U.; Fox, T.D.; Rehling, P. Inventory control: Cytochrome c oxidase assembly regulates mitochondrial translation. Nat. Rev. Mol. Cell Biol. 2011, 12, 14–20. [Google Scholar] [CrossRef]
- Limongelli, G.; Masarone, D.; Pacileo, G. Mitochondrial disease and the heart. Heart 2017, 103, 390–398. [Google Scholar] [CrossRef]
- Lee, W.S.; Sokol, R.J. Mitochondrial hepatopathies: Advances in genetics, therapeutic approaches, and outcomes. J. Pediatr. 2013, 163, 942–948. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, J.F. Renal manifestations of genetic mitochondrial disease. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Frank, M. Gastrointestinal manifestations of mitochondrial disorders: A systematic review. Ther. Adv. Gastroenterol. 2017, 10, 142–154. [Google Scholar] [CrossRef]
- McFarland, R.; Taylor, R.W.; Turnbull, D.M. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010, 9, 829–840. [Google Scholar] [CrossRef]
- Visapää, I.; Fellman, V.; Vesa, J.; Dasvarma, A.; Hutton, J.L.; Kumar, V.; Payne, G.S.; Makarow, M.; Van Coster, R.; Taylor, R.W.; et al. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am. J. Hum. Genet. 2002, 71, 863–876. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Meunier, B.; Lee, D.; Orengo, C.; Rich, P.R. Yeast cytochrome c oxidase: A model system to study mitochondrial forms of the haem-copper oxidase superfamily. Biochim. Biophys. Acta 2012, 1817, 620–628. [Google Scholar] [CrossRef]
- Hartley, A.M.; Meunier, B.; Pinotsis, N.; Maréchal, A. Rcf2 revealed in cryo-EM structures of hypoxic isoforms of mature mitochondrial III-IV supercomplexes. Proc. Natl. Acad. Sci. USA 2020, 117, 9329–9337. [Google Scholar] [CrossRef] [PubMed]
- Timón-Gómez, A.; Garlich, J.; Stuart, R.A.; Ugalde, C.; Barrientos, A. Distinct roles of mitochondrial HIGD1A and HIGD2A in respiratory complex and supercomplex biogenesis. Cell Rep. 2020, 31, 107607. [Google Scholar] [CrossRef] [PubMed]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landázuri, M.O.; Enríquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.S.; Taanman, J.-W. NDUFA4 (Renamed COXFA4) Is a Cytochrome-c Oxidase Subunit. Trends Endocrinol. Metab. 2018, 29, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B. Regulation of Mammalian 13-Subunit Cytochrome c Oxidase and Binding of other Proteins: Role of NDUFA4. Trends Endocrinol. Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef]
- Morgenstern, M.; Stiller, S.B.; Lübbert, P.; Peikert, C.D.; Dannenmaier, S.; Drepper, F.; Weill, U.; Höß, P.; Feuerstein, R.; Gebert, M.; et al. Definition of a High-Confidence Mitochondrial Proteome at Quantitative Scale. Cell Rep. 2017, 19, 2836–2852. [Google Scholar] [CrossRef]
- McStay, G.P.; Su, C.-H.; Thomas, S.M.; Xu, J.T.; Tzagoloff, A. Characterization of assembly intermediates containing subunit 1 of yeast cytochrome oxidase. J. Biol. Chem. 2013, 288, 26546–26556. [Google Scholar] [CrossRef]
- Hallmann, K.; Kudin, A.P.; Zsurka, G.; Kornblum, C.; Reimann, J.; Stüve, B.; Waltz, S.; Hattingen, E.; Thiele, H.; Nürnberg, P.; et al. Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain 2016, 139, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, G.; Makino, S.; Hayashi, M.; Abe, A.; Numakura, C.; Ueki, M.; Tanaka, A.; Ito, C.; Toshimori, K.; Ogawa, N.; et al. A mutation of COX6A1 causes a recessive axonal or mixed form of Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2014, 95, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Laššuthová, P.; Beharka, R.; Krůtová, M.; Neupauerová, J.; Seeman, P. COX6A1 mutation causes axonal hereditary motor and sensory neuropathy—The confirmation of the primary report. Clin. Genet. 2016, 89, 512–514. [Google Scholar] [CrossRef]
- Inoue, M.; Uchino, S.; Iida, A.; Noguchi, S.; Hayashi, S.; Takahashi, T.; Fujii, K.; Komaki, H.; Takeshita, E.; Nonaka, I.; et al. COX6A2 variants cause a muscle-specific cytochrome c oxidase deficiency. Ann. Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.S.; Rahman, S.; Wedatilake, Y.; Polke, J.M.; Cirak, S.; Foley, A.R.; Sailer, A.; Hurles, M.E.; Stalker, J.; Hargreaves, I.; et al. UK10K Consortium NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013, 3, 1795–1805. [Google Scholar] [CrossRef]
- Pierron, D.; Wildman, D.E.; Hüttemann, M.; Markondapatnaikuni, G.C.; Aras, S.; Grossman, L.I. Cytochrome c oxidase: Evolution of control via nuclear subunit addition. Biochim. Biophys. Acta 2012, 1817, 590–597. [Google Scholar] [CrossRef]
- Su, C.-H.; McStay, G.P.; Tzagoloff, A. The Cox3p assembly module of yeast cytochrome oxidase. Mol. Biol. Cell 2014, 25, 965–976. [Google Scholar] [CrossRef]
- McStay, G.P.; Su, C.H.; Tzagoloff, A. Modular assembly of yeast cytochrome oxidase. Mol. Biol. Cell 2013, 24, 440–452. [Google Scholar] [CrossRef]
- Franco, L.V.R.; Su, C.-H.; McStay, G.P.; Yu, G.J.; Tzagoloff, A. Cox2p of yeast cytochrome oxidase assembles as a stand-alone subunit with the Cox1p and Cox3p modules. J. Biol. Chem. 2018, 293, 16899–16911. [Google Scholar] [CrossRef]
- Su, C.H.; Tzagoloff, A. Cox16 protein is physically associated with Cox1p assembly intermediates and with cytochrome oxidase. J. Biol. Chem. 2017, 292, 16277–16283. [Google Scholar] [CrossRef]
- Jia, L.; Dienhart, M.K.; Stuart, R.A. Oxa1 directly interacts with Atp9 and mediates its assembly into the mitochondrial F1Fo-ATP synthase complex. Mol. Biol. Cell 2007, 18, 1897–1908. [Google Scholar] [CrossRef] [PubMed]
- Stiburek, L.; Fornuskova, D.; Wenchich, L.; Pejznochova, M.; Hansikova, H.; Zeman, J. Knockdown of human Oxa1l impairs the biogenesis of F1Fo-ATP synthase and NADH: Ubiquinone oxidoreductase. J. Mol. Biol. 2007, 374, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Hell, K.; Neupert, W.; Stuart, R.A. Oxa1p acts as a general membrane insertion machinery for proteins encoded by mitochondrial DNA. EMBO J. 2001, 20, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Herrmann, J.M. Co-translational membrane insertion of mitochondrially encoded proteins. Biochim. Biophys. Acta 2010, 1803, 767–775. [Google Scholar] [CrossRef]
- Fiumera, H.L.; Broadley, S.A.; Fox, T.D. Translocation of mitochondrially synthesized Cox2 domains from the matrix to the intermembrane space. Mol. Cell. Biol. 2007, 27, 4664–4673. [Google Scholar] [CrossRef]
- Saracco, S.A.; Fox, T.D. Cox18p is required for export of the mitochondrially encoded Saccharomyces cerevisiae Cox2p C-tail and interacts with Pnt1p and Mss2p in the inner membrane. Mol. Biol. Cell 2002, 13, 1122–1131. [Google Scholar] [CrossRef]
- Lorenzi, I.; Oeljeklaus, S.; Ronsör, C.; Bareth, B.; Warscheid, B.; Rehling, P.; Dennerlein, S. The ribosome-associated Mba1 escorts Cox2 from insertion machinery to maturing assembly intermediates. Mol. Cell. Biol. 2016, 36, 2782–2793. [Google Scholar] [CrossRef]
- Pratje, E.; Mannhaupt, G.; Michaelis, G.; Beyreuther, K. A nuclear mutation prevents processing of a mitochondrially encoded membrane protein in Saccharomyces cerevisiae. EMBO J. 1983, 2, 1049–1054. [Google Scholar] [CrossRef]
- Thompson, K.; Mai, N.; Oláhová, M.; Scialó, F.; Formosa, L.E.; Stroud, D.A.; Garrett, M.; Lax, N.Z.; Robertson, F.M.; Jou, C.; et al. OXA1L mutations cause mitochondrial encephalopathy and a combined oxidative phosphorylation defect. EMBO Mol. Med. 2018, 10, e9060. [Google Scholar] [CrossRef]
- Molina-Gomes, D.; Viegas-Pequignot, E.; Bonnefoy, N.; Dujardin, G. The OXA1L gene that controls cytochrome oxidase assembly maps to the 14q11.2 region of the human genome. Genomics 1995, 30. Available online: https://www.osti.gov/biblio/446994 (accessed on 24 September 2020).
- Mick, D.U.; Dennerlein, S.; Wiese, H.; Reinhold, R.; Pacheu-Grau, D.; Lorenzi, I.; Sasarman, F.; Weraarpachai, W.; Shoubridge, E.A.; Warscheid, B.; et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 2012, 151, 1528–1541. [Google Scholar] [CrossRef]
- Dennerlein, S.; Oeljeklaus, S.; Jans, D.; Hellwig, C.; Bareth, B.; Jakobs, S.; Deckers, M.; Warscheid, B.; Rehling, P. MITRAC7 Acts as a COX1-Specific Chaperone and Reveals a Checkpoint during Cytochrome c Oxidase Assembly. Cell Rep. 2015, 12, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Fontanesi, F.; Clemente, P.; Barrientos, A. Cox25 teams up with Mss51, Ssc1, and Cox14 to regulate mitochondrial cytochrome c oxidase subunit 1 expression and assembly in Saccharomyces cerevisiae. J. Biol. Chem. 2011, 286, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, A.; Zambrano, A.; Tzagoloff, A. Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J. 2004, 23, 3472–3482. [Google Scholar] [CrossRef]
- Mick, D.U.; Wagner, K.; van der Laan, M.; Frazier, A.E.; Perschil, I.; Pawlas, M.; Meyer, H.E.; Warscheid, B.; Rehling, P. Shy1 couples Cox1 translational regulation to cytochrome c oxidase assembly. EMBO J. 2007, 26, 4347–4358. [Google Scholar] [CrossRef]
- Pierrel, F.; Bestwick, M.L.; Cobine, P.A.; Khalimonchuk, O.; Cricco, J.A.; Winge, D.R. Coa1 links the Mss51 post-translational function to Cox1 cofactor insertion in cytochrome c oxidase assembly. EMBO J. 2007, 26, 4335–4346. [Google Scholar] [CrossRef] [PubMed]
- McStay, G.P.; Su, C.H.; Tzagoloff, A. Stabilization of Cox1p intermediates by the Cox14p-Coa3p complex. FEBS Lett. 2013, 587, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Soto, I.C.; Fontanesi, F.; Myers, R.S.; Hamel, P.; Barrientos, A. A heme-sensing mechanism in the translational regulation of mitochondrial cytochrome c oxidase biogenesis. Cell Metab. 2012, 16, 801–813. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef]
- Aras, S.; Pak, O.; Sommer, N.; Finley, R.; Hüttemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 2013, 41, 2255–2266. [Google Scholar] [CrossRef]
- Tanigaki, K.; Honjo, T. Two opposing roles of RBP-J in Notch signaling. Curr. Top. Dev. Biol. 2010, 92, 231–252. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.V.; Poyton, R.O. Structure/function of oxygen-regulated isoforms in cytochrome c oxidase. J. Exp. Biol. 1998, 201, 1163–1175. [Google Scholar] [PubMed]
- Bestwick, M.; Khalimonchuk, O.; Pierrel, F.; Winge, D.R. The role of Coa2 in hemylation of yeast Cox1 revealed by its genetic interaction with Cox10. Mol. Cell. Biol. 2010, 30, 172–185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bareth, B.; Dennerlein, S.; Mick, D.U.; Nikolov, M.; Urlaub, H.; Rehling, P. The heme a synthase Cox15 associates with cytochrome c oxidase assembly intermediates during Cox1 maturation. Mol. Cell. Biol. 2013, 33, 4128–4137. [Google Scholar] [CrossRef]
- Glerum, D.M.; Tzagoloff, A. Isolation of a human cDNA for heme A: Farnesyltransferase by functional complementation of a yeast cox10 mutant. Proc. Natl. Acad. Sci. USA 1994, 91, 8452–8456. [Google Scholar] [CrossRef]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar] [CrossRef]
- Taylor, N.G.; Swenson, S.; Harris, N.J.; Germany, E.M.; Fox, J.L.; Khalimonchuk, O. The assembly factor pet117 couples heme a synthase activity to cytochrome oxidase assembly. J. Biol. Chem. 2017, 292, 1815–1825. [Google Scholar] [CrossRef]
- Khalimonchuk, O.; Ostermann, K.; Rödel, G. Evidence for the association of yeast mitochondrial ribosomes with Cox11p, a protein required for the Cu(B) site formation of cytochrome c oxidase. Curr. Genet. 2005, 47, 223–233. [Google Scholar] [CrossRef]
- Horng, Y.-C.; Cobine, P.A.; Maxfield, A.B.; Carr, H.S.; Winge, D.R. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J. Biol. Chem. 2004, 279, 35334–35340. [Google Scholar] [CrossRef]
- Barros, M.H.; Johnson, A.; Tzagoloff, A. COX23, a homologue of COX17, is required for cytochrome oxidase assembly. J. Biol. Chem. 2004, 279, 31943–31947. [Google Scholar] [CrossRef]
- Bode, M.; Woellhaf, M.W.; Bohnert, M.; van der Laan, M.; Sommer, F.; Jung, M.; Zimmermann, R.; Schroda, M.; Herrmann, J.M. Redox-regulated dynamic interplay between Cox19 and the copper-binding protein Cox11 in the intermembrane space of mitochondria facilitates biogenesis of cytochrome c oxidase. Mol. Biol. Cell 2015, 26, 2385–2401. [Google Scholar] [CrossRef] [PubMed]
- Horn, D.; Al-Ali, H.; Barrientos, A. Cmc1p is a conserved mitochondrial twin CX9C protein involved in cytochrome c oxidase biogenesis. Mol. Cell. Biol. 2008, 28, 4354–4364. [Google Scholar] [CrossRef] [PubMed]
- Horn, D.; Zhou, W.; Trevisson, E.; Al-Ali, H.; Harris, T.K.; Salviati, L.; Barrientos, A. The conserved mitochondrial twin Cx9C protein Cmc2 Is a Cmc1 homologue essential for cytochrome c oxidase biogenesis. J. Biol. Chem. 2010, 285, 15088–15099. [Google Scholar] [CrossRef]
- Lorenzi, I.; Oeljeklaus, S.; Aich, A.; Ronsör, C.; Callegari, S.; Dudek, J.; Warscheid, B.; Dennerlein, S.; Rehling, P. The mitochondrial TMEM177 associates with COX20 during COX2 biogenesis. Biochim. Biophys. Acta 2017, 1865, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Rigby, K.; Cobine, P.A.; Khalimonchuk, O.; Winge, D.R. Mapping the functional interaction of Sco1 and Cox2 in cytochrome oxidase biogenesis. J. Biol. Chem. 2008, 283, 15015–15022. [Google Scholar] [CrossRef]
- Glerum, D.M.; Shtanko, A.; Tzagoloff, A. SCO1 and SCO2 act as high copy suppressors of a mitochondrial copper recruitment defect in Saccharomyces cerevisiae. J. Biol. Chem. 1996, 271, 20531–20535. [Google Scholar] [CrossRef]
- Cerqua, C.; Morbidoni, V.; Desbats, M.A.; Doimo, M.; Frasson, C.; Sacconi, S.; Baldoin, M.C.; Sartori, G.; Basso, G.; Salviati, L.; et al. COX16 is required for assembly of cytochrome c oxidase in human cells and is involved in copper delivery to COX2. Biochim. Biophys. Acta 2018, 1859, 244–252. [Google Scholar] [CrossRef]
- Aich, A.; Wang, C.; Chowdhury, A.; Ronsör, C.; Pacheu-Grau, D.; Richter-Dennerlein, R.; Dennerlein, S.; Rehling, P. COX16 promotes COX2 metallation and assembly during respiratory complex IV biogenesis. Elife 2018, 7, e32572. [Google Scholar] [CrossRef]
- Pacheu-Grau, D.; Wasilewski, M.; Oeljeklaus, S.; Gibhardt, C.S.; Aich, A.; Chudenkova, M.; Dennerlein, S.; Deckers, M.; Bogeski, I.; Warscheid, B.; et al. COA6 facilitates cytochrome c oxidase biogenesis as thiol-reductase for copper metallochaperones in mitochondria. J. Mol. Biol. 2020, 432, 2067–2079. [Google Scholar] [CrossRef]
- Vidoni, S.; Harbour, M.E.; Guerrero-Castillo, S.; Signes, A.; Ding, S.; Fearnley, I.M.; Taylor, R.W.; Tiranti, V.; Arnold, S.; Fernandez-Vizarra, E.; et al. MR-1S Interacts with PET100 and PET117 in Module-Based Assembly of Human Cytochrome c Oxidase. Cell Rep. 2017, 18, 1727–1738. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, W.; Liu, H.; He, H.; Shao, R. Myofibrillogenesis regulator 1 (MR-1): A potential therapeutic target for cancer and PNKD. J. Drug Target. 2018, 26, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Dawitz, H.; Schäfer, J.; Schaart, J.M.; Magits, W.; Brzezinski, P.; Ott, M. Rcf1 Modulates Cytochrome c Oxidase Activity Especially Under Energy-Demanding Conditions. Front. Physiol. 2019, 10, 1555. [Google Scholar] [CrossRef] [PubMed]
- Hock, D.H.; Reljic, B.; Ang, C.-S.; Muellner-Wong, L.; Mountford, H.S.; Compton, A.G.; Ryan, M.T.; Thorburn, D.R.; Stroud, D.A. HIGD2A is required for assembly of the COX3 module of human mitochondrial complex IV. Mol. Cell. Proteom. 2020. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, R.; Bareth, B.; Balleininger, M.; Wissel, M.; Rehling, P.; Mick, D.U. Mimicking a SURF1 allele reveals uncoupling of cytochrome c oxidase assembly from translational regulation in yeast. Hum. Mol. Genet. 2011, 20, 2379–2393. [Google Scholar] [CrossRef]
- Valnot, I.; von Kleist-Retzow, J.C.; Barrientos, A.; Gorbatyuk, M.; Taanman, J.W.; Mehaye, B.; Rustin, P.; Tzagoloff, A.; Munnich, A.; Rötig, A. A mutation in the human heme A: Farnesyltransferase gene (COX10 ) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 1245–1249. [Google Scholar] [CrossRef]
- Sinkler, C.A.; Kalpage, H.; Shay, J.; Lee, I.; Malek, M.H.; Grossman, L.I.; Hüttemann, M. Tissue- and Condition-Specific Isoforms of Mammalian Cytochrome c Oxidase Subunits: From Function to Human Disease. Oxid. Med. Cell. Longev. 2017, 2017, 1534056. [Google Scholar] [CrossRef]
- Halperin, D.; Drabkin, M.; Wormser, O.; Yogev, Y.; Dolgin, V.; Shorer, Z.; Gradstein, L.; Shelef, I.; Flusser, H.; Birk, O.S. Phenotypic variability and mutation hotspot in COX15-related Leigh syndrome. Am. J. Med. Genet. A 2020, 182, 1506–1512. [Google Scholar] [CrossRef]
- van Bon, B.W.M.; Oortveld, M.A.W.; Nijtmans, L.G.; Fenckova, M.; Nijhof, B.; Besseling, J.; Vos, M.; Kramer, J.M.; de Leeuw, N.; Castells-Nobau, A.; et al. CEP89 is required for mitochondrial metabolism and neuronal function in man and fly. Hum. Mol. Genet. 2013, 22, 3138–3151. [Google Scholar] [CrossRef]
- Weraarpachai, W.; Sasarman, F.; Nishimura, T.; Antonicka, H.; Auré, K.; Rötig, A.; Lombès, A.; Shoubridge, E.A. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am. J. Hum. Genet. 2012, 90, 142–151. [Google Scholar] [CrossRef]
- Ostergaard, E.; Weraarpachai, W.; Ravn, K.; Born, A.P.; Jønson, L.; Duno, M.; Wibrand, F.; Shoubridge, E.A.; Vissing, J. Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity, and short stature. J. Med. Genet. 2015, 52, 203–207. [Google Scholar] [CrossRef]
- Huigsloot, M.; Nijtmans, L.G.; Szklarczyk, R.; Baars, M.J.H.; van den Brand, M.A.M.; Hendriksfranssen, M.G.M.; van den Heuvel, L.P.; Smeitink, J.A.M.; Huynen, M.A.; Rodenburg, R.J.T. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Martinez Lyons, A.; Ardissone, A.; Reyes, A.; Robinson, A.J.; Moroni, I.; Ghezzi, D.; Fernandez-Vizarra, E.; Zeviani, M. COA7 (C1orf163/RESA1) mutations associated with mitochondrial leukoencephalopathy and cytochrome c oxidase deficiency. J. Med. Genet. 2016, 53, 846–849. [Google Scholar] [CrossRef]
- Higuchi, Y.; Okunushi, R.; Hara, T.; Hashiguchi, A.; Yuan, J.; Yoshimura, A.; Murayama, K.; Ohtake, A.; Ando, M.; Hiramatsu, Y.; et al. Mutations in COA7 cause spinocerebellar ataxia with axonal neuropathy. Brain 2018, 141, 1622–1636. [Google Scholar] [CrossRef] [PubMed]
- Mohanraj, K.; Wasilewski, M.; Benincá, C.; Cysewski, D.; Poznanski, J.; Sakowska, P.; Bugajska, Z.; Deckers, M.; Dennerlein, S.; Fernandez-Vizarra, E.; et al. Inhibition of proteasome rescues a pathogenic variant of respiratory chain assembly factor COA7. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Mansour, H.; Sabbagh, S.; Bizzari, S.; El-Hayek, S.; Chouery, E.; Gambarini, A.; Gencik, M.; Mégarbané, A. The Lebanese Allele in the PET100 Gene: Report on Two New Families with Cytochrome c Oxidase Deficiency. J. Pediatr. Genet. 2019, 8, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Oláhová, M.; Haack, T.B.; Alston, C.L.; Houghton, J.A.; He, L.; Morris, A.A.; Brown, G.K.; McFarland, R.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N.; et al. A truncating PET100 variant causing fatal infantile lactic acidosis and isolated cytochrome c oxidase deficiency. Eur. J. Hum. Genet. 2015, 23, 935–939. [Google Scholar] [CrossRef]
- Lim, S.C.; Smith, K.R.; Stroud, D.A.; Compton, A.G.; Tucker, E.J.; Dasvarma, A.; Gandolfo, L.C.; Marum, J.E.; McKenzie, M.; Peters, H.L.; et al. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am. J. Hum. Genet. 2014, 94, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Visser, G.; Baertling, F.; Wintjes, L.T.; Wolters, V.M.; van Montfrans, J.; de Kort, G.A.P.; Nikkels, P.G.J.; van Hasselt, P.M.; van der Crabben, S.N.; et al. Mutated PET117 causes complex IV deficiency and is associated with neurodevelopmental regression and medulla oblongata lesions. Hum. Genet. 2017, 136, 759–769. [Google Scholar] [CrossRef]
- Antonicka, H.; Leary, S.C.; Guercin, G.-H.; Agar, J.N.; Horvath, R.; Kennaway, N.G.; Harding, C.O.; Jaksch, M.; Shoubridge, E.A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar] [CrossRef]
- Coenen, M.J.H.; van den Heuvel, L.P.; Ugalde, C.; Ten Brinke, M.; Nijtmans, L.G.J.; Trijbels, F.J.M.; Beblo, S.; Maier, E.M.; Muntau, A.C.; Smeitink, J.A.M. Cytochrome c oxidase biogenesis in a patient with a mutation in COX10 gene. Ann. Neurol. 2004, 56, 560–564. [Google Scholar] [CrossRef]
- Oquendo, C.E.; Antonicka, H.; Shoubridge, E.A.; Reardon, W.; Brown, G.K. Functional and genetic studies demonstrate that mutation in the COX15 gene can cause Leigh syndrome. J. Med. Genet. 2004, 41, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Alfadhel, M.; Lillquist, Y.P.; Waters, P.J.; Sinclair, G.; Struys, E.; McFadden, D.; Hendson, G.; Hyams, L.; Shoffner, J.; Vallance, H.D. Infantile cardioencephalopathy due to a COX15 gene defect: Report and review. Am. J. Med. Genet. A 2011, 155A, 840–844. [Google Scholar] [CrossRef]
- Bugiani, M.; Tiranti, V.; Farina, L.; Uziel, G.; Zeviani, M. Novel mutations in COX15 in a long surviving Leigh syndrome patient with cytochrome c oxidase deficiency. J. Med. Genet. 2005, 42, e28. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, L.C.; Sue, C.M.; Davidson, M.M.; Tanji, K.; Nishino, I.; Sadlock, J.E.; Krishna, S.; Walker, W.; Selby, J.; Glerum, D.M.; et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999, 23, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, M.; Ogilvie, I.; Yao, J.; Kortenhaus, G.; Bresser, H.G.; Gerbitz, K.D.; Shoubridge, E.A. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, A.P.; Saade, D.; Pereira, C.P.; Farooq, A.; Huff, T.C.; Abreu, L.; Moraes, C.T.; Mnatsakanova, D.; Mathews, K.; Yang, H.; et al. SCO2 mutations cause early-onset axonal Charcot-Marie-Tooth disease associated with cellular copper deficiency. Brain 2018, 141, 662–672. [Google Scholar] [CrossRef]
- Jaksch, M.; Horvath, R.; Horn, N.; Auer, D.P.; Macmillan, C.; Peters, J.; Gerbitz, K.D.; Kraegeloh-Mann, I.; Muntau, A.; Karcagi, V.; et al. Homozygosity (E140K) in SCO2 causes delayed infantile onset of cardiomyopathy and neuropathy. Neurology 2001, 57, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Valnot, I.; Osmond, S.; Gigarel, N.; Mehaye, B.; Amiel, J.; Cormier-Daire, V.; Munnich, A.; Bonnefont, J.P.; Rustin, P.; Rötig, A. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet. 2000, 67, 1104–1109. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Leontari, I.; Martinelli, M.; Palumaa, P.; Sillard, R.; Wang, S. Human Sco1 functional studies and pathological implications of the P174L mutant. Proc. Natl. Acad. Sci. USA 2007, 104, 15–20. [Google Scholar] [CrossRef]
- Melchionda, L.; Haack, T.B.; Hardy, S.; Abbink, T.E.M.; Fernandez-Vizarra, E.; Lamantea, E.; Marchet, S.; Morandi, L.; Moggio, M.; Carrozzo, R.; et al. Mutations in APOPT1, encoding a mitochondrial protein, cause cavitating leukoencephalopathy with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 2014, 95, 315–325. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Subunit | Transcription/mRNA Processing | Translation | Proteolytic Processing/Protease | Membrane Insertion | Copper Association | Haem Association | Chaperone | Unknown |
|---|---|---|---|---|---|---|---|---|
| COX1 | Cox24 | Mss51p/_ _/TACO1 | Oma1 | Oxa1 Mba1/_ | Cox11 Cox17 Cox19 Cox23 Cmc1 Cmc2 | Coa2 Cox10 Cox15 Shy1/SURF1 | Coa1/MITRAC15 Coa3/MITRAC12 Cox14 Mdj1 Ssc1 _/MITRAC7 | |
| COX2 | – | Pet111 | Cox20 Imp1 Imp2 Som1 | Oxa1 Cox18 Pnt1 Mss2 | Cox16 Cox17 Cox19 Cox23 Coa6 Cmc1 Cmc2 Sco1 Sco2 _/TMEM177 | – | Cox20 | |
| COX3 | – | Pet54 Pet122 Pet494 | – | – | – | Rcf1p/HIGD2A | ||
| 2 or more genes | LRPPRC | – | – | – | – | – | ||
| Unknown | _/CEP89 _/COA7 _/COA8 Pet191/Coa5 |
| Protein | Matrix | IMS | Trans-Membrane | Total |
|---|---|---|---|---|
| Cox14p | 9.69 N | 4.58 C | 5.52 | 7.51 |
| Coa1p | 10.94 N | 5.66 C | 5.52 | 10.12 |
| Coa3p | 5.48 C | 11.07 N | 8.43 | 9.83 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watson, S.A.; McStay, G.P. Functions of Cytochrome c Oxidase Assembly Factors. Int. J. Mol. Sci. 2020, 21, 7254. https://doi.org/10.3390/ijms21197254
Watson SA, McStay GP. Functions of Cytochrome c Oxidase Assembly Factors. International Journal of Molecular Sciences. 2020; 21(19):7254. https://doi.org/10.3390/ijms21197254
Chicago/Turabian StyleWatson, Shane A., and Gavin P. McStay. 2020. "Functions of Cytochrome c Oxidase Assembly Factors" International Journal of Molecular Sciences 21, no. 19: 7254. https://doi.org/10.3390/ijms21197254
APA StyleWatson, S. A., & McStay, G. P. (2020). Functions of Cytochrome c Oxidase Assembly Factors. International Journal of Molecular Sciences, 21(19), 7254. https://doi.org/10.3390/ijms21197254