Neuroprotective Effect of Antioxidants in the Brain



Abstract
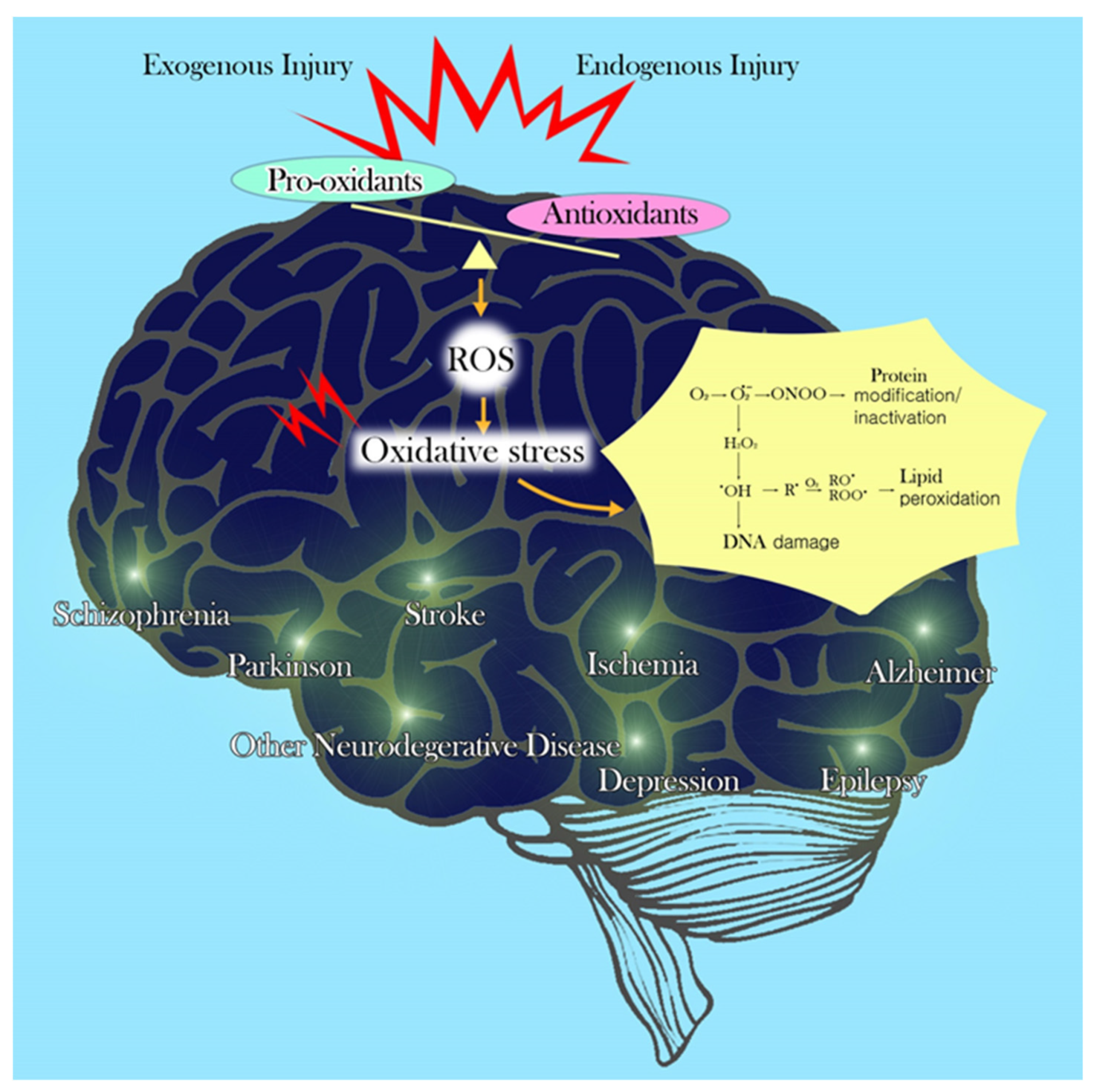
1. Introduction
2. Factors That Contribute to Vulnerability of the Brain to Oxidative Stress
3. Role of Antioxidants in the Brain
3.1. Enzymatic Antioxidants
3.1.1. Superoxide Dismutase
3.1.2. Catalase
3.1.3. Glutathione Peroxidase
3.1.4. Thioredoxin
3.2. Non-Enzymatic Antioxidants
3.2.1. Antioxidant Enzyme Cofactors (Selenium, Coenzyme Q10, Zinc)
Selenium
Coenzyme Q
Zinc and Essential Metals
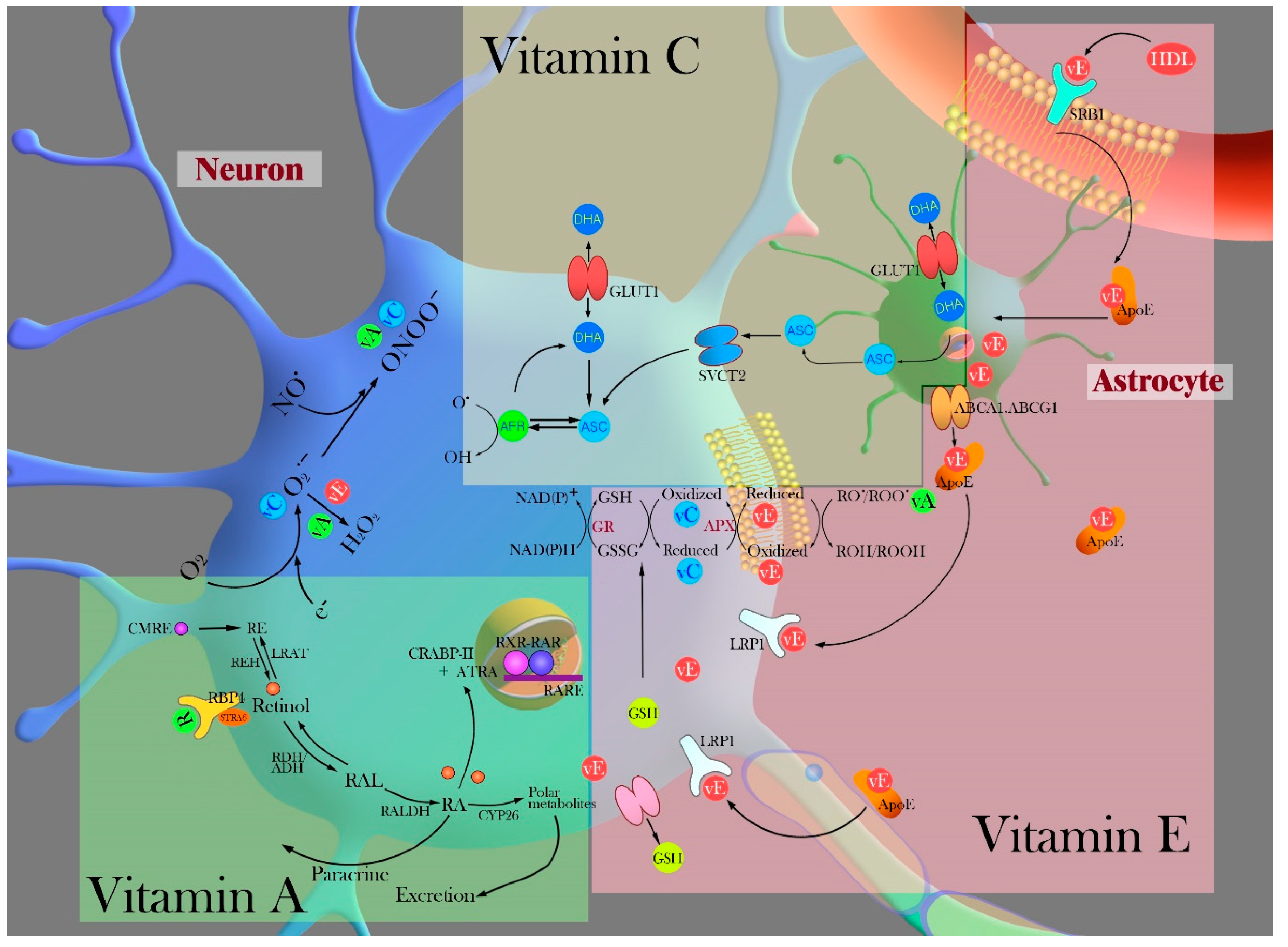
3.2.2. ROS/RNS Scavengers (Vitamin C, E, and A)
Vitamin C
Vitamin E
Vitamin A
3.2.3. Nrf2 Antioxidant System
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer disease |
| ALS | amyotrophic lateral sclerosis |
| ApoE | apolipoprotein E |
| ARE | antioxidant response element |
| ATP | adenosine triphosphate |
| ATPH | alpha-tocopherol |
| ATRA | all-trans-retinoic acid |
| ATTN | alpha-tocotrienol |
| Aβ | beta-amyloid |
| CA3 | cornu ammonis 3 |
| CAT | catalase |
| CNS | central nervous system |
| CoQ | coenzyme Q |
| COX | cyclooxygenase |
| CR | carbonyl reductase |
| CRABP | cellular retinoic acid-binding proteins |
| CRBP | cellular retinol binding proteins |
| DCF | dichlorofluorescein |
| DHA | dehydroascorbate |
| EC-SOD | extracellular superoxide dismutase |
| FADH2 | dihydroflavine-adenine dinucleotide |
| Fe2+ | ferrous ion |
| Fe3+ | ferric ion |
| GCL | glutamate-cysteine ligase |
| GLUT1 | glucose transports |
| GPx | glutathione peroxidase |
| GR | glutathione reductase |
| GSH | glutathione |
| GSSG | glutathione disulfide |
| hAPP | human amyloid precursor protein |
| HD | Huntington’s disease |
| HDL | high density lipoprotein |
| HO-1 | heme oxygenase-1 |
| iNOS | inducible nitric oxide synthase |
| KA | kainic acid |
| LRP | lipoprotein receptor-related protein |
| MAPK | mitogen-activated protein kinase |
| MCAO | middle cerebral artery occlusion |
| Mn-SOD | manganese containing superoxide dismutase |
| MOR | μ-type opioid receptor |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NADH | reduced form of nicotinamide adenine dinucleotide |
| NADPH | reduced form of nicotinamide adenine dinucleotide phosphate |
| NBIA | neurodegeneration brain iron accumulation |
| NF-κB | nuclear factor-κB |
| nNOS | neuronal nitric oxide synthase |
| NOX | NADPH oxidase |
| NQO1 | NADPH quinone oxidoreductase |
| Nrf2 | nuclear factor elytroid 2-related factor 2 |
| 6-OHDA | 6-hydroxydopamine |
| OHSC | organotypic hippocampal slice culture |
| PD | Parkinson disease |
| PI | propidium iodide |
| PTSD | post-traumatic stress disorder |
| RAR | retinoic acid receptors |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| Se | selenium |
| SOD | superoxide dismutase |
| SRB1 | scavenger receptor class B type 1 |
| SVCT | sodium-vitamin C co-transporters |
| TAC | total antioxidant capacity |
| TBARS | thiobarbiturate reactive substance |
| Trx | thioredoxin |
| TrxR | thioredoxin reductase |
| UV | ultra violet |
References
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.M.; Bärtsch, P.; Knauth, M.; Baumgartner, R.W. Emerging concepts in acute mountain sickness and high-altitude cerebral edema: From the molecular to the morphological. Cell. Mol. Life Sci. 2009, 66, 3583–3594. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the balance between ROS and antioxidants: When to use the synthetic antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef]
- Watson, B.D. Evaluation of the concomitance of lipid peroxidation in experimental models of cerebral ischemia and stroke. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 1993; Volume 96, pp. 69–95. [Google Scholar]
- Choi, B.H. Oxygen, antioxidants and brain dysfunction. Yonsei Med. J. 1993, 34, 1–10. [Google Scholar] [CrossRef]
- Poon, H.F.; Calabrese, V.; Scapagnini, G.; Butterfield, D.A. Free radicals: Key to brain aging and heme oxygenase as a cellular response to oxidative stress. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, M478–M493. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Balachandran, B. Role of oxidative stress and antioxidants in neurodegenerative diseases. Nutr. Neurosci. 2002, 5, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Packer, L. Oxidative stress, Antioxidants, Aging and Disease. In Oxidative Stress and Aging; Springer: Berlin/Heidelberg, Germany, 1995; pp. 1–14. [Google Scholar]
- Li, J.; Li, W.; Jiang, Z.G.; Ghanbari, H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.E.; Lee, C. The redox switch that regulates molecular chaperones. Biomol. Concepts 2015, 6, 269–284. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Peltier, J.; Stone, D.; Schaffer, D.V.; Chang, C.J. Nox2 redox signaling maintains essential cell populations in the brain. Nat. Chem. Biol. 2011, 7, 106–112. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric oxide and mitochondrial respiration. Biochim. Biophys. Acta 1999, 1411, 351–369. [Google Scholar] [CrossRef]
- Carballal, S.; Bartesaghi, S.; Radi, R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim. Biophys. Acta 2014, 1840, 768–780. [Google Scholar] [CrossRef]
- Lipton, S.A.; Choi, Y.B.; Pan, Z.H.; Lei, S.Z.; Chen, H.S.; Sucher, N.J.; Loscalzo, J.; Singel, D.J.; Stamler, J.S. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 1993, 364, 626–632. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Jensen, M.B.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T. Suppressors of superoxide-H2O2 production at site IQ of mitochondrial complex I protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef]
- Goncalves, R.L.; Rothschild, D.E.; Quinlan, C.L.; Scott, G.K.; Benz, C.C.; Brand, M.D. Sources of superoxide/H2O2 during mitochondrial proline oxidation. Redox Biol. 2014, 2, 901–909. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Pun, P.B.L.; Logan, A.; Darley-Usmar, V.; Chacko, B.; Johnson, M.S.; Huang, G.W.; Rogatti, S.; Prime, T.A.; Methner, C.; Krieg, T. A mitochondria-targeted mass spectrometry probe to detect glyoxals: Implications for diabetes. Free Radic. Biol. Med. 2014, 67, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S. The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 2017, 18, 251. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Heikkila, R.E. The generation of hydrogen peroxide, superoxide radical, and hydroxyl radical by 6-hydroxydopamine, dialuric acid, and related cytotoxic agents. J. Biol. Chem. 1974, 249, 2447–2452. [Google Scholar]
- Miller, D.M.; Buettner, G.R.; Aust, S.D. Transition metals as catalysts of “autoxidation” reactions. Free Radic. Biol. Med. 1990, 8, 95–108. [Google Scholar] [CrossRef]
- Patra, R.; Swarup, D.; Dwivedi, S. Antioxidant effects of α tocopherol, ascorbic acid and L-methionine on lead induced oxidative stress to the liver, kidney and brain in rats. Toxicology 2001, 162, 81–88. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Borchert, A.; Bräuer, A.U.; Kuhn, H. Role for glutathione peroxidase-4 in brain development and neuronal apoptosis: Specific induction of enzyme expression in reactive astrocytes following brain injury. Free Radic. Biol. Med. 2007, 43, 191–201. [Google Scholar] [CrossRef]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Li, Y. What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: An update. Chem. Rev. 2007, 107, 748–766. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Sueta, G.; Radi, R. Chemical biology of peroxynitrite: Kinetics, diffusion, and radicals. ACS Chem. Biol. 2009, 4, 161–177. [Google Scholar] [CrossRef]
- Thomas, D.D. Breathing new life into nitric oxide signaling: A brief overview of the interplay between oxygen and nitric oxide. Redox Biol. 2015, 5, 225–233. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J. Oxidative DNA damage & repair: An introduction. Free Radic. Biol. Med. 2017, 107, 2–12. [Google Scholar]
- Gutteridge, J. Biological origin of free radicals, and mechanisms of antioxidant protection. Chem. Biol. Interact. 1994, 91, 133–140. [Google Scholar] [CrossRef]
- Ellerby, L.M.; Cabelli, D.E.; Graden, J.A.; Valentine, J.S. Copper−zinc superoxide dismutase: Why not pH-dependent? J. Am. Chem. Soc. 1996, 118, 6556–6561. [Google Scholar] [CrossRef]
- Banci, L.; Benedetto, M.; Bertini, I.; Del Conte, R.; Piccioli, M.; Viezzoli, M.S. Solution structure of reduced monomeric Q133M2 copper, zinc superoxide dismutase (SOD). Why is SOD a dimeric enzyme? Biochemistry 1998, 37, 11780–11791. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Hickey, M.J.; Borgstahl, G.E.; Hallewell, R.A.; Lepock, J.R.; O’Connor, D.; Hsieh, Y.; Nick, H.S.; Silverman, D.N.; Tainer, J.A. Crystal structure of Y34F mutant human mitochondrial manganese superoxide dismutase and the functional role of tyrosine 34. Biochemistry 1998, 37, 4722–4730. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Wang, X.L. Association of extracellular-superoxide dismutase phenotype with the endothelial constitutive nitric oxide synthase polymorphism. FEBS Lett. 1998, 433, 166–168. [Google Scholar] [CrossRef]
- Matés, J.M.; Sánchez-Jiménez, F. Antioxidant enzymes and their implications in pathophysiologic processes. Front. Biosci. 1999, 4, D339–D345. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Velde, C.V.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef]
- Li, Q.X.; Mok, S.S.; Laughton, K.M.; McLean, C.A.; Volitakis, I.; Cherny, R.A.; Cheung, N.S.; White, A.R.; Masters, C.L. Overexpression of Aβ is associated with acceleration of onset of motor impairment and superoxide dismutase 1 aggregation in an amyotrophic lateral sclerosis mouse model. Aging Cell 2006, 5, 153–165. [Google Scholar] [CrossRef]
- Iadecola, C.; Zhang, F.; Niwa, K.; Eckman, C.; Turner, S.K.; Fischer, E.; Younkin, S.; Borchelt, D.R.; Hsiao, K.K.; Carlson, G.A. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat. Neurosci. 1999, 2, 157–161. [Google Scholar] [CrossRef]
- De Haan, J.B.; Newman, J.D.; Kola, I. Cu/Zn superoxide dismutase mRNA and enzyme activity, and susceptibility to lipid peroxidation, increases with aging in murine brains. Brain Res. Mol. Brain Res. 1992, 13, 179–187. [Google Scholar] [CrossRef]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef]
- Kim, U.J.; Lee, B.H.; Lee, K.H. Neuroprotective effects of a protein tyrosine phosphatase inhibitor against hippocampal excitotoxic injury. Brain Res. 2019, 1719, 133–139. [Google Scholar] [CrossRef]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.-Q.; Bien-Ly, N.; Puoliväli, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 2006, 26, 5167–5179. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Wood, T.R.; Nance, E. Superoxide dismutase reduces monosodium glutamate-induced injury in an organotypic whole hemisphere brain slice model of excitotoxicity. J. Biol. Eng. 2020, 14, 1–12. [Google Scholar]
- Hunt, C.R.; Sim, J.E.; Sullivan, S.J.; Featherstone, T.; Golden, W.; Von Kapp-Herr, C.; Hock, R.A.; Gomez, R.A.; Parsian, A.J.; Spitz, D.R. Genomic instability and catalase gene amplification induced by chronic exposure to oxidative stress. Cancer Res. 1998, 58, 3986–3992. [Google Scholar] [PubMed]
- Fita, I.; Rossmann, M.G. The active center of catalase. J. Mol. Biol. 1985, 185, 21–37. [Google Scholar] [CrossRef]
- Usui, S.; Komeima, K.; Lee, S.Y.; Jo, Y.-J.; Ueno, S.; Rogers, B.S.; Wu, Z.; Shen, J.; Lu, L.; Oveson, B.C. Increased expression of catalase and superoxide dismutase 2 reduces cone cell death in retinitis pigmentosa. Mol. Ther. 2009, 17, 778–786. [Google Scholar] [CrossRef]
- Speranza, M.J.; Bagley, A.; Lynch, R. Cells enriched for catalase are sensitized to the toxicities of bleomycin, adriamycin, and paraquat. J. Biol. Chem. 1993, 268, 19039–19043. [Google Scholar]
- Terlecky, S.R.; Koepke, J.I.; Walton, P.A. Peroxisomes and aging. Biochim. Biophy. Acta 2006, 1763, 1749–1754. [Google Scholar] [CrossRef]
- Sheikh, F.G.; Pahan, K.; Khan, M.; Barbosa, E.; Singh, I. Abnormality in catalase import into peroxisomes leads to severe neurological disorder. Proc. Natl. Acad. Sci. USA 1998, 95, 2961–2966. [Google Scholar] [CrossRef]
- Baxter, P.S.; Bell, K.F.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Zhang, Z.; Rydel, R.E.; Drzewiecki, G.J.; Fuson, K.; Wright, S.; Wogulis, M.; Audia, J.E.; May, P.C.; Hyslop, P.A. Amyloid β-mediated oxidative and metabolic stress in rat cortical neurons: No direct evidence for a role for H2O2 generation. J. Neurochem. 1996, 67, 1595–1606. [Google Scholar] [CrossRef]
- Nell, H.J.; Au, J.L.; Giordano, C.R.; Terlecky, S.R.; Walton, P.A.; Whitehead, S.N.; Cechetto, D.F. Targeted antioxidant, catalase–SKL, reduces beta-amyloid toxicity in the rat brain. Brain Pathol. 2017, 27, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Yakunin, E.; Kisos, H.; Kulik, W.; Grigoletto, J.; Wanders, R.J.; Sharon, R. The regulation of catalase activity by PPAR γ is affected by α-synuclein. Ann. Clin. Transl. Neurol. 2014, 1, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Brigelius-Flohé, R.; Aumann, K.D.; Roveri, A.; Schomburg, D.; Flohé, L. Diversity of glutathione peroxidases. Methods Enzymol. 1995, 252, 38–53. [Google Scholar] [PubMed]
- Kemp, M.; Go, Y.M.; Jones, D.P. Nonequilibrium thermodynamics of thiol/disulfide redox systems: A perspective on redox systems biology. Free Radic. Biol. Med. 2008, 44, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Ali, U.; Iannello, R.C.; Hertzog, P.; Crack, P.J. Diminished Akt phosphorylation in neurons lacking glutathione peroxidase-1 (Gpx1) leads to increased susceptibility to oxidative stress-induced cell death. J. Neurochem. 2005, 92, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Chen, Z.H.; Yoshida, Y.; Saito, Y.; Niki, E. Adaptation to hydrogen peroxide enhances PC12 cell tolerance against oxidative damage. Neurosci. Lett. 2005, 383, 256–259. [Google Scholar] [CrossRef]
- Goss, J.R.; Taffe, K.M.; Kochanek, P.M.; DeKosky, S.T. The antioxidant enzymes glutathione peroxidase and catalase increase following traumatic brain injury in the rat. Exp. Neurol. 1997, 146, 291–294. [Google Scholar] [CrossRef]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.Z.; Kim, H.J.; Kang, S.W.; Rhee, S.G. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res. Clin. Pract. 1999, 45, 101–112. [Google Scholar] [CrossRef]
- Burke-Gaffney, A.; Callister, M.E.; Nakamura, H. Thioredoxin: Friend or foe in human disease? Trends Pharmacol. Sci. 2005, 26, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Silva-Adaya, D.; Gonsebatt, M.E.; Guevara, J. Thioredoxin system regulation in the central nervous system: Experimental models and clinical evidence. Oxid. Med. Cell. Longev. 2014, 2014, 590808. [Google Scholar] [CrossRef]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Lundström, J.; Holmgren, A. Protein disulfide-isomerase is a substrate for thioredoxin reductase and has thioredoxin-like activity. J. Biol. Chem. 1990, 265, 9114–9120. [Google Scholar]
- Soerensen, J.; Jakupoglu, C.; Beck, H.; Förster, H.; Schmidt, J.; Schmahl, W.; Schweizer, U.; Conrad, M.; Brielmeier, M. The role of thioredoxin reductases in brain development. PLoS ONE 2008, 3, e1813. [Google Scholar] [CrossRef]
- May, J.M.; Cobb, C.E.; Mendiratta, S.; Hill, K.E.; Burk, R.F. Reduction of the ascorbyl free radical to ascorbate by thioredoxin reductase. J. Biol. Chem. 1998, 273, 23039–23045. [Google Scholar] [CrossRef]
- Arnér, E.S.; Nordberg, J.; Holmgren, A. Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase. Biochem. Biophys. Res. Commun. 1996, 225, 268–274. [Google Scholar] [CrossRef]
- Xia, L.; Björnstedt, M.; Nordman, T.; Eriksson, L.C.; Olsson, J.M. Reduction of ubiquinone by lipoamide dehydrogenase: An antioxidant regenerating pathway. Eur. J. Biochem. 2001, 268, 1486–1490. [Google Scholar] [CrossRef]
- Lu, J.; Zhong, L.; Lönn, M.E.; Burk, R.F.; Hill, K.E.; Holmgren, A. Penultimate selenocysteine residue replaced by cysteine in thioredoxin reductase from selenium-deficient rat liver. FASEB J. 2009, 23, 2394–2402. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Hattori, I.; Nozaki, K.; Mitsui, A.; Ishikawa, M.; Hashimoto, N.; Yodoi, J. Excitotoxic hippocampal injury is attenuated in thioredoxin transgenic mice. J. Cereb. Blood Flow Metab. 2000, 20, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Mitsui, A.; Nishiyama, A.; Nozaki, K.; Sono, H.; Gon, Y.; Hashimoto, N.; Yodoi, J. Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proc. Natl. Acad. Sci. USA 1999, 96, 4131–4136. [Google Scholar] [CrossRef]
- Björnstedt, M.; Xue, J.; Huang, W.; Akesson, B.; Holmgren, A. The thioredoxin and glutaredoxin systems are efficient electron donors to human plasma glutathione peroxidase. J. Biol. Chem. 1994, 269, 29382–29384. [Google Scholar] [PubMed]
- Munemasa, Y.; Kim, S.H.; Ahn, J.H.; Kwong, J.M.; Caprioli, J.; Piri, N. Protective effect of thioredoxins 1 and 2 in retinal ganglion cells after optic nerve transection and oxidative stress. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3535–3543. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zeng, X.S.; Jia, J.J.; Kwon, Y.; Wang, S.D.; Bai, J. The role of thioredoxin-1 in suppression of endoplasmic reticulum stress in Parkinson disease. Free Radic. Biol. Med. 2014, 67, 10–18. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xie, C.; Gabbita, S.P.; Markesbery, W.R. Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer’s disease brain. Free Radic. Biol. Med. 2000, 28, 418–427. [Google Scholar] [CrossRef]
- Sánchez-López, F.; Tasset, I.; Agüera, E.; Feijóo, M.; Fernández-Bolaños, R.; Sánchez, F.M.; Ruiz, M.C.; Cruz, A.H.; Gascón, F.; Túnez, I. Oxidative stress and inflammation biomarkers in the blood of patients with Huntington’s disease. Neurol. Res. 2012, 34, 721–724. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, P.P.; Ying, G.Y.; Zhu, X.D.; Shen, H.; Chen, G. Effects of thioredoxin-1 on neurogenesis after brain ischemia/reperfusion injury. CNS Neurosci. Ther. 2013, 19, 204. [Google Scholar] [CrossRef]
- Hwang, I.K.; Yoo, K.-Y.; Kim, D.W.; Lee, C.H.; Choi, J.H.; Kwon, Y.-G.; Kim, Y.-M.; Choi, S.Y.; Won, M.-H. Changes in the expression of mitochondrial peroxiredoxin and thioredoxin in neurons and glia and their protective effects in experimental cerebral ischemic damage. Free Radic. Biol. Med. 2010, 48, 1242–1251. [Google Scholar] [CrossRef]
- Miller, J.; Brzezinska-Slebodzinska, E.; Madsen, F. Oxidative stress, antioxidants, and animal function. J. Dairy Sci. 1993, 76, 2812–2823. [Google Scholar] [CrossRef]
- Hardy, G.; Hardy, I.; Manzanares, W. Selenium supplementation in the critically ill. Nutr. Clin. Pract. 2012, 27, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.J. Review of selenium toxicity in the aquatic food chain. Sci. Total Environ. 2004, 326, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, N.D. Importance of selenium and selenoprotein for brain function: From antioxidant protection to neuronal signalling. J. Inorg. Biochem. 2015, 153, 1–12. [Google Scholar] [CrossRef]
- Santamaría, A.; Vázquez-Román, B.; La cruz, V.P.D.; González-Cortés, C.; Trejo-Solís, M.C.; Galván-Arzate, S.; Jara-Prado, A.; Guevara-Fonseca, J.; Ali, S.F. Selenium reduces the proapoptotic signaling associated to NF-κB pathway and stimulates glutathione peroxidase activity during excitotoxic damage produced by quinolinate in rat corpus striatum. Synapse 2005, 58, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Zachara, B.A.; Pawluk, H.; Bloch-Boguslawska, E.; Śliwka, K.M.; Korenkiewicz, J.; Skok, Ź.; Ryć, K. Tissue level, distribution and total body selenium content in healthy and diseased humans in Poland. Arch. Environ. Health 2001, 56, 461–466. [Google Scholar] [CrossRef]
- Kühbacher, M.; Bartel, J.; Hoppe, B.; Alber, D.; Bukalis, G.; Bräuer, A.U.; Behne, D.; Kyriakopoulos, A. The brain selenoproteome: Priorities in the hierarchy and different levels of selenium homeostasis in the brain of selenium-deficient rats. J. Neurochem. 2009, 110, 133–142. [Google Scholar] [CrossRef]
- Dalla Puppa, L.; Savaskan, N.E.; Braeuer, A.U.; Behne, D.; Kyriakopoulos, A. The role of selenite on microglial migration. Ann. N. Y. Acad. Sci. 2007, 1096, 179–183. [Google Scholar] [CrossRef]
- Uğuz, A.C.; Nazıroğlu, M. Effects of selenium on calcium signaling and apoptosis in rat dorsal root ganglion neurons induced by oxidative stress. Neurochem. Res. 2012, 37, 1631–1638. [Google Scholar] [CrossRef]
- Haratake, M.; Yoshida, S.; Mandai, M.; Fuchigami, T.; Nakayama, M. Elevated amyloid-β plaque deposition in dietary selenium-deficient Tg2576 transgenic mice. Metallomics 2013, 5, 479–483. [Google Scholar] [CrossRef]
- Ishrat, T.; Parveen, K.; Khan, M.M.; Khuwaja, G.; Khan, M.B.; Yousuf, S.; Ahmad, A.; Shrivastav, P.; Islam, F. Selenium prevents cognitive decline and oxidative damage in rat model of streptozotocin-induced experimental dementia of Alzheimer’s type. Brain Res. 2009, 1281, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Ellwanger, J.H.; Molz, P.; Dallemole, D.R.; dos Santos, A.P.; Müller, T.E.; Cappelletti, L.; da Silva, M.G.; Franke, S.I.R.; Prá, D.; Henriques, J.A.P. Selenium reduces bradykinesia and DNA damage in a rat model of Parkinson’s disease. Nutrition 2015, 31, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Marks, E.; Chen, J.; Moline, J.; Barrows, L.; Raisbeck, M.; Volitakis, I.; Cherny, R.A.; Chopra, V.; Bush, A.I. Altered selenium status in Huntington’s disease: Neuroprotection by selenite in the N171-82Q mouse model. Neurobiol. Dis. 2014, 71, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.E.; Kang, S.K. Selenium effectively inhibits ROS-mediated apoptotic neural precursor cell death in vitro and in vivo in traumatic brain injury. Biochim. Biophys. Acta 2007, 1772, 1199–1210. [Google Scholar] [CrossRef]
- Khalili, H.; Ahl, R.; Cao, Y.; Paydar, S.; Sjölin, G.; Niakan, A.; Dabiri, G.; Mohseni, S. Early selenium treatment for traumatic brain injury: Does it improve survival and functional outcome? Injury 2017, 48, 1922–1926. [Google Scholar] [CrossRef] [PubMed]
- Nohl, H.; Kozlov, A.V.; Staniek, K.; Gille, L. The multiple functions of coenzyme Q. Bioorg. Chem. 2001, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1995, 1271, 195–204. [Google Scholar] [CrossRef]
- Bhagavan, H.N.; Chopra, R.K. Coenzyme Q10: Absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic. Res. 2006, 40, 445–453. [Google Scholar] [CrossRef]
- Hargreaves, I.P. Ubiquinone: Cholesterol’s reclusive cousin. Ann. Clin. Biochem. 2003, 40, 207–218. [Google Scholar] [CrossRef]
- Xia, L.; Nordman, T.; Olsson, J.M.; Damdimopoulos, A.; Björkhem-Bergman, L.; Nalvarte, I.; Eriksson, L.C.; Arnér, E.S.; Spyrou, G.; Björnstedt, M. The mammalian cytosolic selenoenzyme thioredoxin reductase reduces ubiquinone a novel mechanism for defense against oxidative stress. J. Biol. Chem. 2003, 278, 2141–2146. [Google Scholar] [CrossRef]
- Montini, G.; Malaventura, C.; Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008, 358, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Sacconi, S.; Murer, L.; Zacchello, G.; Franceschini, L.; Laverda, A.M.; Basso, G.; Quinzii, C.; Angelini, C.; Hirano, M.; et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: A CoQ10-responsive condition. Neurology 2005, 65, 606–608. [Google Scholar] [CrossRef]
- Hargreaves, I.; Mantle, D. Supplementation with selenium and coenzyme Q10 in critically ill patients. Br. J. Hosp. Med. 2019, 80, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Won, R.; Lee, K.H.; Lee, B.H. Coenzyme Q10 protects neurons against neurotoxicity in hippocampal slice culture. Neuroreport 2011, 22, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Park, C.G.; Park, M.; Lee, S.H.; Park, H.R.; Lim, J.; Paek, S.H.; Choy, Y.B. Intrastriatal administration of coenzyme Q10 enhances neuroprotection in a Parkinson’s disease rat model. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, M.; Sahraei, Z.; Simani, L.; Heydari, K.; Shahidi, F. Coenzyme Q10 supplementation in acute ischemic stroke: Is it beneficial in short-term administration? Nutr. Neurosci. 2020, 23, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Yoritaka, A.; Kawajiri, S.; Yamamoto, Y.; Nakahara, T.; Ando, M.; Hashimoto, K.; Nagase, M.; Saito, Y.; Hattori, N. Randomized, double-blind, placebo-controlled pilot trial of reduced coenzyme Q10 for Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Sawaddiruk, P.; Apaijai, N.; Paiboonworachat, S.; Kaewchur, T.; Kasitanon, N.; Jaiwongkam, T.; Kerdphoo, S.; Chattipakorn, N.; Chattipakorn, S.C. Coenzyme Q10 supplementation alleviates pain in pregabalin-treated fibromyalgia patients via reducing brain activity and mitochondrial dysfunction. Free Radic. Res. 2019, 53, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Kloubert, V.; Rink, L. Zinc as a micronutrient and its preventive role of oxidative damage in cells. Food Funct. 2015, 6, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Oteiza, P.I. Zinc and the modulation of redox homeostasis. Free Radic. Biol. Med. 2012, 53, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Tanaka, K.-i.; Kato-Negishi, M. Zinc, carnosine, and neurodegenerative diseases. Nutrients 2018, 10, 147. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [CrossRef] [PubMed]
- Farbood, Y.; Sarkaki, A.; Mahdavinia, M.; Ghadiri, A.; Teimoori, A.; Seif, F.; Dehghani, M.A.; Navabi, S.P. Protective effects of co-administration of zinc and selenium against streptozotocin-induced Alzheimer’s disease: Behavioral, mitochondrial oxidative stress, and GPR39 expression alterations in rats. Neurotox. Res. 2020, 38, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Jafari, F.; Amani, R.; Tarrahi, M.J. Effect of zinc supplementation on physical and psychological symptoms, biomarkers of inflammation, oxidative stress, and brain-derived neurotrophic factor in young women with premenstrual syndrome: A randomized, double-blind, placebo-controlled trial. Biol. Trace Elem. Res. 2020, 194, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Carocci, A.; Catalano, A.; Sinicropi, M.S.; Genchi, G. Oxidative stress and neurodegeneration: The involvement of iron. Biometals 2018, 31, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Avila, D.S.; Da Rocha, J.B.T.; Aschner, M. Metals, oxidative stress and neurodegeneration: A focus on iron, manganese and mercury. Neurochem. Int. 2013, 62, 575–594. [Google Scholar] [CrossRef] [PubMed]
- Ballaz, S.J.; Rebec, G.V. Neurobiology of vitamin C: Expanding the focus from antioxidant to endogenous neuromodulator. Pharmacol. Res. 2019, 146, 104321. [Google Scholar] [CrossRef]
- Moretti, M.; Fraga, D.B.; Rodrigues, A.L.S. Preventive and therapeutic potential of ascorbic acid in neurodegenerative diseases. CNS Neurosci. Ther. 2017, 23, 921–929. [Google Scholar] [CrossRef]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef]
- García-Krauss, A.; Ferrada, L.; Astuya, A.; Salazar, K.; Cisternas, P.; Martínez, F.; Ramírez, E.; Nualart, F. Dehydroascorbic acid promotes cell death in neurons under oxidative stress: A protective role for astrocytes. Mol. Neurobiol. 2016, 53, 5847–5863. [Google Scholar] [CrossRef]
- Astuya, A.; Caprile, T.; Castro, M.; Salazar, K.; García, M.d.l.A.; Reinicke, K.; Rodríguez, F.; Vera, J.C.; Millán, C.; Ulloa, V. Vitamin C uptake and recycling among normal and tumor cells from the central nervous system. J. Neurosci. Res. 2005, 79, 146–156. [Google Scholar] [CrossRef]
- Ulloa, V.; García-Robles, M.; Martínez, F.; Salazar, K.; Reinicke, K.; Pérez, F.; Godoy, D.F.; Godoy, A.S.; Nualart, F. Human choroid plexus papilloma cells efficiently transport glucose and vitamin C. J. Neurochem. 2013, 127, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Ziylan, Y.Z.; Diler, A.S.; Lefauconnier, J.-M.; Bourre, J.-M. Evidence for ascorbic acid transport system in rat brain capillaries. Int. J. Neurosci. 2006, 116, 25–38. [Google Scholar] [CrossRef]
- Covarrubias-Pinto, A.; Acuna, A.; Boncompain, G.; Papic, E.; Burgos, P.; Perez, F.; Castro, M. Ascorbic acid increases SVCT2 localization at the plasma membrane by accelerating its trafficking from early secretory compartments and through the endocytic-recycling pathway. Free Radic. Biol. Med. 2018, 120, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Getoff, N. Vitamin C: Electron emission, free radicals and biological versatility. In Vivo 2013, 27, 565–570. [Google Scholar] [PubMed]
- Lane, D.J.; Lawen, A. Ascorbate and plasma membrane electron transport—Enzymes vs efflux. Free Radic. Biol. Med. 2009, 47, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Joksovic, P.M.; Su, P.; Kang, H.-W.; Van Deusen, A.; Baumgart, J.P.; David, L.S.; Snutch, T.P.; Barrett, P.Q.; Lee, J.-H. Molecular mechanisms of subtype-specific inhibition of neuronal T-type calcium channels by ascorbate. J. Neurosci. 2007, 27, 12577–12583. [Google Scholar] [CrossRef]
- Smythies, J. Redox aspects of signaling by catecholamines and their metabolites. Antioxid. Redox Signal. 2000, 2, 575–583. [Google Scholar] [CrossRef]
- Sandstrom, M.I.; Rebec, G.V. Extracellular ascorbate modulates glutamate dynamics: Role of behavioral activation. BMC Neurosci. 2007, 8, 1–6. [Google Scholar] [CrossRef]
- Karanth, S.; Wen, H.Y.; Walczewska, A.; Mastronardi, C.A.; McCann, S.M. Ascorbic acid stimulates gonadotropin release by autocrine action by means of NO. Proc. Natl. Acad. Sci. USA 2001, 98, 11783–11788. [Google Scholar] [CrossRef]
- Harrison, F.; Dawes, S.; Meredith, M.; Babaev, V.; Li, L.; May, J. Low vitamin C and increased oxidative stress and cell death in mice that lack the sodium-dependent vitamin C transporter SVCT2. Free Radic. Biol. Med. 2010, 49, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Naseer, M.; Ullah, N.; Ullah, I.; Koh, P.; Lee, H.; Park, M.; Kim, M. Vitamin C protects against ethanol and PTZ-induced apoptotic neurodegeneration in prenatal rat hippocampal neurons. Synapse 2011, 65, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Barak, O.F.; Caljkusic, K.; Hoiland, R.L.; Ainslie, P.N.; Thom, S.R.; Yang, M.; Jovanov, P.; Dujic, Z. Differential influence of vitamin C on the peripheral and cerebral circulation after diving and exposure to hyperoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R759–R767. [Google Scholar] [CrossRef]
- Jain, S.K.; Dar, M.Y.; Kumar, S.; Yadav, A.; Kearns, S.R. Role of anti-oxidant (Vitamin-C) in post-operative pain relief in foot and ankle trauma surgery: A prospective randomized trial. Foot Ankle Surg. 2019, 25, 542–545. [Google Scholar] [CrossRef]
- Kim, E.J.; Park, Y.G.; Baik, E.J.; Jung, S.J.; Won, R.; Nahm, T.S.; Lee, B.H. Dehydroascorbic acid prevents oxidative cell death through a glutathione pathway in primary astrocytes. J. Neurosci. Res. 2005, 79, 670–679. [Google Scholar] [CrossRef]
- Siqueira, I.R.; Elsner, V.R.; Leite, M.C.; Vanzella, C.; dos Santos Moysés, F.; Spindler, C.; Godinho, G.; Battú, C.; Wofchuk, S.; Souza, D.O. Ascorbate uptake is decreased in the hippocampus of ageing rats. Neurochem. Int. 2011, 58, 527–532. [Google Scholar] [CrossRef]
- Naziroglu, M.; Butterworth, P.J.; Sonmez, T.T. Dietary vitamin C and E modulates antioxidant levels in blood, brain, liver, muscle, and testes in diabetic aged rats. Int. J. Vitam Nutr. Res. 2011, 81, 347. [Google Scholar] [CrossRef]
- Kim, E.J.; Won, R.; Sohn, J.-H.; Chung, M.-A.; Nam, T.S.; Lee, H.-J.; Lee, B.H. Anti-oxidant effect of ascorbic and dehydroascorbic acids in hippocampal slice culture. Biochem. Biophys. Res. Commun. 2008, 366, 8–14. [Google Scholar] [CrossRef]
- Rebec, G.V.; Barton, S.J.; Ennis, M.D. Dysregulation of ascorbate release in the striatum of behaving mice expressing the Huntington’s disease gene. J. Neurosci. 2002, 22, RC202. [Google Scholar] [CrossRef]
- Rebec, G.V.; Barton, S.J.; Marseilles, A.M.; Collins, K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport 2003, 14, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Ulatowski, L.M. Vitamin E: Mechanism of transport and regulation in the CNS. IUBMB Life 2019, 71, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.; Epand, R.F.; Epand, R.M. Tocopherols and tocotrienols in membranes: A critical review. Free Radic. Biol. Med. 2008, 44, 739–764. [Google Scholar] [CrossRef] [PubMed]
- Goti, D.; Hrzenjak, A.; Levak-Frank, S.; Frank, S.; Van Der Westhuyzen, D.R.; Malle, E.; Sattler, W. Scavenger receptor class B, type I is expressed in porcine brain capillary endothelial cells and contributes to selective uptake of HDL-associated vitamin E. J. Neurochem. 2001, 76, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Balazs, Z.; Panzenboeck, U.; Hammer, A.; Sovic, A.; Quehenberger, O.; Malle, E.; Sattler, W. Uptake and transport of high-density lipoprotein (HDL) and HDL-associated α-tocopherol by an in vitro blood–brain barrier model. J. Neurochem. 2004, 89, 939–950. [Google Scholar] [CrossRef]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef]
- Vance, J.E.; Hayashi, H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim. Biophys. Acta 2010, 1801, 806–818. [Google Scholar] [CrossRef]
- Hayashi, H.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Apolipoprotein E-containing lipoproteins protect neurons from apoptosis via a signaling pathway involving low-density lipoprotein receptor-related protein-1. J. Neurosci. 2007, 27, 1933–1941. [Google Scholar] [CrossRef]
- Lane, M.A.; Bailey, S.J. Role of retinoid signalling in the adult brain. Prog. Neurobiol. 2005, 75, 275–293. [Google Scholar] [CrossRef]
- Ambrogini, P.; Torquato, P.; Bartolini, D.; Albertini, M.C.; Lattanzi, D.; Di Palma, M.; Marinelli, R.; Betti, M.; Minelli, A.; Cuppini, R. Excitotoxicity, neuroinflammation and oxidant stress as molecular bases of epileptogenesis and epilepsy-derived neurodegeneration: The role of vitamin E. Biochim. Biophys. Acta 2019, 1865, 1098–1112. [Google Scholar] [CrossRef]
- Mohn, E.S.; Kuchan, M.J.; Erdman, J.W.; Neuringer, M.; Matthan, N.R.; Chen, C.-Y.O.; Johnson, E.J. The subcellular distribution of alpha-tocopherol in the adult primate brain and its relationship with membrane arachidonic acid and its oxidation products. Antioxidants 2017, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Khanna, S.; Rink, C.; Roy, S. Tocotrienols: The Emerging Face of Natural Vitamin E. In Vitam. Horm; Academic Press: Cambridge, MA, USA, 2007; Volume 76, pp. 203–261. [Google Scholar]
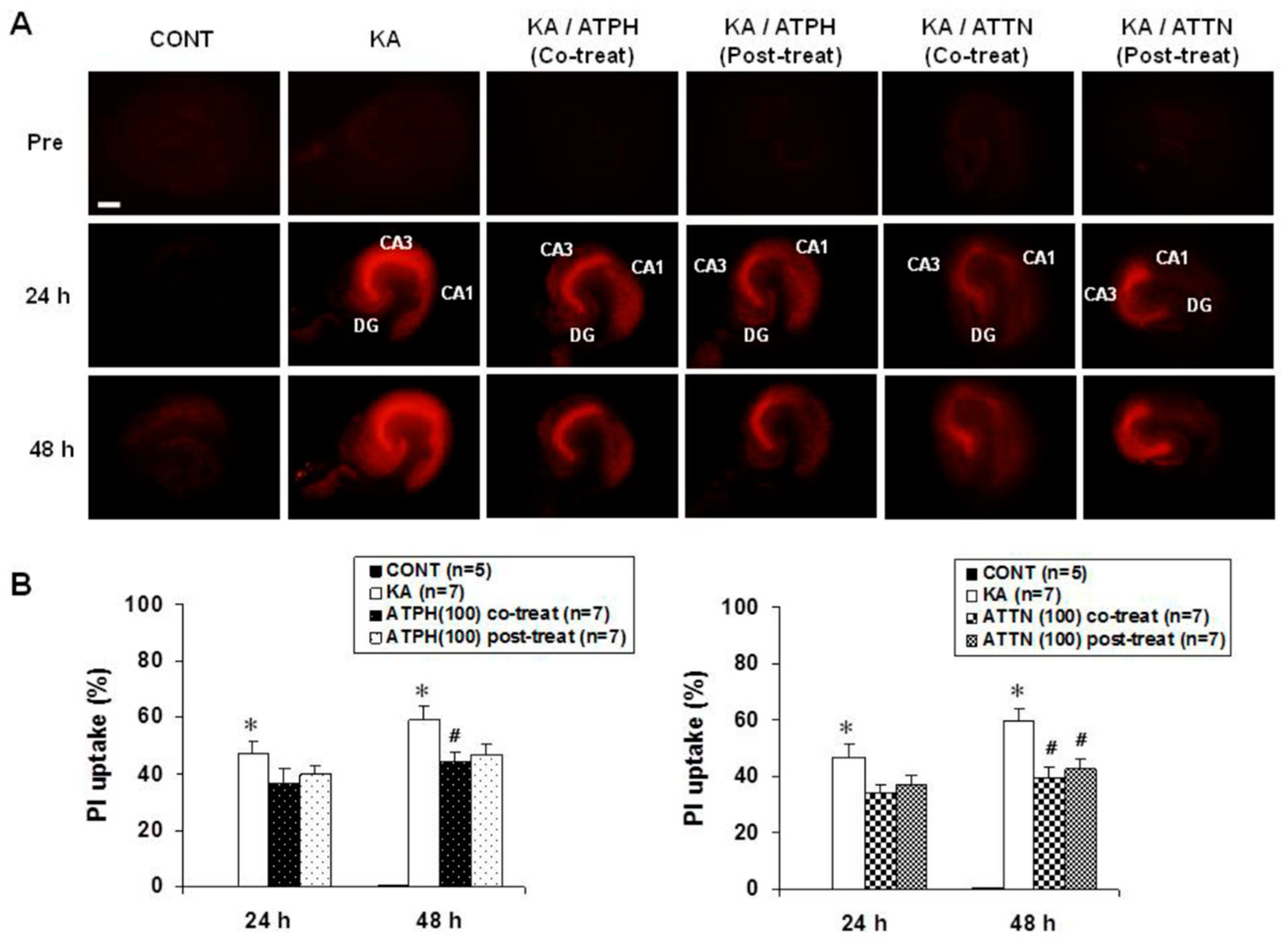
- Jung, N.Y.; Lee, K.H.; Won, R.; Lee, B.H. Neuroprotective effects of α-tocotrienol on kainic acid-induced neurotoxicity in organotypic hippocampal slice cultures. Int. J. Mol. Sci. 2013, 14, 18256–18268. [Google Scholar] [CrossRef]
- Mehrabadi, S.; Sadr, S.S. Administration of Vitamin D3 and E supplements reduces neuronal loss and oxidative stress in a model of rats with Alzheimer’s disease. Neurol. Res. 2020, 38, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Alzoubi, K.H.; Khabour, O.F. Vitamin E prevents the cognitive impairments in post-traumatic stress disorder rat model: Behavioral and molecular study. Psychopharmacology (Berlin) 2020, 237, 599–607. [Google Scholar] [CrossRef]
- Taghizadeh, M.; Tamtaji, O.R.; Dadgostar, E.; Kakhaki, R.D.; Bahmani, F.; Abolhassani, J.; Aarabi, M.H.; Kouchaki, E.; Memarzadeh, M.R.; Asemi, Z. The effects of omega-3 fatty acids and vitamin E co-supplementation on clinical and metabolic status in patients with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Neurochem. Int. 2017, 108, 183–189. [Google Scholar] [CrossRef]
- Shannon, S.R.; Moise, A.R.; Trainor, P.A. New insights and changing paradigms in the regulation of vitamin A metabolism in development. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e264. [Google Scholar] [CrossRef]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef]
- Mueller, L.; Boehm, V. Antioxidant activity of β-carotene compounds in different in vitro assays. Molecules 2011, 16, 1055–1069. [Google Scholar] [CrossRef]
- Honarvar, N.M.; Saedisomeolia, A.; Abdolahi, M.; Shayeganrad, A.; Sangsari, G.T.; Rad, B.H.; Muench, G. Molecular anti-inflammatory mechanisms of retinoids and carotenoids in Alzheimer’s disease: A review of current evidence. J. Mol. Neurosci. 2017, 61, 289–304. [Google Scholar] [CrossRef]
- Woodall, A.A.; Lee, S.W.-M.; Weesie, R.J.; Jackson, M.J.; Britton, G. Oxidation of carotenoids by free radicals: Relationship between structure and reactivity. Biochim. Biophys. Acta 1997, 1336, 33–42. [Google Scholar] [CrossRef]
- Ben-Dor, A.; Steiner, M.; Gheber, L.; Danilenko, M.; Dubi, N.; Linnewiel, K.; Zick, A.; Sharoni, Y.; Levy, J. Carotenoids activate the antioxidant response element transcription system. Mol. Cancer Ther. 2005, 4, 177–186. [Google Scholar] [PubMed]
- Palozza, P.; Serini, S.; Torsello, A.; Nicuolo, F.D.; Piccioni, E.; Ubaldi, V.; Pioli, C.; Wolf, F.I.; Calviello, G. Nutrient-gene interactions-b-carotene regulates NF-kB DNA-binding activity by a redox mechanism in human leukemia and colon adenocarcinoma cells. J. Nutr. 2003, 133, 381–388. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, P.N.; Bok, D.; Ong, D.E. Localization of cellular retinol-binding protein and retinol-binding protein in cells comprising the blood-brain barrier of rat and human. Proc. Natl. Acad. Sci. USA 1990, 87, 4265–4269. [Google Scholar] [CrossRef]
- Craft, N.; Haitema, T.; Garnett, K.; Fitch, K.; Dorey, C. Carotenoid, tocopherol, and retinol concentrations in elderly human brain. Exp. Anim. 2004, 21, 22. [Google Scholar]
- de Oliveira, B.F.; Veloso, C.A.; Nogueira-Machado, J.A.; de Moraes, E.N.; dos Santos, R.R.; Cintra, M.T.G.; Chaves, M.M. Ascorbic acid, alpha-tocopherol, and beta-carotene reduce oxidative stress and proinflammatory cytokines in mononuclear cells of Alzheimer’s disease patients. Nutr. Neurosci. 2012, 15, 244–251. [Google Scholar] [CrossRef]
- Shudo, K.; Fukasawa, H.; Nakagomi, M.; Yamagata, N. Towards retinoid therapy for Alzheimer’s disease. Curr. Alzheimer Res. 2009, 6, 302–311. [Google Scholar] [CrossRef]
- Bonhomme, D.; Minni, A.M.; Alfos, S.; Roux, P.; Richard, E.; Higueret, P.; Moisan, M.-P.; Pallet, V.; Touyarot, K. Vitamin A status regulates glucocorticoid availability in Wistar rats: Consequences on cognitive functions and hippocampal neurogenesis? Front. Behav. Neurosci. 2014, 8, 20. [Google Scholar] [CrossRef]
- Zetterström, R.H.; Lindqvist, E.; De Urquiza, A.M.; Tomac, A.; Eriksson, U.; Perlmann, T.; Olson, L. Role of retinoids in the CNS: Differential expression of retinoid binding proteins and receptors and evidence for presence of retinoic acid. Eur. J. Neurosci. 1999, 11, 407–416. [Google Scholar] [CrossRef]
- Sodhi, R.K.; Singh, N. Retinoids as potential targets for Alzheimer’s disease. Pharmacol. Biochem. Behav. 2014, 120, 117–123. [Google Scholar] [CrossRef]
- Ahlemeyer, B.; Krieglstein, J. Inhibition of glutathione depletion by retinoic acid and tocopherol protects cultured neurons from staurosporine-induced oxidative stress and apoptosis. Neurochem. Int. 2000, 36, 1–5. [Google Scholar] [CrossRef]
- Ahlemeyer, B.; Bauerbach, E.; Plath, M.; Steuber, M.; Heers, C.; Tegtmeier, F.; Krieglstein, J. Retinoic acid reduces apoptosis and oxidative stress by preservation of SOD protein level. Free Radic. Biol. Med. 2001, 30, 1067–1077. [Google Scholar] [CrossRef]
- McCaffery, P.; Dräger, U.C. High levels of a retinoic acid-generating dehydrogenase in the meso-telencephalic dopamine system. Proc. Natl. Acad. Sci. USA 1994, 91, 7772–7776. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, D.; Rouillard, C. Nur77 and retinoid X receptors: Crucial factors in dopamine-related neuroadaptation. Trends Neurosci. 2007, 30, 22–30. [Google Scholar] [CrossRef]
- Crandall, J.; Sakai, Y.; Zhang, J.; Koul, O.; Mineur, Y.; Crusio, W.E.; McCaffery, P. 13-cis-retinoic acid suppresses hippocampal cell division and hippocampal-dependent learning in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 5111–5116. [Google Scholar] [CrossRef]
- Pan, J.; Yu, J.; Sun, L.; Xie, C.; Chang, L.; Wu, J.; Hawes, S.; Saez–Atienzar, S.; Zheng, W.; Kung, J. ALDH1A1 regulates postsynaptic μ–opioid receptor expression in dorsal striatal projection neurons and mitigates dyskinesia through transsynaptic retinoic acid signaling. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Esteves, M.; Cristóvão, A.C.; Saraiva, T.; Rocha, S.M.; Baltazar, G.; Ferreira, L.; Bernardino, L. Retinoic acid-loaded polymeric nanoparticles induce neuroprotection in a mouse model for Parkinson’s disease. Front. Aging Neurosci. 2015, 7, 20. [Google Scholar] [CrossRef]
- Prema, A.; Janakiraman, U.; Manivasagam, T.; Thenmozhi, A.J. Neuroprotective effect of lycopene against MPTP induced experimental Parkinson’s disease in mice. Neurosci. Lett. 2015, 599, 12–19. [Google Scholar] [CrossRef]
- Wen, X.; Huang, A.; Hu, J.; Zhong, Z.; Liu, Y.; Li, Z.; Pan, X.; Liu, Z. Neuroprotective effect of astaxanthin against glutamate-induced cytotoxicity in HT22 cells: Involvement of the Akt/GSK-3β pathway. Neuroscience 2015, 303, 558–568. [Google Scholar] [CrossRef]
- Bitarafan, S.; Saboor-Yaraghi, A.; Sahraian, M.-A.; Soltani, D.; Nafissi, S.; Togha, M.; Moghadam, N.B.; Roostaei, T.; Honarvar, N.M.; Harirchian, M.-H. Effect of vitamin A supplementation on fatigue and depression in multiple sclerosis patients: A double-blind placebo-controlled clinical trial. Iran. J. Allergy Asthma Immunol. 2016, 15, 13–19. [Google Scholar]
- Corbett, A.; Pickett, J.; Burns, A.; Corcoran, J.; Dunnett, S.B.; Edison, P.; Hagan, J.J.; Holmes, C.; Jones, E.; Katona, C. Drug repositioning for Alzheimer’s disease. Nat. Rev. Drug Discov. 2012, 11, 833–846. [Google Scholar] [CrossRef] [PubMed]
- McGarry, A.; McDermott, M.; Kieburtz, K.; de Blieck, E.A.; Beal, F.; Marder, K.; Ross, C.; Shoulson, I.; Gilbert, P.; Mallonee, W.M. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 2017, 88, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Kryscio, R.J.; Abner, E.L.; Caban-Holt, A.; Lovell, M.; Goodman, P.; Darke, A.K.; Yee, M.; Crowley, J.; Schmitt, F.A. Association of antioxidant supplement use and dementia in the prevention of Alzheimer’s disease by vitamin E and selenium trial (PREADViSE). JAMA Neurol. 2017, 74, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Salari, S.; Khomand, P.; Arasteh, M.; Yousefzamani, B.; Hassanzadeh, K. Zinc sulphate: A reasonable choice for depression management in patients with multiple sclerosis: A randomized, double-blind, placebo-controlled clinical trial. Pharmacol. Rep. 2015, 67, 606–609. [Google Scholar] [CrossRef]
- Endres, K.; Fahrenholz, F.; Lotz, J.; Hiemke, C.; Teipel, S.; Lieb, K.; Tüscher, O.; Fellgiebel, A. Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 2014, 83, 1930–1935. [Google Scholar] [CrossRef]
- Thorsen, E.; Haave, H.; Hofsø, D.; Ulvik, R.J. Exposure to hyperoxia in diving and hyperbaric medicine--effects on blood cell counts and serum ferritin. Undersea Hyperb. Med. 2001, 28, 57–62. [Google Scholar]
- Group, P.S. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N. Engl. J. Med. 1993, 328, 176–183. [Google Scholar]
- Zhang, D.; Hannink, D.D. Distinct Cysteine residues in keap1 are required for keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1–Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef]
- Strom, J.; Xu, B.; Tian, X.; Chen, Q.M. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016, 30, 66–80. [Google Scholar] [CrossRef]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nuñez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Ikeda, Y.; Ohta, Y.; Deguchi, K.; Tian, F.; Shang, J.; Matsuura, T.; Abe, K. Expression of Keap1–Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011, 1370, 246–253. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, G.; Zhang, J.; Strong, R.; Dash, P.K.; Kan, Y.W.; Grotta, J.C.; Aronowski, J. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke 2007, 38, 3280–3286. [Google Scholar] [CrossRef] [PubMed]
- Burton, N.C.; Kensler, T.W.; Guilarte, T.R. In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology 2006, 27, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Leem, Y.H.; Park, J.E.; Kim, D.Y.; Kim, H.S. Neuroprotective effect of β-lapachone in MPTP-induced Parkinson’s disease mouse model: Involvement of astroglial p-AMPK/Nrf2/HO-1 signaling pathways. Biomol. Ther. 2019, 27, 178. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antioxidant | Number of Patients | Follow up Period | Dosage | Route | Effects | Disease | Reference |
|---|---|---|---|---|---|---|---|
| CoQ10 | 609 | 60 month | 2400 mg/day | Oral | No | Huntington | [119,195] |
| 40 | 96 week | 300 mg/day | Oral | Y | Parkinson | ||
| Selenium | 7540 | 7 year | 200 µg/day | Oral | No | Alzheimer | [107,196] |
| 6 month | 1000 µg/day | I.V. | Y | Traumatic brain injury | |||
| Zinc | 43 | 12 week | 20 mg/day | Oral | Y | Depression with multiple sclerosis | [197] |
| Vitamin A | 50 | 28 day | 30 mg/day | Oral | Y | Alzheimer | [191,198] |
| 101 | 6 month | 25,000 UI/day | Oral | Y | Multiple sclerosis | ||
| Vitamin C | 12 | 6 day | 2 g/day | Oral | Y | Hyperoxia | [147,199] |
| 60 | 3 month | 500 mg/day | Oral | Y | Trauma surgery | ||
| Vitamin E | 7540 | 7 year | 400 UI/day | Oral | No | Alzheimer | [168,196,200] |
| 60 | 3 month | 400 UI/day | Oral | Y | Parkinson | ||
| 50 | 12 month | 45 UI/day | Oral | Y | Parkinson |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. https://doi.org/10.3390/ijms21197152
Lee KH, Cha M, Lee BH. Neuroprotective Effect of Antioxidants in the Brain. International Journal of Molecular Sciences. 2020; 21(19):7152. https://doi.org/10.3390/ijms21197152
Chicago/Turabian StyleLee, Kyung Hee, Myeounghoon Cha, and Bae Hwan Lee. 2020. "Neuroprotective Effect of Antioxidants in the Brain" International Journal of Molecular Sciences 21, no. 19: 7152. https://doi.org/10.3390/ijms21197152
APA StyleLee, K. H., Cha, M., & Lee, B. H. (2020). Neuroprotective Effect of Antioxidants in the Brain. International Journal of Molecular Sciences, 21(19), 7152. https://doi.org/10.3390/ijms21197152