Drug Development and the Use of Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Disease Modeling and Drug Toxicity Screening

 , , and
, , and 
Abstract
1. Introduction
2. Drug Development, Adverse Drug Reactions, Mechanisms of Cardiotoxicity and the Need for Efficient Drug Screening Protocols
2.1. The Formal Paths for New Drug Approval
- ❖
- Chemical and biological research
- Comprehensive understanding of the disease pathophysiology
- Development of a NCE or repurpose an already-approved drug
- Testing the drug effects/efficacy in in vitro and in vivo applicable models
- ❖
- Pre-clinical development
- Pharmacokinetics and pharmacodynamics characterization
- Acute and late onset toxicology, teratogenicity, mutagenicity and carcinogenicity
- Feasibility—proof-of-concept
- ❖
- Clinical trials
2.2. Failure Rates of Drugs, Drug Development, Marketing and Adverse Drug Reactions (ADRs)
2.3. Drug-Induced Cardiotoxicity
2.4. Safety Screening during Drug Development
2.5. Drug Development—Pharmacogenomics, Pharmacodynamics, Pharmacokinetics and Patient-Specific Effects
2.6. Drugs Adverse Effects—Different Mechanisms
2.6.1. Ion Channels
2.6.2. Intracellular Ca2+ Handling
2.6.3. Apoptosis
3. The Use of iPSC-CMs for Disease Modeling and Developing Novel Drugs for Heart Diseases
3.1. Induced Pluripotent Stem Cells, Pluripotency and Applications
3.1.1. Promoting the Maturation of Immature iPSC-CMs
3.1.2. Generating Isogenic iPSCs Using the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)
3.2. Acquired Heart Diseases
3.2.1. Cardiotoxicity Caused by Anti-Cancer Drugs
Cardiotoxicity Caused by Anthracyclines
Cardiotoxicity Caused by Trastuzumab
Cardiotoxicity Caused by Tyrosine Kinase Inhibitors
3.2.2. Cardiac Hypertrophy Induced by Hormones (AT-II) and Mechanical Stretch
3.3. Inherited Heart Diseases
3.3.1. Structural Mutations
Hypertrophic Cardiomyopathy (HCM)
Mutations in the MYH7 Gene
Mutations in MYBPC3 Gene
Dilated Cardiomyopathy (DCM)
Duchenne Muscular Dystrophy (DMD)
3.3.2. Channelopathies
Long QT Syndrome
Timothy Syndrome—LQTS Type 8
Short QT Syndrome
Brugada Syndrome
3.3.3. Inherited Arrhythmias Resulting from Non-Ion Channel Mutations
3.3.4. Metabolic Mutations
Danon Disease
Familial Wolff–Parkinson–White (WPW)
3.4. Laminopathies
4. Summary
Funding
Conflicts of Interest
References
- Arrowsmith, J.; Miller, P. Trial watch: Phase II and phase III attrition rates 2011-2012. Nat. Rev. Drug Discov. 2013, 12, 569. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.K. Phase II and phase III failures: 2013–2015. Nat. Rev. Drug Discov. 2016, 15, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Festing, M.F.W. Improving toxicity screening and drug development by using genetically defined strains. Methods Mol. Biol. 2010, 602, 1–21. [Google Scholar] [CrossRef] [PubMed]
- FDA. The Drug Development Process. Available online: https://www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process (accessed on 10 August 2020).
- Chen, D.; Qi, E.Y. Innovative highlights of clinical drug trial design. Transl. Res. 2020, 224, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.J.; Carpenter, D.; Lauffenburger, J.C.; Wang, B.; Franklin, J.M.; Kesselheim, A.S. Failure of investigational drugs in late-stage clinical development and publication of trial results. JAMA Intern. Med. 2016, 176, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: A systematic review of the world literature. BMC Med. 2016, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kocadal, K.; Saygi, S.; Alkas, F.; Sardas, S. Drug-associated cardiovascular risks: A retrospective evaluation of withdrawn drugs. North. Clin. Istanbul 2019, 6, 196–202. [Google Scholar]
- Fournier, A.; Zureik, M. Estimate of deaths due to valvular insufficiency attributable to the use of benfluorex in France. Pharmacoepidemiol. Drug Saf. 2012, 21, 343–351. [Google Scholar] [CrossRef]
- Cosyns, B.; Droogmans, S.; Rosenhek, R.; Lancellotti, P. Drug-induced valvular heart disease. Heart 2013, 99, 7–12. [Google Scholar] [CrossRef]
- Bond, C.A.; Raehl, C.L. Adverse drug reactions in United States hospitals. Pharmacotherapy 2006, 26, 601–608. [Google Scholar] [CrossRef]
- FDA. FDA Adverse Event Reporting System (FAERS) Public Dashboard. Available online: https://www.fda.gov/drugs/questions-and-answers-fdas-adverse-event-reporting-system-faers/fda-adverse-event-reporting-system-faers-public-dashboard (accessed on 19 August 2020).
- Magdy, T.; Schuldt, A.J.T.; Wu, J.C.; Bernstein, D.; Burridge, P.W. Human induced pluripotent stem cell (hiPSC)-derived cells to assess drug cardiotoxicity: Opportunities and problems. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Magdy, T.; Burmeister, B.; Burridge, P.W. Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: What is missing? Pharmacol. Ther. 2016, 168, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ray, W.A.; Meredith, S.; Thapa, P.B.; Hall, K.; Murray, K.T. Cyclic antidepressants and the risk of sudden cardiac death. Clin. Pharmacol. Ther. 2004, 75, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Ray, W.A.; Murray, K.T.; Meredith, S.; Narasimhulu, S.S.; Hall, K.; Stein, C.M. Oral erythromycin and the risk of sudden death from cardiac causes. N. Engl. J. Med. 2004, 351, 1089–1096. [Google Scholar] [CrossRef]
- Van der Hooft, C.; Heeringa, J.; Brusselle, G. Corticosteroids and the risk of atrial fibrillation. Arch. Intern. Med. 2006, 166, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.S.; Baillargeon, J.; Kuo, Y.F.; Freeman, J.L.; Goodwin, J.S. Atrial fibrillation and stroke associated with intravenous bisphosphonate therapy in older patients with cancer. J. Clin. Oncol. 2010, 28, 4898–4905. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef]
- Jensen, B.C.; McLeod, H.L. Pharmacogenomics as a risk mitigation strategy for chemotherapeutic cardiotoxicity. Pharmacogenomics 2013, 14, 205–213. [Google Scholar] [CrossRef]
- Safety Guidelines. Available online: https://www.ich.org/page/safety-guidelines (accessed on 1 August 2020).
- S7B and E14 Q&A, Endorsed by the MC with Support of the Assembly on 15 November 2018. Available online: https://database.ich.org/sites/default/files/E14S7B_IWG_Concept_Paper.pdf (accessed on 1 August 2020).
- Redfern, W.S.; Carlsson, L.; Davis, A.S.; Lynch, W.G.; MacKenzie, I.; Palethorpe, S.; Siegl, P.K.S.; Strang, I.; Sullivan, A.T.; Wallis, R.; et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003, 58, 32–45. [Google Scholar] [CrossRef]
- Ando, K.; Hombo, T.; Kanno, A.; Ikeda, H.; Imaizumi, M.; Shimizu, N.; Sakamoto, K.; Kitani, S.; Yamamoto, Y.; Hizume, S.; et al. In vivo QT assay with a conscious monkey for assessment of the potential for drug-induced QT interval prolongation. J. Pharmacol. Sci. 2005, 99, 487–500. [Google Scholar] [CrossRef]
- Toyoshima, S.; Kanno, A.; Kitayama, T.; Sekiya, K.; Nakai, K.; Haruna, M.; Mino, T.; Miyazaki, H.; Yano, K.; Yamamoto, K. In vivo QT assay in the conscious dog for assessing the potential for QT interval prolongation by human pharmaceuticals. J. Pharmacol. Sci. 2005, 99, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.A.; Bass, A.S.; Gintant, G.; Mittelstadt, S.; Rampe, D.; Thomas, K. ILSI-HESI cardiovascular safety subcommittee initiative: Evaluation of three non-clinical models of QT prolongation. J. Pharmacol. Toxicol. Methods 2006, 54, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Guth, B.D. Preclinical cardiovascular risk assessment in modern drug development. Toxicol. Sci. 2007, 97, 4–20. [Google Scholar] [CrossRef]
- Wallis, R.M. Integrated risk assessment and predictive value to humans of non-clinical repolarization assays. Br. J. Pharmacol. 2010, 159, 115–121. [Google Scholar] [CrossRef]
- Roden, D.M. Predicting drug-induced QT prolongation and torsades de pointes. J. Physiol. 2016, 594, 2459–2468. [Google Scholar] [CrossRef] [PubMed]
- Gintant, G. An evaluation of hERG current assay performance: Translating preclinical safety studies to clinical QT prolongation. Pharmacol. Ther. 2011, 129, 109–119. [Google Scholar] [CrossRef]
- Gintant, G.; Sager, P.T.; Stockbridge, N. Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug Discov. 2016, 15, 457–471. [Google Scholar] [CrossRef]
- Kramer, J.; Obejero-Paz, C.A.; Myatt, G.; Kuryshev, Y.A.; Bruening-Wright, A.; Verducci, J.S.; Brown, A.M. MICE models: Superior to the HERG model in predicting torsade de pointes. Sci. Rep. 2013, 3, 1–7. [Google Scholar] [CrossRef]
- Mirams, G.R.; Davies, M.R.; Brough, S.J.; Bridgland-Taylor, M.H.; Cui, Y.; Gavaghan, D.J.; Abi-Gerges, N. Prediction of thorough QT study results using action potential simulations based on ion channel screens. J. Pharmacol. Toxicol. Methods 2014, 70, 246–254. [Google Scholar] [CrossRef]
- Van Opstal, J.M.; Schoenmakers, M.; Verduyn, S.C.; De Groot, S.H.M.; Leunissen, J.D.M.; Van Der Hulst, F.F.; Molenschot, M.M.C.; Wellens, H.J.J.; Vos, M.A. Chronic amiodarone evokes no torsade de pointes arrhythmias despite QT lengthening in an animal model of acquired long-QT syndrome. Circulation 2001, 104, 2722–2727. [Google Scholar] [CrossRef]
- Baczkó, I.; Jost, N.; Virág, L.; Bősze, Z.; Varró, A. Rabbit models as tools for preclinical cardiac electrophysiological safety testing: Importance of repolarization reserve. Prog. Biophys. Mol. Biol. 2016, 121, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Myers, D.D.; Diaz, J.A.; Conte, M.L.; Swindle, M.M. Cardiothoracic and vascular surgery/chronic intravascular catheterization. In Swine in the Laboratory: Surgery, Anesthesia, Imaging, and Experimental Techniques; Swindle, M.M., Smith, A.C., Eds.; CRC Press: Boca Raton, FL, USA, 2016; pp. 283–316. [Google Scholar]
- Sager, P.T.; Gintant, G.; Turner, J.R.; Pettit, S.; Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: A meeting report from the cardiac safety research consortium. Am. Heart J. 2014, 167, 292–300. [Google Scholar] [CrossRef] [PubMed]
- CIPA update workshop CSRC-HESI-SPS-FDA meeting | Cardiac Safety Research Consortium. Available online: https://cardiac-safety.org/think-tanks-meetings/2014-meetings/cipa-update-workshop-csrc-hesi-sps-fda-meeting/ (accessed on 23 September 2020).
- Park, J.; Jeon, J.; Yang, J.; Kim, M. Introduction to in silico model for proarrhythmic risk assessment under the CiPA initiative. Transl. Clin. Pharmacol. 2019, 27, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Colatsky, T.; Fermini, B.; Gintant, G.; Pierson, J.B.; Sager, P.; Sekino, Y.; Strauss, D.G.; Stockbridge, N. The comprehensive in vitro proarrhythmia assay (CiPA) initiative — Update on progress. J. Pharmacol. Toxicol. Methods 2016, 81, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Weinshilboum, R.M.; Wang, L. Pharmacogenomics: Precision medicine and drug response. Mayo Clin. Proc. 2017, 92, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Crotti, L.; Aiba, T.; Spazzolini, C.; Denjoy, I.; Fressart, V.; Hayashi, K.; Nakajima, T.; Ohno, S.; Makiyama, T.; et al. The genetics underlying acquired long QT syndrome: Impact for genetic screening. Eur. Heart J. 2016, 37, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Strauss, D.G.; Vicente, J.; Johannesen, L.; Blinova, K.; Mason, J.W.; Weeke, P.; Behr, E.R.; Roden, D.M.; Woosley, R.; Kosova, G.; et al. Common genetic variant risk score is associated with drug-induced QT prolongation and torsade de pointes risk. Circulation 2017, 135, 1300–1310. [Google Scholar] [CrossRef]
- McNamara, D.M.; Taylor, A.L.; Tam, S.W.; Worcel, M.; Yancy, C.W.; Hanley-Yanez, K.; Cohn, J.N.; Feldman, A.M. G-protein beta-3 subunit genotype predicts enhanced benefit of fixed-dose isosorbide dinitrate and hydralazine: Results of A-HeFT. JACC Heart. Fail. 2014, 2, 551–557. [Google Scholar] [CrossRef]
- Kacevska, M.; Ivanov, M.; Ingelman-Sundberg, M. Perspectives on epigenetics and its relevance to adverse drug reactions. Clin. Pharmacol. Ther. 2011, 89, 902–907. [Google Scholar] [CrossRef]
- Ikeda, M.; Ide, T.; Fujino, T.; Arai, S.; Saku, K.; Kakino, T.; Tyynismaa, H.; Yamasaki, T.; Yamada, K.I.; Kang, D.; et al. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006, 113, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.C.; Cheng, G.; Zhang, L.X.; Tseng, Y.T.; Padbury, J.F. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc. Res. 2007, 76, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Granger, A.; Abdullah, I.; Huebner, F.; Stout, A.; Wang, T.; Huebner, T.; Epstein, J.A.; Gruber, P.J. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008, 22, 3549–3560. [Google Scholar] [CrossRef] [PubMed]
- Madian, A.G.; Wheeler, H.E.; Jones, R.B.; Dolan, M.E. Relating human genetic variation to variation in drug responses. Trends Genet. 2012, 28, 487–495. [Google Scholar] [CrossRef]
- McCallum, L.; Lip, S.; Padmanabhan, S. Pharmacodynamic pharmacogenomics. In Handbook of Pharmacogenomics and Stratified Medicine; Elsevier Inc.: Amsterdam, The Netherlands, 2014; pp. 365–383. [Google Scholar]
- Simon, F.E.R.; Simons, K.J. H1 antihistamines: Current status and future directions. World Allergy Organ. J. 2008, 1, 145–155. [Google Scholar] [CrossRef]
- Kalpaklioglu, F.; Baccioglu, A. Efficacy and safety of H1-antihistamines: An update. Antiinflammation Antiallergy Agents Med. Chem. 2012, 11, 230–237. [Google Scholar] [CrossRef]
- Martinez, L.G.; Pulido, A.; Fleites, A. Negative inotropic action and QT prolongation by azithromycin. Revista Cubana de Cardiologia y Cirugia Cardiovascular 2017, 23, 1–15. [Google Scholar]
- Roden, D.M. Cellular basis of drug-induced torsades de pointes. Br. J. Pharmacol. 2008, 154, 1502–1507. [Google Scholar] [CrossRef]
- Lester, R.M.; Olbertz, J. Early drug development: Assessment of proarrhythmic risk and cardiovascular safety: The age of repolarization cardiac toxicity. Expert Rev. Clin. Pharmacol. 2016, 9, 1611–1618. [Google Scholar] [CrossRef]
- Kramer, D.; Zimetbaum, P. Long-QT syndrome. Cardiol. Rev. 2011, 19, 217–225. [Google Scholar] [CrossRef]
- Soldovieri, M.V.; Miceli, F.; Taglialatela, M. Cardiotoxic effects of antihistamines: From basics to clinics (…and back). Chem. Res. Toxicol. 2008, 21, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Kim, S.Y.; Yoon, K.S.; Shin, H.; Jeong, H.S.; Chung, H.; Kim, Y.H.; Shin, J.; Cha, H.J.; Moon Han, K.; et al. P21 (Cdc42/Rac)-activated kinase 1 (pak1) is associated with cardiotoxicity induced by antihistamines. Arch. Pharm. Res. 2016, 39, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.D.; Lam, A.; Tham, S.; Dulhunty, A.F.; Beard, N.A. Adverse effects of doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Mol. Pharmacol. 2014, 2, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Berdichevski, A.; Meiry, G.; Milman, F.; Reiter, I.; Sedan, O.; Eliyahu, S.; Duffy, H.S.; Youdim, M.B.; Binah, O. TVP1022 protects neonatal rat ventricular myocytes against doxorubicin-induced functional derangements. J. Pharmacol. Exp. Ther. 2010, 332, 413–420. [Google Scholar] [CrossRef]
- Burridge, P.W.; Li Yong, F.; Matsa, E.; Wu, H.; Ong, S.-G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human induced pluripotent stem cell–derived cardiomyocytes recapitulate the predilection of breast cancer patients to Doxorubicin–induced cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef]
- Sharma, A.; Burridge, P.W.; Mckeithan, W.L.; Serrano, R.; Shukla, P.; Sayed, N.; Churko, J.M.; Kitani, T.; Holmström, A.; Matsa, E.; et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 2017, 9, 1–27. [Google Scholar] [CrossRef]
- Wroblewski, H.A.; Kovacs, R.J.; Kingery, J.R.; Overholser, B.R.; Tisdale, J.E. High risk of QT interval prolongation and torsades de pointes associated with intravenous quinidine used for treatment of resistant malaria or babesiosis. Antimicrob. Agents Chemother. 2012, 56, 4495–4499. [Google Scholar] [CrossRef]
- Weeke, P.; Delaney, J.; Mosley, J.D.; Wells, Q.; Van Driest, S.; Norris, K.; Kucera, G.; Stubblefield, T.; Roden, D.M. Qt variability during initial exposure to sotalol: Experience based on a large electronic medical record. Clin. Res. 2013, 15, 1791–1797. [Google Scholar] [CrossRef]
- Jaiswal, A.; Goldbarg, S. Dofetilide induced torsade de pointes: Mechanism, risk factors and management strategies. Indian Heart J. 2014, 66, 640–648. [Google Scholar] [CrossRef]
- Thorburn, A.; Frandel, A.E. Apoptosis and anthracycline cardiotoxicity. Mol. Cancer Ther. 2006, 5, 197–199. [Google Scholar] [CrossRef]
- Shi, J.; Abdelwahid, E.; Wei, L. Apoptosis in anthracycline cardiomyopathy. Curr. Pediatr. Rev. 2011, 7, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chua, C.C.; Gao, J.; Chen, Z.; Landy, C.L.C.; Hamdy, R.; Chua, B.H.L. Pifithrin-α protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, Y.; Bar-Am, O.; Amit, T.; Berdichevski, A.; Liani, E.; Maor, G.; Reiter, I.; Youdim, M.B.H.; Binah, O. TVP1022 and propargylamine protect neonatal rat ventricular myocytes against doxorubicin-induced and serum starvation-induced cardiotoxicity. J. Cardiovasc. Pharmacol. 2008, 52, 268–277. [Google Scholar] [CrossRef]
- Mercurio, V.; Pirozzi, F.; Lazzarini, E.; Marone, G.; Rizzo, P.; Agnetti, G.; Tocchetti, C.G.; Ghigo, A.; Ameri, P. Models of heart failure based on the cardiotoxicity of anticancer drugs. J. Card. Fail. 2016, 22, 449–458. [Google Scholar] [CrossRef]
- Chaudhari, U.; Nemade, H.; Wagh, V.; Gaspar, J.A.; Ellis, J.K.; Srinivasan, S.P.; Spitkovski, D.; Nguemo, F.; Louisse, J.; Bremer, S.; et al. Identification of genomic biomarkers for anthracycline-induced cardiotoxicity in human iPSC-derived cardiomyocytes: An in vitro repeated exposure toxicity approach for safety assessment. Arch. Toxicol. 2016, 90, 2763–2777. [Google Scholar] [CrossRef]
- Tang, H.; Tao, A.; Song, J.; Liu, Q.; Wang, H.; Rui, T. Doxorubicin-induced cardiomyocyte apoptosis: Role of mitofusin 2. Int. J. Biochem. Cell Biol. 2017, 88, 55–59. [Google Scholar] [CrossRef]
- Nemade, H.; Chaudhari, U.; Acharya, A.; Hescheler, J.; Hengstler, J.G.; Papadopoulos, S.; Sachinidis, A. Cell death mechanisms of the anti-cancer drug etoposide on human cardiomyocytes isolated from pluripotent stem cells. Arch. Toxicol. 2018, 92, 1507–1524. [Google Scholar] [CrossRef]
- Gu, J.; Fan, Y.Q.; Zhang, H.L.; Pan, J.A.; Yu, J.Y.; Zhang, J.F.; Wang, C.Q. Resveratrol suppresses doxorubicin-induced cardiotoxicity by disrupting E2F1 mediated autophagy inhibition and apoptosis promotion. Biochem. Pharmacol. 2018, 150, 202–213. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Li, X.; Wu, S. Long noncoding RNA LINC00339 aggravates doxorubicin-induced cardiomyocyte apoptosis by targeting MiR-484. Biochem. Biophys. Res. Commun. 2018, 503, 3038–3043. [Google Scholar] [CrossRef]
- Lue, Y.; Gao, C.; Swerdloff, R.; Hoang, J.; Avetisyan, R.; Jia, Y.; Rao, M.; Ren, S.; Atienza, V.; Yu, J.; et al. Humanin analog enhances the protective effect of dexrazoxane against doxorubicin-induced cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H634–H643. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Xu, T.; Ding, W.; Zhang, X.; Ji, X.; Yu, T.; Yu, W.; Lin, Z.; Wang, J. miR-499-5p attenuates mitochondrial fission and cell apoptosis via p21 in doxorubicin cardiotoxicity. Front. Genet. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.X.; Cheng, L.; Qin, H.L.; Zhang, Y.Z.; Wang, L.Y.; Zhang, Y.G. MiR-15b-5p is involved in Doxorubicin-induced cardiotoxicity via inhibiting Bmpr1a signal in H9c2 cardiomyocyte. Cardiovasc. Toxicol. 2019, 19, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Tscheschner, H.; Meinhardt, E.; Schlegel, P.; Jungmann, A.; Lehmann, L.H.; Müller, O.J.; Most, P.; Katus, H.A.; Raake, P.W. CaMKII activation participates in doxorubicin cardiotoxicity and is attenuated by moderate GRP78 overexpression. PLoS ONE 2019, 14, 1–20. [Google Scholar] [CrossRef]
- Sachinidis, A. Cardiotoxicity and heart failure: Lessons from human-induced pluripotent stem cell-derived cardiomyocytes and anticancer drugs. Cells 2020, 1001. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, M.; Liu, J.; Ye, J.; Jiang, H.; Xu, Y.; Ye, D.; Wan, J. Inhibition of TRPA1 attenuates doxorubicin-induced acute cardiotoxicity by suppressing oxidative stress, the inflammatory response, and endoplasmic reticulum stress. Oxid. Med. Cell. Longev. 2018, 1–9. [Google Scholar] [CrossRef]
- Chen, X.; Peng, X.; Luo, Y.; You, J.; Yin, D.; Xu, Q.; He, H.; He, M. Quercetin protects cardiomyocytes against doxorubicin-induced toxicity by suppressing oxidative stress and improving mitochondrial function via 14-3-3γ. Toxicol Mech. Methods. 2019, 29, 344–354. [Google Scholar] [CrossRef]
- Doherty, K.R.; Wappel, R.L.; Talbert, D.R.; Trusk, P.B.; Moran, D.M.; Kramer, J.W.; Brown, A.M.; Shell, S.A.; Bacus, S. Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol. Appl. Pharmacol. 2013, 272, 245–255. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bot-Flugel, L.; Dorn, T.; Goedel, A.; Hohnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Barad, L.; Zeevi-Levin, N.; Shick, R.; Shtrichman, R.; Lorber, A.; Itskovitz-Eldor, J.; Binah, O. Cardiomyocytes generated from CPVT D307H patients are arrhythmogenic in response to β-adrenergic stimulation. J. Cell. Mol. Med. 2012, 16, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Ben Jehuda, R.; Eisen, B.; Shemer, Y.; Mekies, L.N.; Szantai, A.; Reiter, I.; Cui, H.; Guan, K.; Haron-Khun, S.; Freimark, D.; et al. CRISPR correction of the PRKAG2 gene mutation in the patient’s induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm 2018, 15, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Eisen, B.; Ben Jehuda, R.; Cuttitta, A.J.; Mekies, L.N.; Shemer, Y.; Baskin, P.; Reiter, I.; Willi, L.; Freimark, D.; Gherghiceanu, M.; et al. Electrophysiological abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from Duchenne muscular dystrophy patients. J. Cell. Mol. Med. 2019, 23, 2125–2135. [Google Scholar] [CrossRef]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5, 1–13. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. IPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 1–17. [Google Scholar] [CrossRef]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.P.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef]
- Hua, H.; Shang, L.; Martinez, H.; Freeby, M.; Gallagher, M.P.; Ludwig, T.; Deng, L.; Greenberg, E.; LeDuc, C.; Chung, W.K.; et al. iPSC-derived β cells model diabetes due to glucokinase deficiency. J. Clin. Invest. 2013, 123, 3146–3153. [Google Scholar] [CrossRef]
- Pellegrini, S.; Ungaro, F.; Mercalli, A.; Melzi, R.; Sebastiani, G.; Dotta, F.; Broccoli, V.; Piemonti, L.; Sordi, V. Human induced pluripotent stem cells differentiate into insulin-producing cells able to engraft in vivo. Acta Diabetol. 2015, 52, 1025–1035. [Google Scholar] [CrossRef]
- Yasuno, T.; Osafune, K.; Sakurai, H.; Asaka, I.; Tanaka, A.; Yamaguchi, S.; Yamada, K.; Hitomi, H.; Arai, S.; Kurose, Y.; et al. Functional analysis of iPSC-derived myocytes from a patient with carnitine palmitoyltransferase II deficiency. Biochem. Biophys. Res. Commun. 2014, 448, 175–181. [Google Scholar] [CrossRef]
- Van der Wal, E.; Bergsma, A.J.; van Gestel, T.J.M.; in ‘t Groen, S.L.M.; Zaehres, H.; Araúzo-Bravo, M.J.; Schöler, H.R.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. GAA Deficiency in Pompe Disease Is Alleviated by Exon Inclusion in iPSC-Derived Skeletal Muscle Cells. Mol. Ther. Nucleic Acids 2017, 7, 101–115. [Google Scholar] [CrossRef]
- Severs, N.J.; Shovel, K.S.; Slade, A.M.; Powell, T.; Twist, V.W.; Green, C.R. Fate of gap junctions in isolated adult mammalian cardiomyocytes. Circ. Res. 1989, 65, 22–42. [Google Scholar] [CrossRef] [PubMed]
- Felzen, B.; Berke, G.; Rosen, D.; Binah, O. Mechanisms whereby cytotoxic T lymphocytes damage guinea-pig ventricular myocytes in vitro. Pflügers Arch. Eur. J. Physiol. 1994, 427, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, K.-D.; Schreiber, D. Adult Ventricular Cardiomyocytes: Isolation and culture. Basic Cell Cult. Protoc. 2004, 290, 305–314. [Google Scholar]
- Liao, R.; Jain, M. Isolation, culture, and functional analysis of adult mouse cardiomyocytes. Methods Mol. Med. 2007, 139, 251–262. [Google Scholar] [PubMed]
- Maltsev, V.A.; Reznikov, V.; Undrovinas, N.A.; Sabbah, H.N.; Undrovinas, A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: Similarities and differences. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, 1597–1608. [Google Scholar] [CrossRef]
- Maltsev, V.A.; Kyle, J.W.; Mishra, S.; Undrovinas, A. Molecular identity of the late sodium current in adult dog cardiomyocytes identified by Nav1.5 antisense inhibition. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, 667–676. [Google Scholar] [CrossRef]
- Louch, W.E.; Sheehan, K.A.; Wolska, B.M. Methods in cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell Cardiol. 2011, 51, 288–298. [Google Scholar] [CrossRef]
- Ehler, E.; Moore-Morris, T.; Lange, S. Isolation and culture of neonatal mouse cardiomyocytes. J. Vis. Exp. 2013, 79, 1–10. [Google Scholar] [CrossRef]
- Baker, H.; Lindsy, R.; Wesibroth, S. The Laboratory Rat; Academic Press: Cambridge, MA, USA, 1979. [Google Scholar]
- Ho, D.; Zhao, X.; Gao, S.; Hong, C.; Vatner, D.E.; Vatner, S.F. Heart Rate and Electrocardiography Monitoring in Mice. Curr. Protoc. Mouse Biol. 2011, 1, 123–139. [Google Scholar]
- Yoshida, Y.; Yamanaka, S. Induced Pluripotent Stem Cells 10 Years Later. Circ. Res. 2017, 120, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.G.; Reidell, O.; Stachelscheid, H.; Klose, K.; Gossen, M.; Falk, V.; Roll, W.; Stamm, C. Regenerative medicine/cardiac cell therapy: Pluripotent stem cells. Thorac. Cardiovasc. Surg. 2018, 66, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Gintant, G.; Fermini, B.; Stockbridge, N.; Strauss, D. The evolving roles of human iPSC-derived cardiomyocytes in drug safety and discovery. Cell Stem Cell 2017, 21, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Lundy, S.D.; Zhu, W.Z.; Regnier, M.; Laflamme, M.A. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013, 22, 1991–2002. [Google Scholar] [CrossRef]
- Kamakura, T.; Makiyama, T.; Sasaki, K.; Yoshida, Y.; Wuriyanghai, Y.; Chen, J.; Hattori, T.; Ohno, S.; Kita, T.; Horie, M.; et al. Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ. J. 2013, 77, 1307–1314. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.; Pabon, L.; Fischer, K.A.; Reinecke, H.; Regnier, M.; Sniadecki, N.J.; Ruohola-Baker, H.; Murry, C.E. Tri-iodo-L-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell Cardiol. 2015, 72, 296–304. [Google Scholar] [CrossRef]
- Parikh, S.S.; Blackwell, D.J.; Gomez-Hurtado, N.; Frisk, M.; Wang, L.; Kim, K.; Dahl, C.P.; Fiane, A.; Tonnessen, T.; Kryshtal, D.O.; et al. Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circ. Res. 2017, 121, 1323–1330. [Google Scholar] [CrossRef]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Teixeira, A.; Domian, I.; Serra, M.; Alves, P.M. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci. Rep. 2017, 7, 1–17. [Google Scholar] [CrossRef]
- Hu, D.; Linders, A.; Yamak, A.; Correia, C.; Kijlstra, J.D.; Garakani, A.; Xiao, L.; Milan, D.J.; Van Der Meer, P.; Serra, M.; et al. Metabolic maturation of human pluripotent stem cellderived cardiomyocytes by inhibition of HIF1α and LDHA. Circ. Res. 2018, 123, 1066–1079. [Google Scholar] [CrossRef]
- Horikoshi, Y.; Yan, Y.; Terashvili, M.; Wells, C.; Horikoshi, H.; Fujita, S.; Bosnjak, Z.J.; Bai, X. Fatty acid-treated induced pluripotent stem cell-derived human cardiomyocytes exhibit adult cardiomyocyte-like energy metabolism phenotypes. Cells 2019, 8, 1095. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C.; Pabon, L.; et al. Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell 2017, 21, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Silbernagel, N.; Körner, A.; Balitzki, J.; Jaggy, M.; Bertels, S.; Richter, B.; Hippler, M.; Hellwig, A.; Hecker, M.; Bastmeyer, M.; et al. Shaping the heart: Structural and functional maturation of iPSC-cardiomyocytes in 3D-micro-scaffolds. Biomaterials 2020, 227, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kolanowski, T.J.; Busek, M.; Schubert, M.; Dmitrieva, A.; Binnewerg, B.; Pöche, J.; Fisher, K.; Schmieder, F.; Grünzner, S.; Hansen, S.; et al. Enhanced structural maturation of human induced pluripotent stem cell-derived cardiomyocytes under a controlled microenvironment in a microfluidic system. Acta Biomaterialia 2020, 102, 273–386. [Google Scholar] [CrossRef] [PubMed]
- Ben Jehuda, R.; Shemer, Y.; Binah, O. Genome editing in induced pluripotent stem cells using CRISPR/Cas9. Stem Cell Rev. Rep. 2018, 14, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Matre, P.R.; Mu, X.; Wu, J.; Danila, D.; Hall, M.A.; Kolonin, M.G.; Darabi, R.; Huard, J. CRISPR/Cas9-based Dystrophin restoration reveals a novel role for Dystrophin in bioenergetics and stress resistance of muscle progenitors. Stem Cells 2019, 37, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.P.T.; Broeders, M.; Herrero-Hernandez, P.; Oussoren, E.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Ready for repair? gene editing enters the clinic for the treatment of human disease. Mol. Ther. Methods Clin. Dev. 2020, 18, 532–557. [Google Scholar] [CrossRef]
- Jin, Y.; Shen, Y.; Su, X.; Weintraub, N.L.; Tang, Y. Effective restoration of dystrophin expression in iPSC Mdx-derived muscle progenitor cells using the CRISPR/Cas9 system and homology-directed repair technology. Comput. Struct. Biotechnol. J. 2020, 18, 765–773. [Google Scholar] [CrossRef]
- Tay, L.S.; Palmer, N.; Panwala, R.; Chew, W.L.; Mali, P. Translating CRISPR-Cas therapeutics: Approaches and challenges. Cris. J. 2020, 3, 253–275. [Google Scholar] [CrossRef]
- Seeger, T.; Shrestha, R.; Lam, C.K.; Chen, C.; Mckeithan, W.L.; Lau, E.; Wnorowski, A.; Greenhaw, M.; Lee, J.; Lee, S.; et al. A premature termination codon mutation in MYBPC3 causes hypertrophic cardiomyopathy via chronic activation of nonsense-mediated decay. Circulation 2020, 139, 799–811. [Google Scholar] [CrossRef]
- Shinnawi, R.; Shaheen, N.; Huber, I.; Shiti, A.; Arbel, G.; Gepstein, A.; Ballan, N.; Setter, N.; Tijsen, A.J.; Borggrefe, M.; et al. Modeling reentry in the short QT syndrome with human-induced pluripotent stem cell–derived cardiac cell sheets. J. Am. Coll. Cardiol. 2019, 73, 2310–2324. [Google Scholar] [CrossRef] [PubMed]
- Brana, I.; Tabernero, J. Cardiotoxicity. Ann. Oncol. 2010, 21, vii173–vii179. [Google Scholar] [CrossRef] [PubMed]
- Y, O.; CG, T.; KL, G.; S, J.; HJ, C.; AL, M. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef]
- Armenian, S.H.; Lacchetti, C.; Barac, A.; Carver, J.; Constine, L.S.; Denduluri, N.; Dent, S.; Douglas, P.S.; Durand, J.B.; Ewer, M.; et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2017, 35, 893–911. [Google Scholar] [CrossRef] [PubMed]
- L’Abbate, S.; Russo, I.; Kusmic, C. The role of metabolic diseases in cardiotoxicity associated with cancer therapy: What we know, what we would know. Life Sci. 2020, 255, 1–15. [Google Scholar] [CrossRef]
- Ruggiero, A.; De Rosa, G.; Rizzo, D.; Leo, A.; Maurizi, P.; De Nisco, A.; Vendittelli, F.; Zuppi, C.; Mordente, A.; Riccardi, R. Myocardial performance index and biochemical markers for early detection of doxorubicin-induced cardiotoxicity in children with acute lymphoblastic leukaemia. Int. J. Clin. Oncol. 2013, 18, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Batist, G.; Barton, J.; Chaikin, P.; Swenson, C.; Welles, L. Myocet (liposome-encapsulated doxorubicin citrate): A new approach in breast cancer therapy. Expert Opin. Pharmacother. 2002, 3, 1739–1751. [Google Scholar] [CrossRef]
- Guo, B.; Zhu, H.L.; Li, S.X.; Lu, X.C.; Fan, H. Individualized liposomal doxorubicin-based treatment in elderly patients with non-Hodgkin’s lymphoma. Oncol. Res. Treat. 2011, 34, 184–188. [Google Scholar] [CrossRef]
- Ferrans, V.J.; Clark, J.R.; Zhang, J.; Yu, X.; Herman, E.H. Pathogenesis and prevention of doxorubicin cardiomyopathy. Tsitologiia 1997, 39, 928–937. [Google Scholar]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Shakir, D. Chemotherapy induced cardiomyopathy: Pathogenesis, monitoring and management. J. Clin. Med. Res. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Kotamraju, S.; Konorev, E.A.; Joseph, J.; Kalyanaraman, B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J. Biol. Chem. 2000, 275, 33585–33592. [Google Scholar] [CrossRef] [PubMed]
- Aries, A.; Paradis, P.; Lefebvre, C.; Schwartz, R.J.; Nemer, M. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc. Natl. Acad. Sci. USA 2004, 101, 6975–6980. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Fujiwara, H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef] [PubMed]
- Karhu, S.T.; Kinnunen, S.M.; Tölli, M.; Välimäki, M.J.; Szabó, Z.; Talman, V.; Ruskoaho, H. GATA4-targeted compound exhibits cardioprotective actions against doxorubicin-induced toxicity in vitro and in vivo: Establishment of a chronic cardiotoxicity model using human iPSC-derived cardiomyocytes. Arch. Toxicol. 2020, 94, 2113–2130. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative stress and cellular response to Doxorubicin: A common factor in the complex milieu of anthracycline cardiotoxicity. Oxid. Med. Cell. Longev. 2017, 1–13. [Google Scholar] [CrossRef]
- Farías, J.G.; Molina, V.M.; Carrasco, R.A.; Zepeda, A.B.; Figueroa, E.; Letelier, P.; Castillo, R.L. Antioxidant therapeutic strategies for cardiovascular conditions associated with oxidative stress. Nutrients 2017, 9, 966. [Google Scholar] [CrossRef]
- Štěrba, M.; Popelová, O.; Vávrová, A.; Jirkovský, E.; Kovaříková, P.; Geršl, V.; Šimůnek, T. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid. Redox Signal. 2013, 18, 899–929. [Google Scholar] [CrossRef]
- Maillet, A.; Tan, K.; Chai, X.; Sadananda, S.N.; Mehta, A.; Ooi, J.; Hayden, M.R.; Pouladi, M.A.; Ghosh, S.; Shim, W.; et al. Modeling Doxorubicin-induced cardiotoxicity in human pluripotent stem cell derived-cardiomyocytes. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Han, D.; Wang, Y.; Zhang, J.; Dai, X.; Zhou, T.; Chen, J.; Tao, B.; Wang, Y.; Cao, F. The tumor-suppressive human circular RNA CircITCH sponges miR-330-5p to ameliorate Doxorubicin-induced cardiotoxicity yhrough upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127. [Google Scholar] [CrossRef] [PubMed]
- Brown, C. Targeted therapy: An elusive cancer target. Nature 2016, 537, S106–S108. [Google Scholar] [CrossRef] [PubMed]
- AL-Busairi, W.; Khajah, M. The Principles behind targeted therapy for cancer treatment. In Tumor Progression and Metastasis; IntechOpen: London, UK, 2020; pp. 1–23. [Google Scholar]
- Lamerato, L.; Havstad, S.; Gandhi, S.; Jones, D.; Chlebowski, R. Breast cancer recurrence and related mortality in U.S. pts with early breast cancer. J. Clin. Oncol. 2005, 23, 738. [Google Scholar] [CrossRef]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef]
- Murray, L.J.; Ramakrishnan, S.; O’Toole, L.; Manifold, I.H.; Purohit, O.P.; Coleman, R.E. Adjuvant trastuzumab in routine clinical practice and the impact of cardiac monitoring guidelines on treatment delivery. Breast 2010, 19, 339–344. [Google Scholar] [CrossRef]
- Kitani, T.; Ong, S.G.; Lam, C.K.; Rhee, J.W.; Zhang, J.Z.; Oikonomopoulos, A.; Ma, N.; Tian, L.; Lee, J.; Telli, M.L.; et al. Human-induced pluripotent stem cell model of Trastuzumab-induced cardiac dysfunction in patients with breast cancer. Circulation 2019, 139, 2451–2465. [Google Scholar] [CrossRef]
- Kurokawa, Y.K.; Shang, M.R.; Yin, R.T.; George, S.C. Modeling trastuzumab-related cardiotoxicity in vitro using human stem cell-derived cardiomyocytes. Toxicol. Lett. 2018, 285, 74–80. [Google Scholar] [CrossRef]
- Illouz, F.; Laboureau-Soares, S.; Dubois, S.; Rohmer, V.; Rodien, P. Tyrosine kinase inhibitors and modifications of thyroid function tests: A review. Eur. J. Endocrinol. 2009, 160, 331–336. [Google Scholar] [CrossRef]
- Sócrates, A.-V.; Antonieta, C.-G.; Héctor, M. Tyrosine kinase inhibitors (TKI): A new revolution in the treatment of chronic myeloid leukemia (CML). Gac. Med. Mex. 2013, 149, 646–654. [Google Scholar]
- Resteghini, C.; Cavalieri, S.; Galbiati, D.; Granata, R.; Alfieri, S.; Bergamini, C.; Bossi, P.; Licitra, L.; Locati, L.D. Management of tyrosine kinase inhibitors (TKI) side effects in differentiated and medullary thyroid cancer patients. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 349–361. [Google Scholar] [CrossRef]
- Orphanos, G.S.; Ioannidis, G.N.; Ardavanis, A.G. Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol. (Madr.) 2009, 48, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Lamore, S.D.; Kohnken, R.A.; Peters, M.F.; Kolaja, K.L. Cardiovascular toxicity induced by kinase inhibitors: Mechanisms and preclinical approaches. Chem. Res. Toxicol. 2020, 33, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sheehan, R.P.; Palmer, A.C.; Everley, R.A.; Boswell, S.A.; Ron-Harel, N.; Ringel, A.E.; Holton, K.M.; Jacobson, C.A.; Erickson, A.R.; et al. Adaptation of human iPSC-derived cardiomyocytes to tyrosine kinase inhibitors reduces acute cardiotoxicity via metabolic reprogramming. Cell Syst. 2019, 8, 412–426.e7. [Google Scholar] [CrossRef] [PubMed]
- Talbert, D.R.; Doherty, K.R.; Trusk, P.B.; Moran, D.M.; Shell, S.A.; Bacus, S. A multi-parameter in vitro screen in human stem cell-derived cardiomyocytes identifies ponatinib-induced structural and functional cardiac toxicity. Toxicol. Sci. 2015, 143, 147–155. [Google Scholar] [CrossRef]
- Yoshida, K.; Holmes, J.W. Computational Models of Cardiac Hypertrophy; Elsevier Ltd.: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Fulghum, K.; Hill, B.G. Metabolic mechanisms of exercise-induced cardiac remodeling. Front. Cardiovasc. Med. 2018, 5, 1–17. [Google Scholar] [CrossRef]
- Redondo-Angulo, I.; Mas-Stachurska, A.; Sitges, M.; Giralt, M.; Villarroya, F.; Planavila, A. Fgf21 is required for cardiac remodeling in pregnancy. Cardio 2017, 113, 1574–1584. [Google Scholar] [CrossRef]
- Savu, O.; Jurcuţ, R.; Giuşcǎ, S.; Van Mieghem, T.; Gussi, I.; Popescu, B.A.; Ginghinǎ, C.; Rademakers, F.; Deprest, J.; Voigt, J.U. Morphological and functional adaptation of the maternal heart during pregnancy. Circ. Cardiovasc. Imaging 2012, 5, 289–297. [Google Scholar] [CrossRef]
- Hunter, S.; Robson, S.C. Adaptation of the maternal heart in pregnancy. Heart 1992, 68, 540–543. [Google Scholar] [CrossRef]
- Massie, B.M. Myocardial hypertrophy and cardiac failure: A complex interrelationship. Am. J. Med. 1983, 75, 67–74. [Google Scholar] [CrossRef]
- Wolf, C.M. Hypertrophic cardiomyopathy: Genetics and clinical perspectives. Cardiovasc. Diagn. Ther. 2019, 9, 388–415. [Google Scholar] [CrossRef]
- Gallo, S.; Vitacolonna, A.; Bonzano, A.; Comoglio, P.; Crepaldi, T. ERK: A key player in the pathophysiology of cardiac hypertrophy. Int. J. Mol. Sci. 2019, 20, 2164. [Google Scholar] [CrossRef] [PubMed]
- Wehbe, N.; Nasser, S.A.; Pintus, G.; Badran, A.; Eid, A.H.; Baydoun, E. MicroRNAs in cardiac hypertrophy. Int. J. Mol. Sci. 2019, 20, 4714. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Wang, K.; Li, P. The role of post-translational modifications in cardiac hypertrophy. J. Cell. Mol. Med. 2019, 23, 3795–3807. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Adiarto, S.; Heiden, S.; Vignon-Zellweger, N.; Nakayama, K.; Yagi, K.; Yanagisawa, M.; Emoto, N. ET-1 from endothelial cells is required for complete angiotensin II-induced cardiac fibrosis and hypertrophy. Life Sci. 2012, 91, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Narang, N.; Chen, P.; Yu, B.; Knapp, M.; Janardanan, J.; Blair, J.; Liao, J.K. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2017, 2, 1–21. [Google Scholar] [CrossRef]
- Seo, K.; Parikh, V.N.; Ashley, E.A. Stretch-induced biased signaling in Angiotensin II type 1 and apelin receptors for the mediation of cardiac contractility and hypertrophy. Front. Physiol. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Archer, C.R.; Robinson, E.L.; Drawnel, F.M.; Roderick, H.L. Endothelin-1 promotes hypertrophic remodelling of cardiac myocytes by activating sustained signalling and transcription downstream of endothelin type A receptors. Cell. Signal. 2017, 36, 240–254. [Google Scholar] [CrossRef]
- Miyata, S.; Haneda, T.; Osaki, J. Renin-angiotensin system in stretch-induceed hypertrophy of cultured neonatal rat heart cells. Eur. J. Pharmacol. 1996, 307, 81–88. [Google Scholar] [CrossRef]
- Tanaka, A.; Yuasa, S.; Mearini, G.; Egashira, T.; Seki, T.; Kodaira, M.; Kusumoto, D.; Kuroda, Y.; Okata, S.; Suzuki, T.; et al. Endothelin-1 induces myofibrillar disarray and contractile vector variability in hypertrophic cardiomyopathy-induced pluripotent stem cell-derived cardiomyocytes. J. Am. Heart Assoc. 2014, 3, 1–25. [Google Scholar] [CrossRef]
- Ruan, J.-L.; Tulloch, N.L.; Razumova, M.V.; Saiget, M.; Muskheli, V.; Pabon, L.; Reinecke, H.; Regnier, M.; Murry, C.E. Mechanical stress conditioning and electrical stimulation promote contractility and force maturation of induced pluripotent stem cell-derived human cardiac tissue. Circulation 2016, 134, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Rupert, C.E.; Chang, H.H.; Coulombe, K.L.K. Hypertrophy changes 3D shape of hiPSC-cardiomyocytes: Implications for cellular maturation in regenerative medicine. Cell. Mol. Bioeng. 2017, 10, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Gintant, G.; Traebert, M. The roles of human induced pluripotent stem cell-derived cardiomyocytes in drug discovery: Managing in vitro safety study expectations. Expert Opin. Drug Discov. 2020, 15, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Alain Hagege, A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 2733–2779. [Google Scholar] [CrossRef]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary definitions and classification of the cardiomyopathies. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Bowles, N.E.; Bowles, K.R.; Towbin, J.A. The “final common pathway” hypothesis and inherited cardiovascular disease. Herz 2000, 25, 168–175. [Google Scholar] [CrossRef]
- Towbin, J.A.; Bowles, N.E. The failing heart. Nature 2002, 415, 227–233. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Van Driest, S.L.; Ommen, S.R.; Will, M.L.; Nishimura, R.A.; Tajik, A.J.; Gersh, B.J. Prevalence and severity of “benign” mutations in the β-myosin heavy chain, cardiac troponin T, and α-tropomyosin genes in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2002, 39, 2042–2048. [Google Scholar] [CrossRef]
- Morimoto, S. Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc. Res. 2008, 77, 659–666. [Google Scholar] [CrossRef]
- Frazier, A.; Ramirez-Correa, G.; Murphy, A. Molecular mechanisms of sarcomere dysfunction in dilated and hypertrophic cardiomyopathy. Prog. Pediatr. Cardiol. 2011, 31, 29–33. [Google Scholar] [CrossRef]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, D.; McKenna, W.J. Genetics of inherited cardiomyopathy. Eur. Heart J. 2012, 33, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Shirani, J.; Poliak, L.C.; Mathenge, R.; Roberts, W.C.; Mueller, F.O. Sudden Death in Young Competitive Athletes. JAMA 1996, 276, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Alcalai, R.; Seidman, J.G.; Seidman, C.E. Genetic basis of hypertrophic cardiomyopathy: From bench to the clinics. J. Cardiovasc. Electrophysiol. 2008, 19, 104–110. [Google Scholar] [CrossRef]
- Teekakirikul, P.; Kelly, M.A.; Rehm, H.L.; Lakdawala, N.K.; Funke, B.H. Inherited cardiomyopathies: Molecular genetics and clinical genetic testing in the postgenomic era. J. Mol. Diagn. 2013, 15, 158–170. [Google Scholar] [CrossRef]
- Maron, B.J. Hypertrophic cardiomyopathy: A systematic review. JAMA 2002, 287, 1308–1320. [Google Scholar] [CrossRef]
- Gustafson, T.A.; Bahl, J.J.; Markham, B.E.; Roeske, W.R.; Morkin, E. Hormonal regulation of myosin heavy chain and alpha-actin gene expression in cultured fetal rat heart myocytes. J. Biol. Chem. 1987, 262, 13316–13322. [Google Scholar]
- Tsuchimochi, H.; Sugi, M.; Kuro-o, M. Isozymic changes in myosin of human atrial myocardium induced by overload. Immunohistochemical study using monoclonal antibodies. J. Clin. Invest. 1984, 74, 662–665. [Google Scholar] [CrossRef]
- Bouvagnet, P.; Mairhofer, H.; Leger, J.O.C.; Puech, P.; Leger, J.J. Distribution pattern of α and β myosin in normal and diseased human ventricular myocardium. Basic Res. Cardiol. 1989, 84, 91–102. [Google Scholar] [CrossRef]
- Nakao, K.; Minobe, W.; Roden, R.; Bristow, M.R.; Leinwand, L.A. Myosin heavy chain gene expression in human heart failure. J. Clin. Invest. 1997, 100, 2362–2370. [Google Scholar] [CrossRef]
- Lompre, A.M.; Schwartz, K.; D’Albis, A.; Lacombe, G.; Van Thiem, N.; Swynghedauw, B. Myosin isoenzyme redistribution in chronic heart overload. Nature 1979, 282, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Mercadier, J.J.; Lompre, A.M.; Wisnewsky, C. Myosin isoenzymic changes in several models of rat cardiac hypertrophy. Circ. Res. 1981, 49, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Izumo, S.; Lompré, A.M.; Matsuoka, R.; Koren, G.; Schwartz, K.; Nadal-Ginard, B.; Mahdavi, V. Myosin heavy chain messenger RNA and protein isoform transitions during cardiac hypertrophy. Interaction between hemodynamic and thyroid hormone-induced signals. J. Clin. Invest. 1987, 79, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Schmidt-Traub, H.; Hoffmann, B.; Prager, M.; Bit-Avragim, N.; Rudenko, R.I.; Usupbaeva, D.A.; Kabaeva, Z.; Imanov, B.; Mirrakhimov, M.M.; et al. Prevalence of cardiac beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J. Mol. Med. 2005, 83, 468–477. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Carrier, L. Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes—a systematic review. Pflugers Arch. Eur. J. Physiol. 2019, 471, 755–768. [Google Scholar] [CrossRef]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Abilez, O.J.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific Induced Pluripotent Stem Cells. Cell Stem Cell 2014, 12, 101–113. [Google Scholar] [CrossRef]
- Han, L.; Li, Y.; Tchao, J.; Kaplan, A.D.; Lin, B.; Li, Y.; Mich-Basso, J.; Lis, A.; Hassan, N.; London, B.; et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 2014, 104, 258–269. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, T.; Piao, C.; Li, X.; Guo, J.; Zheng, S.; Zhang, X.; Cai, T.; Du, J. Screening mutations of MYBPC3 in 114 unrelated patients with hypertrophic cardiomyopathy by targeted capture and next-generation sequencing. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- Mohamed, I.A.; Krishnamoorthy, N.T.; Nasrallah, G.K.; Da’as, S.I. The role of cardiac myosin binding protein C3 in hypertrophic cardiomyopathy-progress and novel therapeutic opportunities. J. Cell. Physiol. 2017, 232, 1650–1659. [Google Scholar] [CrossRef]
- Luk, A.; Ahn, E.; Soor, G.S.; Butany, J. Dilated cardiomyopathy: A review. J. Clin. Pathol. 2009, 62, 219–225. [Google Scholar] [CrossRef]
- Elliott, P. Cardiomyopathy, Diagnosis and management of dilated cardiomyopathy. Heart 2000, 84, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Burkett, E.L.; Hershberger, R.E. Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D.; CSANZ Cardiac Genetic Diseases Council Writing Group. Guidelines for the diagnosis and management of damilial dilated cardiomyopathy. Heart Lung Circ. 2011, 20, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Givertz, M.M.; Mann, D.L. Epidemiology and natural history of recovery of left ventricular function in recent onset dilated cardiomyopathies. Curr. Heart Fail. Rep. 2013, 10, 321–330. [Google Scholar] [CrossRef]
- Dec, G.W.; Fuster, V. Idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1994, 331, 1564–1575. [Google Scholar] [CrossRef]
- Cahill, T.J.; Ashrafian, H.; Watkins, H. Genetic cardiomyopathies causing heart failure. Circ. Res. 2013, 113, 660–675. [Google Scholar] [CrossRef]
- Morales, A.; Hershberger, R.E. Genetic evaluation of dilated cardiomyopathy. Curr. Cardiol. Rep. 2013, 15, 1–8. [Google Scholar] [CrossRef]
- Mestroni, L.; Taylor, M.R.G. Genetics and genetic testing of dilated cardiomyopathy: A new perspective. Discov. Med. 2013, 15, 43–49. [Google Scholar]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef]
- Ahn, A.H.; Kunkel, L.M. The structural and functional diversity of dystrophin. Nat. Genet. 1993, 3, 283–291. [Google Scholar] [CrossRef]
- Tennyson, C.N.; Klamut, H.J.; Worton, R.G. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat. Genet. 1995, 9, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Shin, W.S.; Suzuki, J.-I.; Nakajima, T.; Kawada, T.; Uehara, Y.; Nakazawa, M.; Toyo-Oka, T. Dystrophin disruption might be related to myocardial cell apoptosis caused by isoproterenol. J. Cardiovasc. Pharmacol. 2000, 36, S25–S29. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.C.; Fassati, A.; Popplewell, L.; Page, A.M.; Henry, M.D.; Campbell, K.P.; Dickson, G. Dystrophic phenotype induced in vitro by antibody blockade of muscle alpha-dystroglycan-laminin interaction. J. Cell Sci. 1999, 112, 209–216. [Google Scholar] [PubMed]
- Sciandra, F.; Bozzi, M.; Bianchi, M.; Pavoni, E.; Giardina, B.; Brancaccio, A. Dystroglycan and muscular dystrophies related to the dystrophin-glycoprotein complex. Ann. Ist. Super. Sanita 2003, 39, 173–181. [Google Scholar]
- Deconinck, N.; Dan, B. Pathophysiology of duchenne muscular dystrophy: Current hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef]
- Allard, B. Sarcolemmal ion channels in dystrophin-deficient skeletal muscle fibres. J. Muscle Res. Cell Motil. 2006, 27, 367–373. [Google Scholar] [CrossRef]
- Constantin, B.; Sebille, S.; Cognard, C. New insights in the regulation of calcium transfers by muscle dystrophin-based cytoskeleton: Implications in DMD. J. Muscle Res. Cell Motil. 2006, 27, 375–386. [Google Scholar] [CrossRef]
- Romfh, A.; McNally, E.M. Cardiac Assessment in Duchenne and Becker Muscular Dystrophies. Curr. Heart Fail. Rep. 2010, 7, 212–218. [Google Scholar] [CrossRef]
- McNally, E.M.; Kaltman, J.R.; Benson, D.W.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Groh, W.J.; Hoffman, E.P.; Judge, D.P.; et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Working Group of the National Heart, Lung, and Blood Institute in collaboration with Parent Project Muscular Dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- Segawa, K.; Komaki, H.; Mori-Yoshimura, M.; Oya, Y.; Kimura, K.; Tachimori, H.; Kato, N.; Sasaki, M.; Takahashi, Y. Cardiac conduction disturbances and aging in patients with Duchenne muscular dystrophy. Medicine (Baltimore) 2017, 96, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.O.; Jefferies, J.L.; Lorts, A.; Anderson, J.B.; Gao, Z.; Benson, D.W.; Hor, K.N.; Cripe, L.H.; Urbina, E.M.; Woodrow Benson, D.; et al. Autonomic dysfunction: A driving force for myocardial fibrosis in young Duchenne muscular dystrophy patients? Pediatr. Cardiol. 2015, 36, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Yotsukura, M.; Sasaki, K.; Kachi, E.; Sasaki, A.; Ishihara, T.; Ishikawa, K. Circadian rhythm and variability of heart rate in Duchenne-type progressive muscular dystrophy. Am. J. Cardiol. 1995, 76, 947–951. [Google Scholar] [CrossRef]
- Yotsukura, M.; Fujii, K.; Katayama, A.; Tomono, Y.; Ando, H.; Sakata, K.; Ishihara, T.; Ishikawa, K. Nine-year follow-up study of heart rate variability in patients with Duchenne-type progressive muscular dystrophy. Am. Heart J. 1998, 136, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.A.; Dello Russo, A.; Giglio, V.; De Luca, L.; Messano, L.; Santini, C.; Ricci, E.; Damiani, A.; Fumagalli, G.; De Martino, G.; et al. Impairment of cardiac autonomic function in patients with Duchenne muscular dystrophy: Relationship to myocardial and respiratory function. Am. Heart J. 2001, 141, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Li, Y.; Han, L.; Kaplan, A.D.; Ao, Y.; Kalra, S.; Bett, G.C.L.; Rasmusson, R.L.; Denning, C.; Yang, L. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis. Model. Mech. 2015, 8, 457–466. [Google Scholar] [CrossRef]
- Song, T.J.; Lee, K.A.; Kang, S.W.; Cho, H.; Choi, Y.C. Three cases of manifesting female carriers in patients with Duchenne muscular dystrophy. Yonsei Med. J. 2011, 52, 192–195. [Google Scholar] [CrossRef]
- Abriel, H.; Zaklyazminskaya, E.V. Cardiac channelopathies: Genetic and molecular mechanisms. Gene 2013, 517, 1–11. [Google Scholar] [CrossRef]
- Waddell-Smith, K.E.; Skinner, J.R. Update on the diagnosis and management of familial Long QT Syndrome. Heart Lung Circ. 2016, 25, 769–776. [Google Scholar] [CrossRef]
- Fernández-Falgueras, A.; Sarquella-Brugada, G.; Brugada, J.; Brugada, R.; Campuzano, O. Cardiac channelopathies and sudden death: Recent clinical and genetic advances. Biology (Basel) 2017, 6, 7. [Google Scholar] [CrossRef]
- Garcia-Elias, A.; Benito, B. Ion channel disorders and sudden cardiac death. Int. J. Mol. Sci. 2018, 19, 692. [Google Scholar] [CrossRef] [PubMed]
- Kline, J.; Costantini, O. Inherited cardiac arrhythmias and channelopathies. Med. Clin. North Am. 2019, 103, 809–820. [Google Scholar] [CrossRef]
- Wu, J.C.; Garg, P.; Yoshida, Y.; Yamanaka, S.; Gepstein, L.; Hulot, J.S.; Knollmann, B.C.; Schwartz, P.J. Towards precision medicine with human iPSCs for cardiac channelopathies. Circ. Res. 2019, 125, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Chahal, C.A.A.; Salloum, M.N.; Alahdab, F.; Gottwald, J.A.; Tester, D.J.; Anwer, L.A.; So, E.L.; Murad, M.H.; St Louis, E.K.; Ackerman, M.J.; et al. Systematic review of the genetics of sudden unexpected death in epilepsy: Potential overlap with sudden cardiac death and arrhythmia-related genes. J. Am. Heart Assoc. 2020, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Torrente, A.G.; Mesirca, P.; Bidaud, I.; Mangoni, M.E. Channelopathies of voltage-gated L-type Cav1.3/α1D and T-type Cav3.1/α1G Ca2+ channels in dysfunction of heart automaticity. Pflugers Arch. Eur. J. Physiol. 2020, 472, 817–830. [Google Scholar] [CrossRef]
- Skinner, J.R.; Winbo, A.; Abrams, D.; Vohra, J.; Wilde, A.A. Channelopathies that lead to sudden cardiac death: Clinical and genetic aspects. Heart Lung Circ. 2019, 28, 22–30. [Google Scholar] [CrossRef]
- Singh, M.; Morin, D.P.; Link, M.S. Sudden cardiac death in Long QT syndrome (LQTS), Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia (CPVT). Prog. Cardiovasc. Dis. 2019, 62, 227–234. [Google Scholar] [CrossRef]
- Goldenberg, I.; Moss, A.J. Long QT syndrome. J. Am. Coll. Cardiol. 2008, 51, 2291–2300. [Google Scholar] [CrossRef]
- Lieve, K.V.; Wilde, A.A. Inherited ion channel diseases: A brief review. Europace 2015. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Belardinelli, L.; Zygmunt, A.C.; Burashnikov, A.; Di Diego, J.M.; Fish, J.M.; Cordeiro, J.M.; Thomas, G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 2004, 110, 904–910. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, P.; Lan, F.; Wu, H.; Lisowski, L.; Gu, M.; Hu, S.; Kay, M.A.; Urnov, F.D.; Shinnawi, R.; et al. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J. Am. Coll. Cardiol. 2014, 64, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 2011, 471, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Yarotskyy, V.; Elmslie, K.S. Roscovitine, a cyclin-dependent kinase inhibitor, affects several gating mechanisms to inhibit cardiac L-type (Ca (V)1.2) calcium channels. Br. J. Pharmacol. 2007, 152, 386–395. [Google Scholar] [CrossRef]
- Yarotskyy, V.; Gao, G.; Peterson, B.Z.; Elmslie, K.S. The Timothy syndrome mutation of cardiac CaV1.2 (L-type) channels: Multiple altered gating mechanisms and pharmacological restoration of inactivation. J. Physiol. 2009, 587, 551–565. [Google Scholar] [CrossRef]
- Yarotskyy, V.; Gao, G.; Du, L.; Ganapathi, S.B.; Peterson, B.Z.; Elmslie, K.S. Roscovitine binds to novel L-channel (Cav1.2) sites that separately affect activation and inactivation. J. Biol. Chem. 2010, 285, 43–53. [Google Scholar] [CrossRef]
- Gaita, F.; Giustetto, C.; Bianchi, F.; Wolpert, C.; Schimpf, R.; Riccardi, R.; Grossi, S.; Richiardi, E.; Borggrefe, M. Short QT Syndrome: A familial cause of sudden death. Circulation 2003, 108, 965–970. [Google Scholar] [CrossRef]
- Rudic, B.; Schimpf, R.; Borggrefe, M. Short QT syndrome – Review of diagnosis and treatment. Arrhythmia Electrophysiol. Rev. 2014, 3, 76. [Google Scholar] [CrossRef]
- Wolpert, C.; Vogel, M.; Nagel, C.; Rüb, N. Short QT syndrome: An update. Rom. J. Cardiol. 2017, 27, 353–358. [Google Scholar]
- Guerrier, K.; Kwiatkowski, D.; Czosek, R.J.; Spar, D.S.; Anderson, J.B.; Knilans, T.K. Short QT interval prevalence and clinical outcomes in a pediatric population. Circ. Arrhythmia Electrophysiol. 2015, 8, 1460–1464. [Google Scholar] [CrossRef]
- Mazzanti, A.; Priori, S.G. Inherited arrhythmias: LQTS/SQTS/CPVT. In Cardiovascular Genetics and Genomics: Principles and Clinical Practice; Springer International Publishing: Cham, Switzerland, 2018; pp. 413–435. [Google Scholar]
- Templin, C.; Ghadri, J.; Rougier, J.; Baumer, A.; Kaplan, V.; Albesa, M.; Sticht, H.; Rauch, A.; Puleo, C.; Hu, D.; et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J. 2011, 32, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Schimpf, R.; Wolpert, C.; Gaita, F.; Giustetto, C.; Borggrefe, M. Short QT syndrome. Cardiovasc. Res. 2005, 67, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Mizobuchi, M.; Enjoji, Y.; Yamamoto, R.; Ono, T.; Funatsu, A.; Kambayashi, D.; Kobayashi, T.; Nakamura, S. Nifekalant and disopyramide in a patient with short QT syndrome: Evaluation of pharmacological effects and electrophysiological properties. Pacing Clin. Electrophysiol. 2008, 31, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Giustetto, C.; Schimpf, R.; Mazzanti, A.; Scrocco, C.; Maury, P.; Anttonen, O.; Probst, V.; Blanc, J.J.; Sbragia, P.; Dalmasso, P.; et al. Long-term follow-up of patients with short QT syndrome. J. Am. Coll. Cardiol. 2011, 58, 587–595. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Lan, H.; Cyganek, L.; Zhao, Z.; Li, X.; Buljubasic, F.; Lang, S.; Yücel, G.; Sattler, K.; Zimmermann, W.H.; et al. Modeling short QT syndrome using human-induced pluripotent stem cell-derived cardiomyocytes. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, X.; El-Battrawy, I.; Lan, H.; Zhong, R.; Xu, Q.; Huang, M.; Liao, Z.; Lang, S.; Zimmermann, W.; et al. Drug testing in human-induced pluripotent stem cell–derived cardiomyocytes from a patient With short QT syndrome type 1. Clin. Pharmacol. Ther. 2019, 106, 642–651. [Google Scholar] [CrossRef]
- Brugada, P.; Brugada, J.; Roy, D. Brugada syndrome 1992-2012: 20 years of scientific excitement, and more. Eur. Heart J. 2013, 34, 3610–3615. [Google Scholar] [CrossRef]
- Nademanee, K.; Veerakul, G.; Nimmannit, S.; Chaowakul, V.; Bhuripanyo, K.; Likittanasombat, K.; Tunsanga, K.; Kuasirikul, S.; Malasit, P.; Tansupasawadikul, S.; et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation 1997, 96, 2595–2600. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Brugada, P.; Brugada, J.; Brugada, R. Brugada syndrome: From cell to bedside. Curr. Probl. Cardiol. 2005, 30, 9–54. [Google Scholar] [CrossRef]
- Mellor, G.; Raju, H.; De Noronha, S.V.; Papadakis, M.; Sharma, S.; Behr, E.R.; Sheppard, M.N. Clinical characteristics and circumstances of death in the sudden arrhythmic death syndrome. Circ. Arrhythmia Electrophysiol. 2014, 7, 1078–1083. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Yan, G.X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Europace 2017, 19, 665–694. [Google Scholar] [PubMed]
- Antzelevich, C.; Brugada, P.; Borggrefe, M.; Brugada, J.; Brugada, R.; Corrado, D.; Gussak, I.; LeMarec, H.; Nademanee, K.; Perez Riera, A.R.; et al. Brugada syndrome: Report of the second consensus conference. Heart Rhythm 2005, 2, 429–440. [Google Scholar] [CrossRef]
- Campuzano, O.; Brugada, R.; Iglesias, A. Genetics of Brugada syndrome. Curr. Opin. Cardiol. 2010, 25, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Schimpf, R.; Veltmann, C.; Wolpert, C.; Borggrefe, M. Channelopathies: Brugada syndrome, long QT syndrome, short QT syndrome, and CPVT. Herz 2009, 34, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Sarquella-Brugada, G.; Campuzano, O.; Arbelo, E.; Brugada, J.; Brugada, R. Brugada syndrome: Clinical and genetic findings. Genet. Med. 2016, 18, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Le Scouarnec, S.; Karakachoff, M.; Gourraud, J.B.; Lindenbaum, P.; Bonnaud, S.; Portero, V.; Duboscq-Bidot, L.; Daumy, X.; Simonet, F.; Teusan, R.; et al. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for brugada syndrome. Hum. Mol. Genet. 2015, 24, 2757–2763. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S.; et al. Reappraisal of reported genes for sudden arrhythmic death: Evidence-based evaluation of gene validity for brugada syndrome. Circulation 2018, 138, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Stauske, M.; Luo, X.; Wagner, S.; Vollrath, M.; Mehnert, C.; Cyganek, L.; Chen, S.; Hasheminasab, S.-M.; Wulf, G.; et al. Disease phenotypes and mechanisms of iPSC-derived cardiomyocytes from Brugada syndrome patients with a loss-of-function SCN5A mutation. SSRN Electron. J. 2020. A review of papers under review. [Google Scholar] [CrossRef]
- Gurabi, Z.; Koncz, I.; Patocskai, B.; Nesterenko, V.V.; Antzelevitch, C. Cellular mechanism underlying hypothermia-induced ventricular tachycardia/ventricular fibrillation in the setting of early repolarization and the protective effect of quinidine, cilostazol, and milrinone. Circ. Arrhythmia Electrophysiol. 2014, 7, 134–142. [Google Scholar] [CrossRef]
- Leenhardt, A.; Thomas, O.; Cauchemez, B.; Maison-Blanche, P.; Denjoy, I.; de Jode, P.; Kedra, W.; Coumel, P. Value of the exercise test in the study of arrhythmia. Arch. Mal. Coeur Vaiss. 1995, 88, 59–66. [Google Scholar]
- Napolitano, C. Flecainide monotherapy for catecholaminergic polymorphic ventricular tachycardia: Perspectives and limitations. Heart Rhythm 2016, 13, 614–615. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Barad, L.; Lorber, A.; Gherghiceanu, M.; Reiter, I.; Eisen, B.; Eldor, L.; Itskovitz-Eldor, J.; Eldar, M.; Arad, M.; et al. Functional abnormalities in iPSC-derived cardiomyocytes generated from CPVT1 and CPVT2 patients carrying ryanodine or calsequestrin mutations. J. Cell. Mol. Med. 2015, 19, 2006–2018. [Google Scholar] [CrossRef] [PubMed]
- Preininger, M.K.; Jha, R.; Maxwell, J.T.; Wu, Q.; Singh, M.; Wang, B.; Dalal, A.; McEachin, Z.T.; Rossoll, W.; Hales, C.M.; et al. A human pluripotent stem cell model of catecholaminergic polymorphic ventricular tachycardia recapitulates patient-specific drug responses. Dis. Model. Mech. 2016, 9, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Pölönen, R.P.; Swan, H.; Aalto-Setälä, K. Mutation-specific differences in arrhythmias and drug responses in CPVT patients: Simultaneous patch clamp and video imaging of iPSC derived cardiomyocytes. Mol. Biol. Rep. 2020, 47, 1067–1077. [Google Scholar] [CrossRef]
- Itzhaki, I.; Maizels, L.; Huber, I.; Gepstein, A.; Arbel, G.; Caspi, O.; Miller, L.; Belhassen, B.; Nof, E.; Glikson, M.; et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J. Am. Coll. Cardiol. 2012, 60, 990–1000. [Google Scholar] [CrossRef]
- Darras, B.T.; Friedman, N.R. Metabolic myopathies: A clinical approach; Part I. Pediatr. Neurol. 2000, 22, 87–97. [Google Scholar] [CrossRef]
- Guertl, B.; Noehammer, C.; Hoefler, G. Metabolic cardiomyopathies. Int. J. Exp. Pathol. 2000, 81, 349–372. [Google Scholar] [CrossRef]
- George, A.; Figueredo, V.M. Alcoholic cardiomyopathy: A review. J. Card. Fail. 2011, 17, 844–849. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef]
- Laforêt, P.; Richard, P.; Said, M.A.; Romero, N.B.; Lacene, E.; Leroy, J.P.; Baussan, C.; Hogrel, J.Y.; Lavergne, T.; Wahbi, K.; et al. A new mutation in PRKAG2 gene causing hypertrophic cardiomyopathy with conduction system disease and muscular glycogenosis. Neuromuscul. Disord. 2006, 16, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Cenacchi, G.; Papa, V.; Pegoraro, V.; Marozzo, R.; Fanin, M.; Angelini, C. Review: Danon disease: Review of natural history and recent advances. Neuropathol. Appl. Neurobiol. 2020, 46, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Cuervo, A.M.; Taylor, M.R.G.; Nishino, I.; Blum, J.S.; Dice, J.F.; Sandoval, I.V.; Lippincott-Schwartz, J.; August, J.T.; Saftig, P. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic 2005, 6, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Cuervo, A.M. Chaperone-mediated autophagy: Molecular mechanisms and physiological relevance. Semin. Cell Dev. Biol. 2010, 21, 719–726. [Google Scholar] [CrossRef]
- Nucifora, G.; Miani, D.; Piccoli, G.; Proclemer, A. Cardiac magnetic resonance imaging in Danon disease. Cardiology 2012, 121, 27–30. [Google Scholar] [CrossRef]
- D’souza, R.S.; Levandowski, C.; Slavov, D.; Graw, S.L.; Allen, L.A.; Adler, E.; Mestroni, L.; Taylor, M.R.G. Danon disease clinical features, evaluation, and management. Circ. Heart. Fail. 2014, 7, 843–849. [Google Scholar] [CrossRef]
- He, J.; Xu, J.; Chen, L.; Ji, K.; Fan, X.; Zhao, S.; Lu, M. Clinical features and cardiovascular magnetic resonance characteristics in Danon disease. Clin. Radiol. 2020, 75, 1–11. [Google Scholar] [CrossRef]
- Majer, F.; Vlaskova, H.; Krol, L.; Kalina, T.; Kubanek, M.; Stolnaya, L.; Dvorakova, L.; Elleder, M.; Sikora, J. Danon disease: A focus on processing of the novel LAMP2 mutation and comments on the beneficial use of peripheral white blood cells in the diagnosis of LAMP2 deficiency. Gene 2012, 498, 183–195. [Google Scholar] [CrossRef]
- Hashem, S.I.; Perry, C.N.; Bauer, M.; Han, S.; Clegg, S.D.; Ouyang, K.; Deacon, D.C.; Spinharney, M.; Panopoulos, A.D.; Belmonte, J.C.I.; et al. Brief Report: Oxidative stress mediates cardiomyocyte apoptosis in a human model of Danon disease and heart failure. Stem Cells 2015, 33, 2343–2350. [Google Scholar] [CrossRef]
- Ng, K.M.; Mok, P.Y.; Butler, A.W.; Ho, J.C.Y.; Choi, S.W.; Lee, Y.K.; Lai, W.H.; Au, K.W.; Lau, Y.M.; Wong, L.Y.; et al. Amelioration of X-linked related autophagy failure in Danon disease with DNA methylation inhibitor. Circulation 2016, 134, 1373–1389. [Google Scholar] [CrossRef]
- Yoshida, S.; Nakanishi, C.; Okada, H.; Mori, M.; Yokawa, J.; Yoshimuta, T.; Ohta, K.; Konno, T.; Fujino, N.; Kawashiri, M.A.; et al. Characteristics of induced pluripotent stem cells from clinically divergent female monozygotic twins with Danon disease. J. Mol. Cell Cardiol. 2018, 114, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Leonard, A.; Knight, W.E.; Beussman, K.M.; Zhao, Y.; Cao, Y.; Londono, P.; Aune, E.; Trembley, M.A.; Small, E.M.; et al. LAMP-2B regulates human cardiomyocyte function by mediating autophagosome–lysosome fusion. Proc. Natl. Acad. Sci. USA 2019, 116, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Gollob, M.H.; Robert, R. AMP-activated protein kinase and familial Wolff-Parkinson-White syndrome: New perspectives on heart development and arrhythmogenesis. Eur. Heart J. 2002, 23, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.K.; Wells, D.J.; Liu, K.; Whitrow, H.R.; Daniel, T.D.; Grignani, R.; Lygate, C.A.; Schneider, J.E.; Noël, G.; Watkins, H.; et al. Characterization of the role of γ2 R531G mutation in AMP-activated protein kinase in cardiac hypertrophy and Wolff-Parkinson-White syndrome. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, 1942–1951. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.M.J.; Ehtisham, J.; Redwood, C.S.; Ostman-Smith, I.; Blair, E.M.; Watkins, H. Mutation analysis of AMP-activated protein kinase subunits in inherited cardiomyopathies: Implications for kinase function and disease pathogenesis. J. Mol. Cell Cardiol. 2003, 35, 1251–1255. [Google Scholar] [CrossRef]
- Miyamoto, L. Molecular pathogenesis of familial wolff-parkinson-white syndrome. ~molecular mechanisms of cardiac glycogen regulation by AMPK. J. Med. Investig. 2018, 65, 1–8. [Google Scholar] [CrossRef]
- Arad, M.; Maron, B.J.; Gorham, J.M.; Johnson, W.H.; Saul, J.P.; Perez-Atayde, A.R.; Spirito, P.; Wright, G.B.; Kanter, R.J.; Seidman, C.E.; et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 2005, 352, 362–372. [Google Scholar] [CrossRef]
- Zhan, Y.; Sun, X.; Li, B.; Cai, H.; Xu, C.; Liang, Q.; Lu, C.; Qian, R.; Chen, S.; Yin, L.; et al. Establishment of a PRKAG2 cardiac syndrome disease model and mechanism study using human induced pluripotent stem cells. J. Mol. Cell Cardiol. 2018, 117, 49–61. [Google Scholar] [CrossRef]
- Capell, B.C.; Collins, F.S. Human laminopathies: Nuclei gone genetically awry. Nat. Rev. Genet. 2006, 7, 940–952. [Google Scholar] [CrossRef]
- Van Berlo, J.H.; De Voogt, W.G.; Van Der Kooi, A.J.; Van Tintelen, J.P.; Bonne, G.; Yaou, R.B.; Duboc, D.; Rossenbacker, T.; Heidbüchel, H.; De Visser, M.; et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med. 2005, 83, 79–83. [Google Scholar] [CrossRef]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Brodt, C.; Siegfried, J.D.; Hofmeyer, M.; Martel, J.; Rampersaud, E.; Li, D.; Morales, A.; Hershberger, R.E. Temporal relationship of conduction system disease and ventricular dysfunction in LMNA cardiomyopathy. J Card. Fail. 2013, 19, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Baldinger, S.H.; Gandjbakhch, E.; Maury, P.; Sellal, J.M.; Androulakis, A.F.A.; Waintraub, X.; Charron, P.; Rollin, A.; Richard, P.; et al. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J. Am. Coll. Cardiol. 2016, 68, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Van Rijsingen, I.A.W.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-Van Ast, J.F.; Van Der Kooi, A.J.; Van Tintelen, J.P.; Van Den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk factors for malignant ventricular arrhythmias in Lamin A/C mutation carriers: A European cohort study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar]
- Hutchison, C.J.; Worman, H.J. A-type lamins: Guardians of the soma? Nat. Cell Biol. 2004, 6, 1062–1067. [Google Scholar] [CrossRef]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef]
- Puckelwartz, M.J.; Depreux, F.F.S.; McNally, E.M. Gene expression, chromosome position and lamin A/C mutations. Nucleus 2011, 2, 162–167. [Google Scholar] [CrossRef]
- Davidson, P.M.; Lammerding, J. Broken nuclei – lamins, nuclear mechanics and disease Patricia. Trends Cell Biol. 2014, 24, 247–256. [Google Scholar] [CrossRef]
- Siu, C.W.; Lee, Y.K.; Ho, J.C.Y.; Lai, W.H.; Chan, Y.C.; Ng, K.M.; Wong, L.Y.; Au, K.W.; Lau, Y.M.; Zhang, J.; et al. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging (Albany. NY) 2012, 4, 803–822. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lau, Y.M.; Cai, Z.J.; Lai, W.H.; Wong, L.Y.; Tse, H.F.; Ng, K.M.; Siu, C.W. Modeling treatment response for Lamin A/C related dilated cardiomyopathy in human induced pluripotent stem cells. J. Am. Heart Assoc. 2017, 6, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Termglinchan, V.; Diecke, S.; Itzhaki, I.; Lam, C.K.; Garg, P.; Lau, E.; Greenhaw, M.; Seeger, T.; Wu, H.; et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 2019, 572, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Bertero, A.; Fields, P.A.; Smith, A.S.T.; Leonard, A.; Beussman, K.; Sniadecki, N.J.; Kim, D.H.; Tse, H.F.; Pabon, L.; Shendure, J.; et al. Chromatin compartment dynamics in a haploinsufficient model of cardiac laminopathy. J. Cell Biol. 2019, 218, 2919–2944. [Google Scholar] [CrossRef] [PubMed]







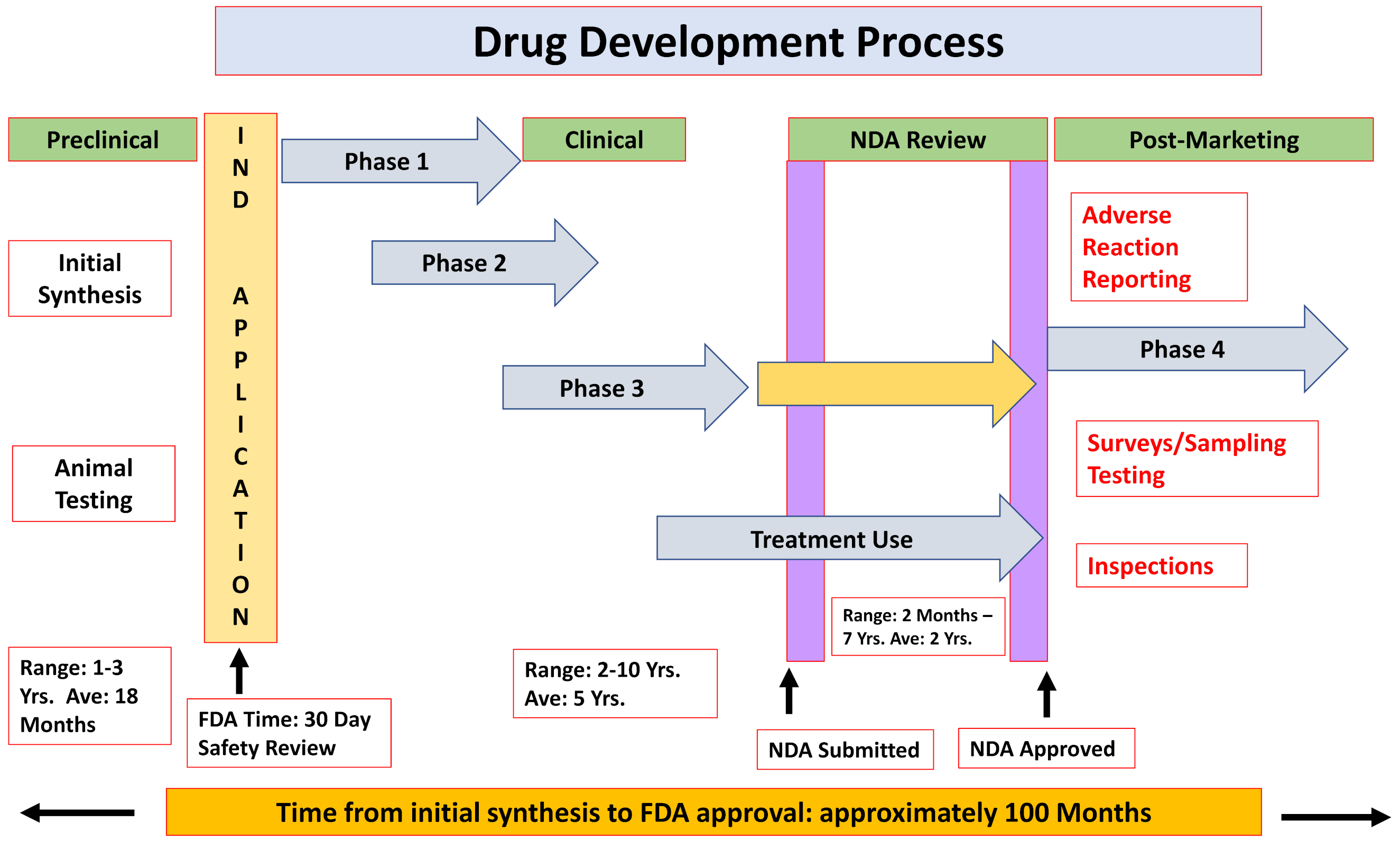{kind=link}
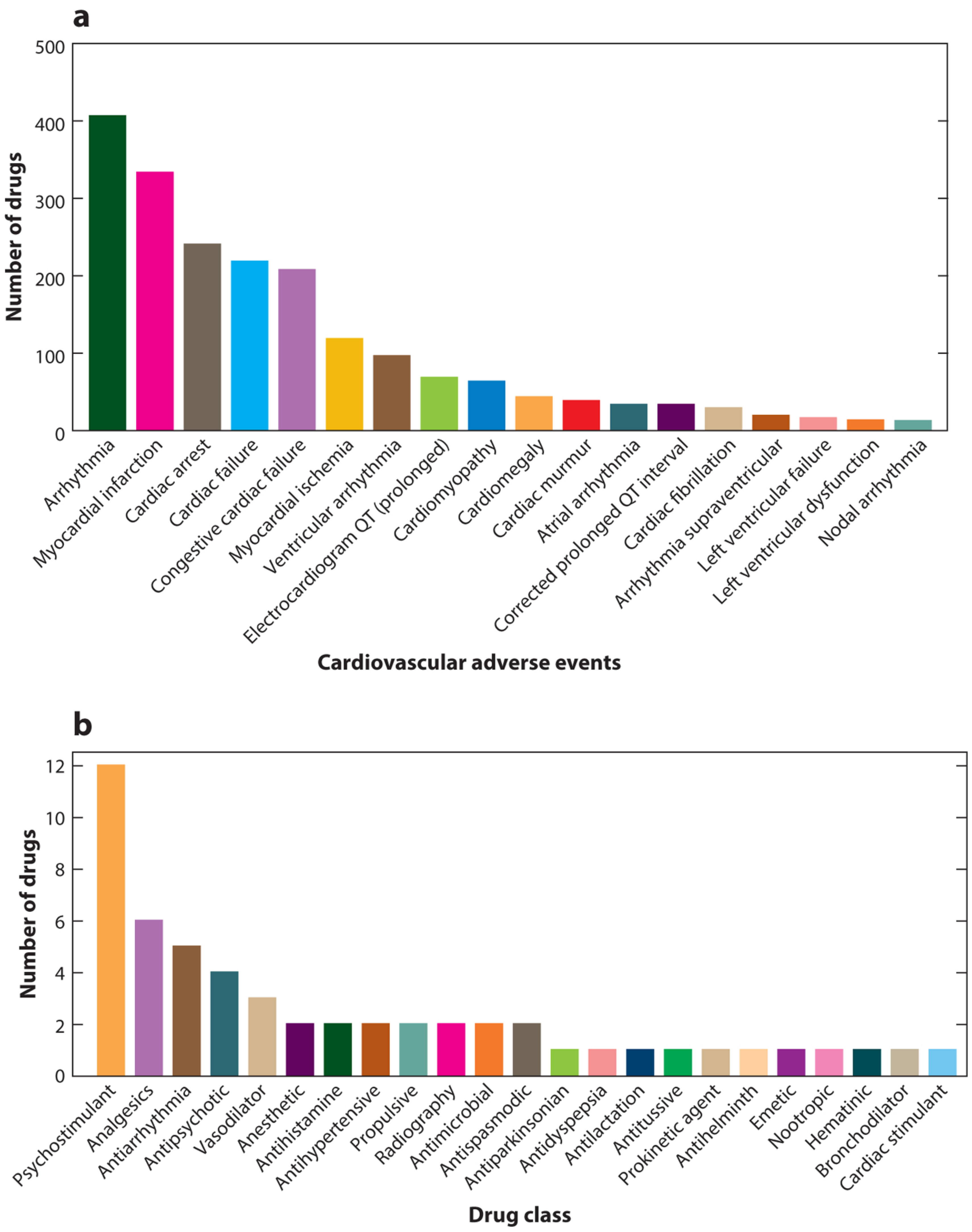{kind=link}
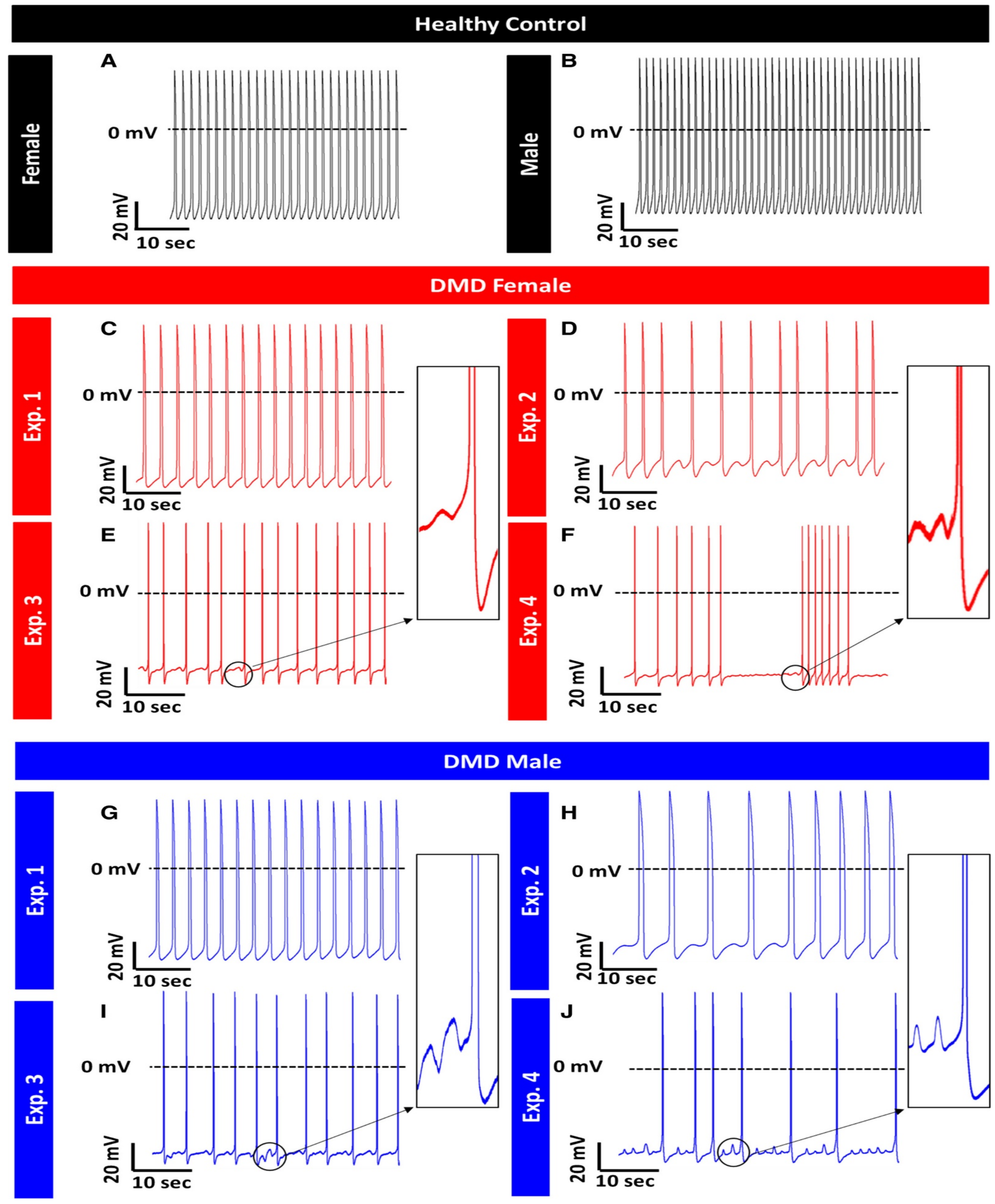{kind=link}
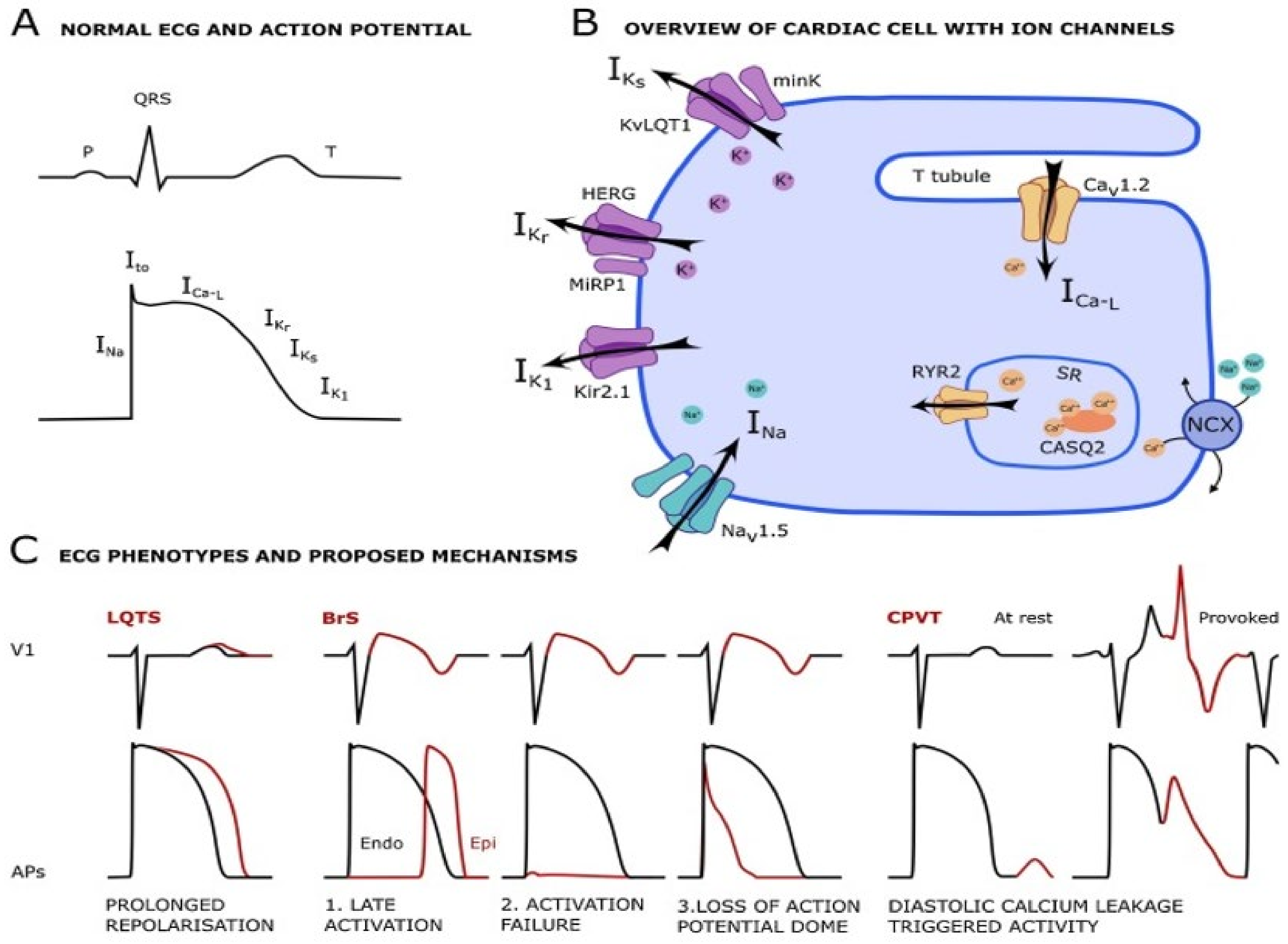{kind=link}
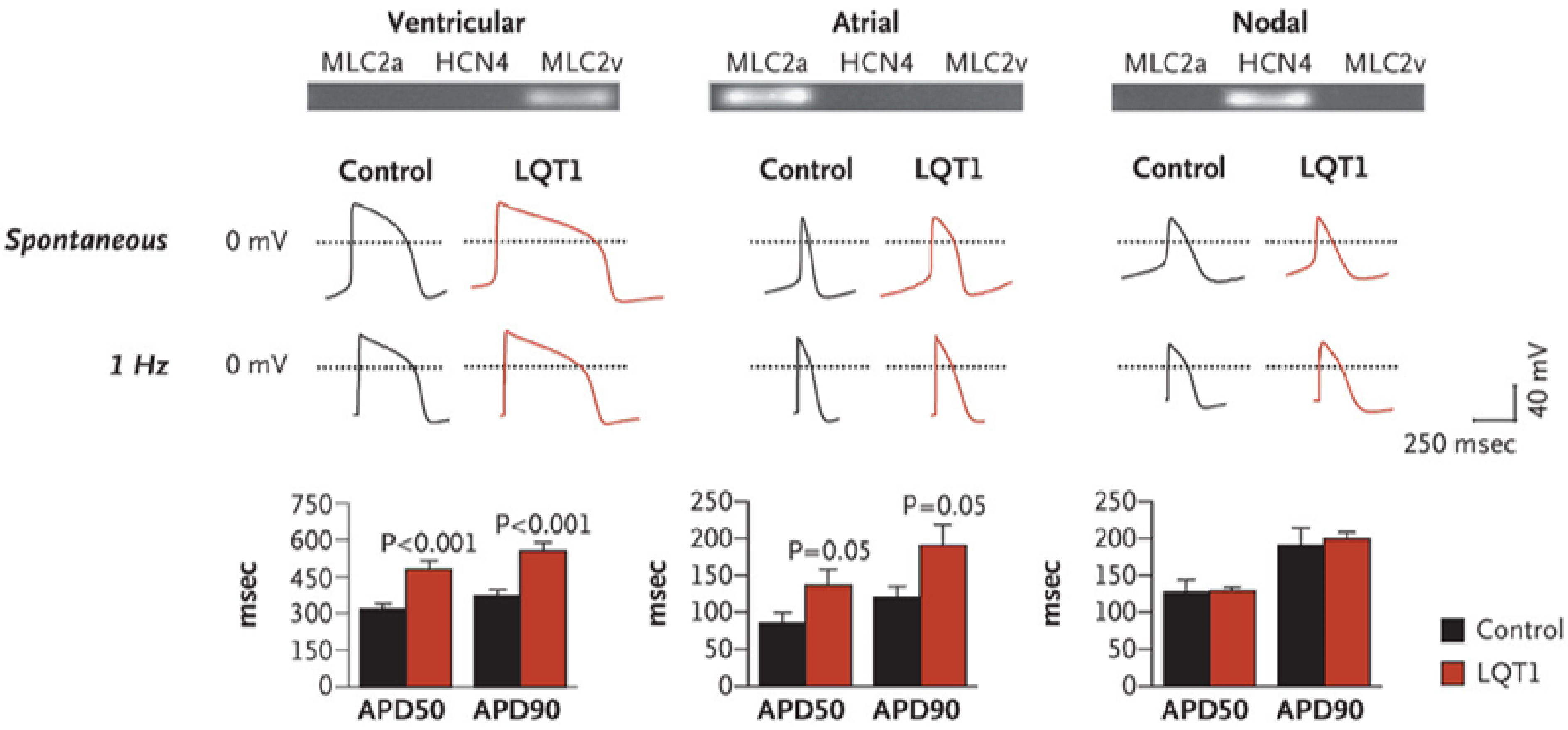{kind=link}
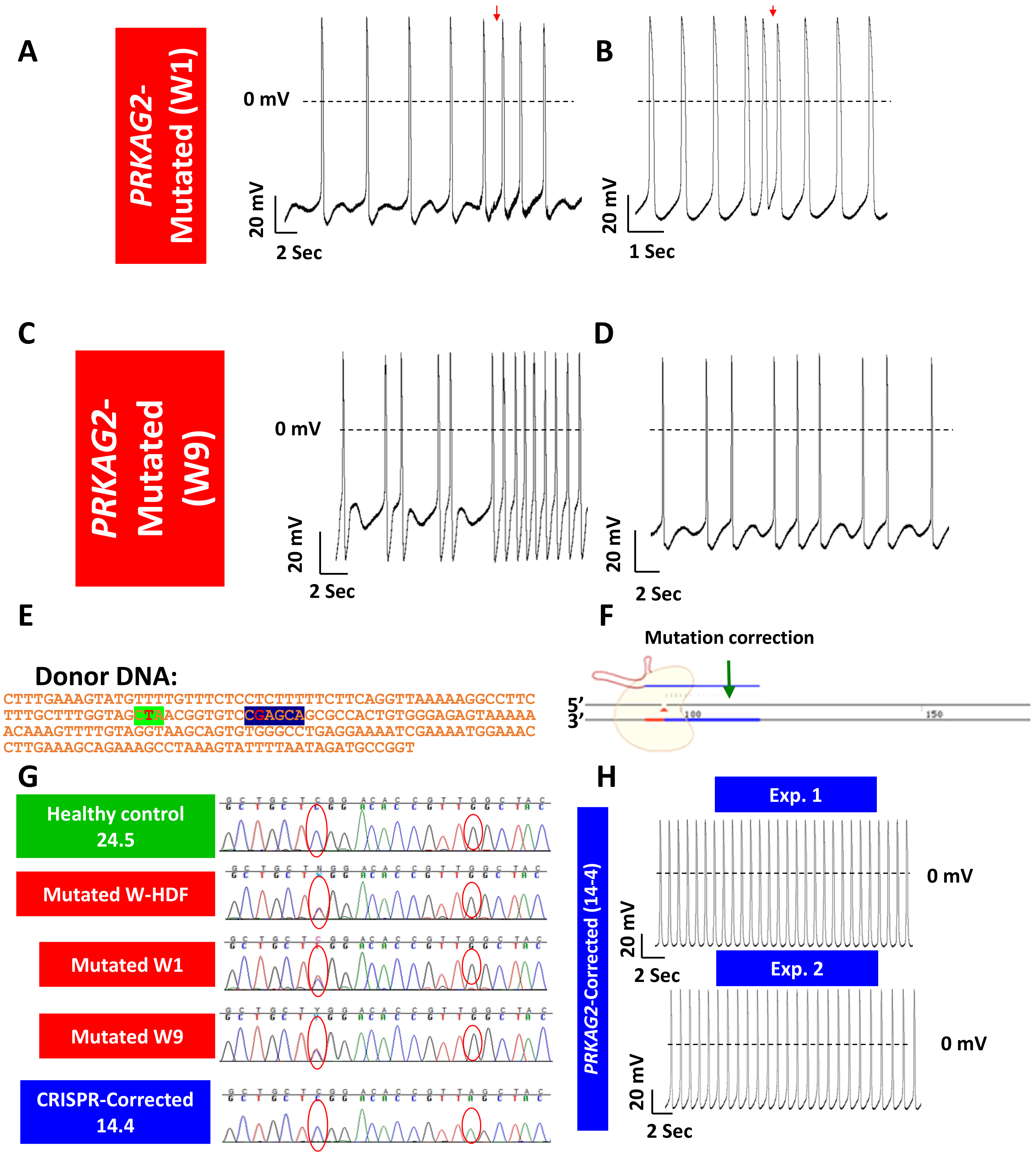{kind=link}
| Drug Group | Example |
|---|---|
| Antiarrhythmics | Amiodarone, Disopyramide, Dofetilide, Ibutilide, Procainamide, Quinidine, Sotalol |
| Antibiotics | Chloroquine, Clarithromycin, Erythromycin, Halofantrine, Pentamidine, Sparfloxacin, Antipsychotics, Chlorpromazine, Haloperidol, Mesoridazine, Pimozide, Thioridazine |
| Antinauseants/Antiemetic | Domperidone, Droperidol |
| Antineoplastic | Arsenic trioxide |
| Calcium channel blocker | Bepridil, Lidoflazine |
| Gastric promotility | Cisapride |
| Opiates | Methadone, Levomethadyl |
| Antihistamines | Terfenadine, Astemizole |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ovics, P.; Regev, D.; Baskin, P.; Davidor, M.; Shemer, Y.; Neeman, S.; Ben-Haim, Y.; Binah, O. Drug Development and the Use of Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Disease Modeling and Drug Toxicity Screening. Int. J. Mol. Sci. 2020, 21, 7320. https://doi.org/10.3390/ijms21197320
Ovics P, Regev D, Baskin P, Davidor M, Shemer Y, Neeman S, Ben-Haim Y, Binah O. Drug Development and the Use of Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Disease Modeling and Drug Toxicity Screening. International Journal of Molecular Sciences. 2020; 21(19):7320. https://doi.org/10.3390/ijms21197320
Chicago/Turabian StyleOvics, Paz, Danielle Regev, Polina Baskin, Mor Davidor, Yuval Shemer, Shunit Neeman, Yael Ben-Haim, and Ofer Binah. 2020. "Drug Development and the Use of Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Disease Modeling and Drug Toxicity Screening" International Journal of Molecular Sciences 21, no. 19: 7320. https://doi.org/10.3390/ijms21197320
APA StyleOvics, P., Regev, D., Baskin, P., Davidor, M., Shemer, Y., Neeman, S., Ben-Haim, Y., & Binah, O. (2020). Drug Development and the Use of Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Disease Modeling and Drug Toxicity Screening. International Journal of Molecular Sciences, 21(19), 7320. https://doi.org/10.3390/ijms21197320