3.1.2. Synthetic Procedures and Experimental Characterisation Data
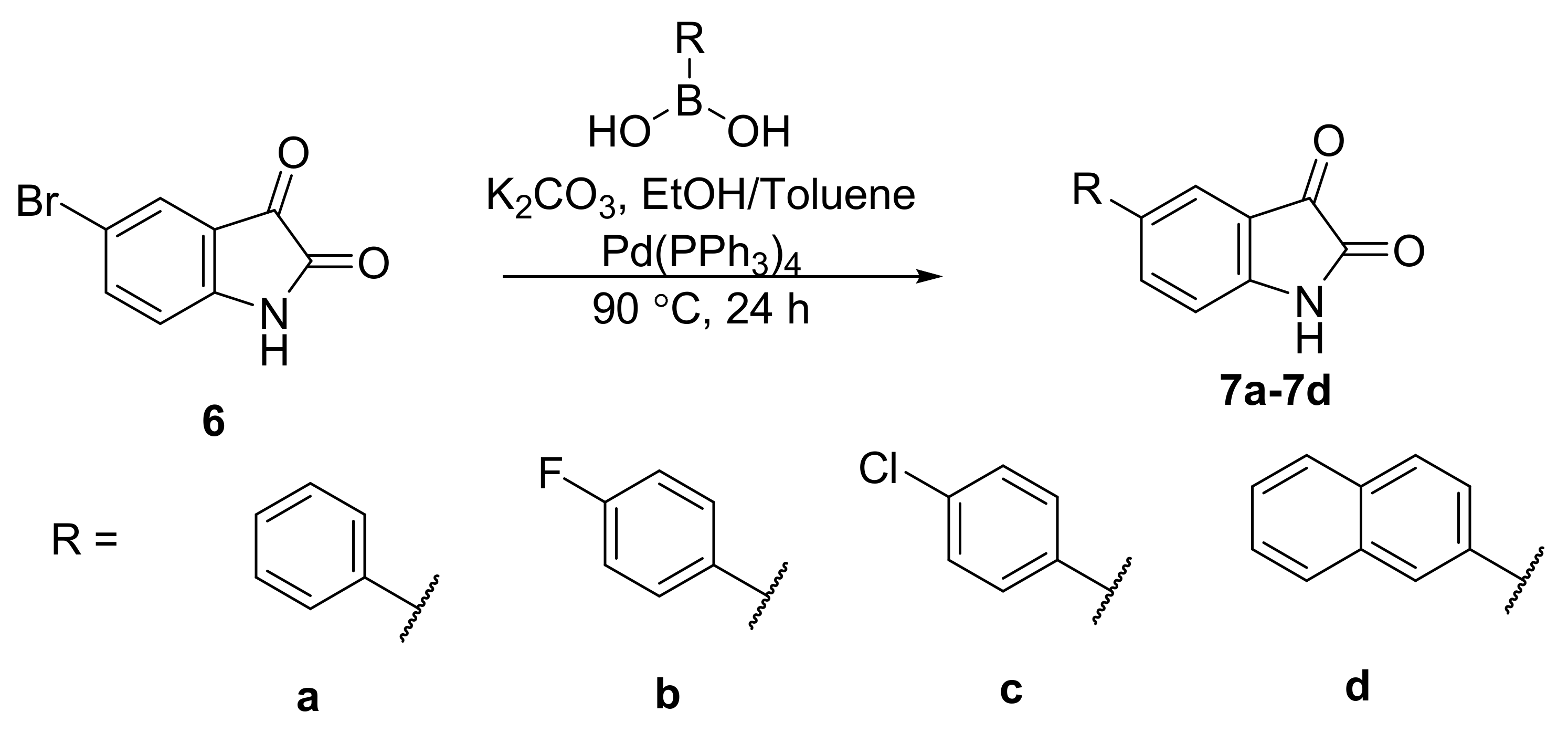
General Synthetic Procedure A for 5-arylisatins
To a solution mixture of 5-bromoisatin (1.0 equivalent) and the appropriate boronic acid (1.1 equivalents) in degassed (for 30 min) 1:1 toluene/ethanol solution (40 mL), 2 M potassium carbonate solution (2.0 equivalents; degassed for 30 min prior to addition) was added. The dark brown solution was degassed for 30 min. Pd(PPh3)4 (0.01 equivalents) was then added to the solution mixture and the reaction was heated at 90 °C under nitrogen atmosphere for 24 h. The brownish-black solution was concentrated in vacuo. Water was then added to the reaction mixture and the resulting solution was then acidified to pH 1 with HCl (2 M). The reddish-orange organic layer was extracted thrice with dichloromethane (3 × 30 mL), washed with brine, dried over sodium sulphate and concentrated in vacuo to give the crude product as a red solid. The crude product was purified by flash column chromatography on silica to afford the product.
5-Phenylindoline-2,3-dione (7a)
The titled compound was synthesised from 5-bromoisatin (2.02 g, 8.92 mmol), phenylboronic acid (1.20 g, 9.83 mmol), potassium carbonate (2.50 g, 18.06 mmol) and Pd(PPh3)4 (116 mg, 0.100 mmol) following general synthetic procedure A. The product was obtained as a red solid (1.72 g, 86%); mp 250.3–250.4 °C; 1H NMR (400 MHz, DMSO-d6): δ 11.29 (bs, 1H, NH), 7.91 (dd, J = 8.2, 2.0 Hz, 1H, ArH), 7.76 (d, J = 1.9 Hz, 1H, ArH), 7.67–7.63 (m, 2H, ArH), 7.48–7.42 (m. 2H, ArH), 7.39–7.33 (m, 1H, ArH), 7.01 (d, J = 8.2 Hz, 1H, ArH); 13C NMR (100 MHz, DMSO-d6): δ 184.4 (CO), 159.6 (CO), 150.0 (ArC), 138.7 (ArC), 136.5 (ArCH), 134.9 (ArC), 129.0 (ArCH), 127.5 (ArCH), 126.2 (ArCH), 122.5 (ArCH), 118.4 (ArC), 112.7 (ArCH); IR (ATR): νmax 3273, 1759, 1729, 1617, 1508, 1473, 1432, 1308, 1254, 1203, 1183, 1099, 1022, 966, 910, 846, 770, 752, 730, 697, 654, 576, 517, 456, 424 cm−1; MS (+ ESI): m/z 246.08, [M + Na]+.
5-(4-Fluorophenyl)indoline-2,3-dione (7b)
The titled compound was synthesised from 5-bromoisatin (1.06 g, 4.70 mmol), 4-fluorophenylboronic acid (0.76 g, 5.40 mmol), potassium carbonate (1.31 g, 9.45 mmol) and Pd(PPh3)4 (55 mg, 0.048 mmol) following general synthetic procedure A. The product was obtained as a red solid (0.68 g, 60%); mp 238.9–239.3 °C; 1H NMR (400 MHz, DMSO-d6): δ 11.13 (bs, 1H, NH), 7.87 (dd, J = 8.2, 2.0 Hz, 1H, ArH), 7.74 (d, J = 1.8 Hz, 1H, ArH), 7.72–7.65 (m, 2H, ArH), 7.31–7.23 (m, 2H, ArH), 6.99 (d, J = 8.2 Hz, ArH); 13C NMR (100 MHz, DMSO-d6): δ 184.3 (CO), 161.8 (ArC), 159.5 (CO), 149.9 (ArC), 136.4 (ArCH), 135.2 (ArC), 133.9 (ArC), 128.3 (ArCH), 122.5 (ArCH), 118.4 (ArC), 115.8 (ArCH), 112.7 (ArCH); IR (ATR): νmax 3324, 3273, 1765, 1739, 1621, 1601, 1589, 1505, 1475, 1454, 1366, 1349, 1307, 1275, 1225, 1192, 1159, 1127, 1098, 1013, 967, 935, 914, 891, 843, 821, 752, 703, 652, 590, 566, 514, 497, 460, 448 cm−1; MS (+ ESI): m/z 264.00, [M + Na]+.
5-(4-Chlorophenyl)indoline-2,3-dione (7c)
The titled compound was synthesised from 5-bromoisatin (1.10 g, 4.87 mmol), 4-chlorophenylboronic acid (0.84 g, 5.40 mmol), potassium carbonate (1.37 g, 9.91 mmol) and Pd(PPh3)4 (78 mg, 0.068 mmol) following general synthetic procedure A. The product was obtained as a red solid (0.72 g, 57%); mp 244.2–244.3 °C; 1H NMR (400 MHz, DMSO-d6): δ 11.15 (bs, 1H, NH), 7.90 (dd, J = 8.2, 2.0 Hz, 1H, ArH), 7.78 (d, J = 1.9 Hz, 1H, ArH), 7.71–7.67 (m, 2H, ArH), 7.52–7.47 (m, 2H, ArH), 7.00 (d, J = 8.2 Hz, 1H, ArH); 13C NMR (100 MHz, DMSO-d6): δ 184.3 (CO), 159.5 (CO), 150.2 (ArC), 137.6 (ArC), 136.4 (ArCH), 133.5 (ArC), 132.3 (ArC), 128.9 (ArCH), 128.0 (ArCH), 122.5 (ArCH), 118.5 (ArC), 112.7 (ArCH); IR (ATR): νmax 3327, 3279, 1763, 1742, 1620, 1506, 1474, 1452, 1367, 1349, 1308, 1272, 1258, 1220, 1194, 1160, 1122, 1090, 1012, 966, 915, 842, 821, 811, 753, 742, 703, 655, 584, 567, 541, 513, 497, 460, 448 cm−1; MS (+ESI): m/z 280.08, [M + Na]+.
5-(Naphthalen-2-yl)indoline-2,3-dione (7d)
The titled compound was synthesised from 5-bromoisatin (1.05 g, 4.66 mmol), 4-naphthyllboronic acid (0.94 g, 5.19 mmol), potassium carbonate (1.32 g, 9.51 mmol) and Pd(PPh3)4 (58 mg, 0.050 mmol) following general synthetic procedure A. The product was obtained as a red solid (0.58 g, 46%); mp 293.9–294.0 °C; 1H NMR (600 MHz, DMSO-d6): δ 11.17 (bs, 1H, NH), 8.24 (d, J = 1.7 Hz, 1H, ArH), 8.07 (dd, J = 8.2, 2.1 Hz, 1H, ArH), 8.01–7.97 (m, 2H, ArH), 7.95–7.92 (m, 2H, ArH), 7.84 (dd, J = 8.6, 1.9 Hz, 1H, ArH), 7.56–7.50 (m, 2H, ArH), 7.05 (dd, J = 8.2, 0.5 Hz, 1H, ArH); 13C NMR (150 MHz, DMSO-d6): δ 184.4 (CO), 159.6 (CO), 150.0 (ArC), 136.7 (ArCH), 136.0 (ArC), 134.7 (ArC), 133.3 (ArC), 132.2 (ArC), 128.6 (ArCH), 128.2 (ArCH), 127.5 (ArCH), 126.5 (ArCH), 126.2 (ArCH), 124.7 (ArCH), 124.6 (ArCH), 122.7 (ArCH), 118.6 (ArC), 112.8 (ArCH); IR (ATR): νmax 3252, 1753, 1732, 1616, 1490, 1459, 1429, 1389, 1344, 1305, 1270, 1250, 1195, 1154, 1119, 980, 905, 891, 863, 846, 808, 752, 698, 636, 622, 598, 577, 560, 525, 496, 474, 460, 443 cm−1; HRMS (+ ESI): Found m/z 296.0682 [M + Na]+, C18H11NO2Na required 296.0682.
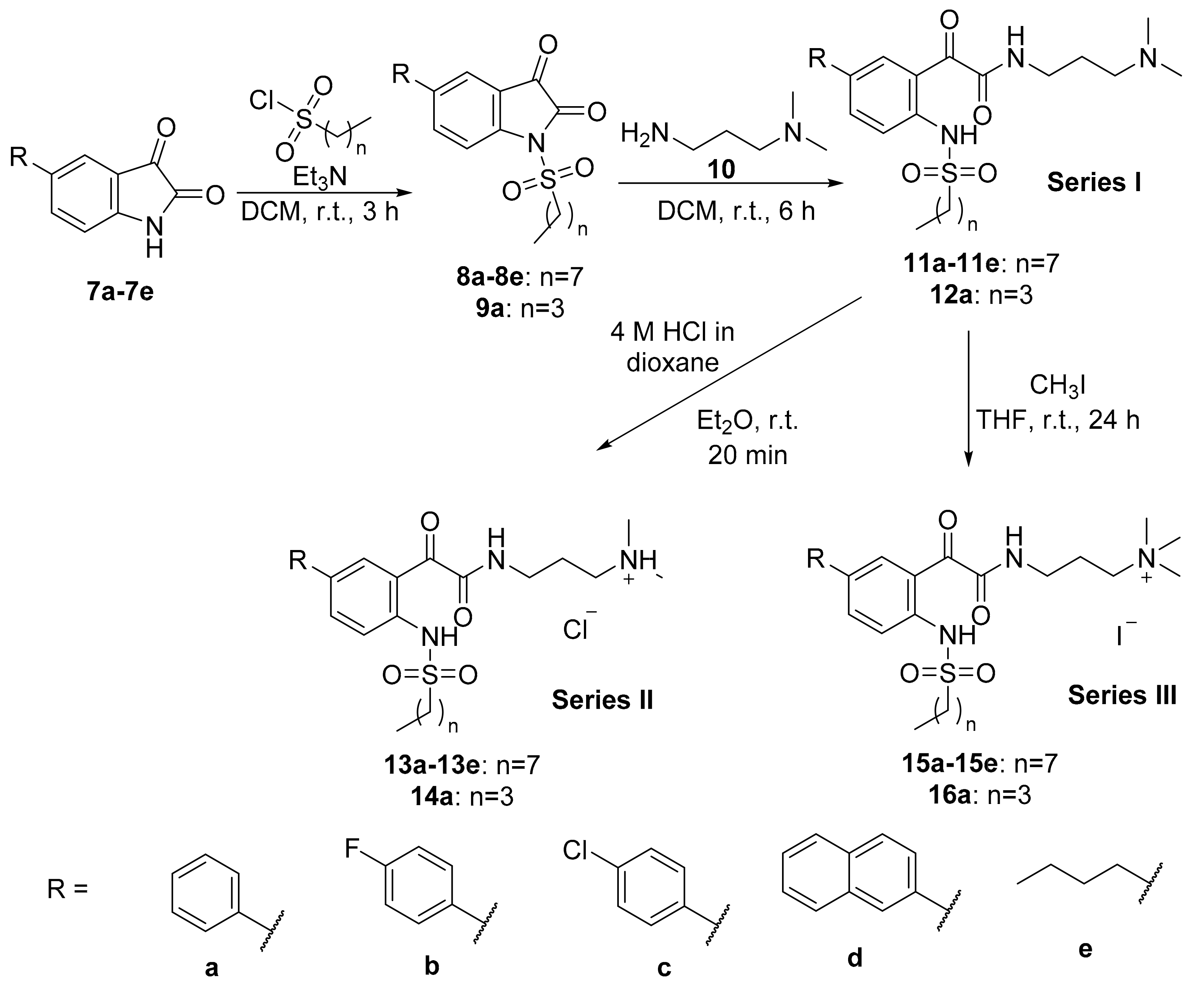
General Synthetic Procedure B for N-sulfonylisatins
To a solution of 5-substituted isatin (1.0 equivalent) in dichloromethane (20 mL), triethylamine (1.1 equivalents) was added at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at 0 °C for 20 min. 1-Octanesulfonyl chloride or 1-butanesulfonyl chloride (1.0 equivalent) was then added slowly dropwise to the reaction mixture at 0 °C with stirring. The reaction mixture was then stirred at room temperature for 3 h. After completion of the reaction, the resulting mixture was concentrated in vacuo and washed with methanol to afford the product.
1-(Octylsulfonyl)-5-phenylindoline-2,3-dione (8a)
The titled compound was synthesised from 5-phenylindoline-2,3-dione 7a (1.70 g, 7.62 mmol), triethylamine (1.20 mL, 8.61 mmol) and 1-octanesulfonyl chloride (1.50 mL, 7.67 mmol) following general synthetic procedure B. The product was obtained as a yellow solid (1.58 g, 52%); mp 131.9–132.2 °C; 1H NMR (400 MHz, DMSO-d6): δ 8.08 (dd, J = 8.5, 2.1 Hz, 1H, ArH), 7.98 (d, J = 2.0 Hz, 1H, ArH), 7.80 (d, J = 8.5 Hz, 1H, ArH), 7.75–7.71 (m, 2H, ArH), 7.50 (t, J = 7.8 Hz, 2H, ArH), 7.41 (t, J = 7.4 Hz, 1H, ArH), 3.67–3.59 (m, 2H, CH2), 1.86–1.76 (m, 2H, CH2), 1.43–1.15 (m, 10H, CH2), 0.83 (t, J = 7.0 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 178.7 (CO), 156.6 (CO), 146.1 (ArC), 138.0 (ArC), 137.0 (ArC), 135.9 (ArCH), 129.1 (ArCH), 128.1 (ArCH), 126.5 (ArCH), 122.4 (ArCH), 120.1 (ArC), 114.6 (ArCH), 53.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.2 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3326, 3274, 2915, 2049, 1765, 1737, 1615, 1505, 1472, 1454, 1373, 1349, 1308, 1259, 1222, 1191, 1175, 1160, 1120, 1096, 1012, 967, 945, 913, 850, 821, 762, 754, 703, 651, 609, 592, 566, 532, 513, 497, 461, 448 cm−1; HRMS (+ ESI): Found m/z 422.1398 [M + Na]+, C22H25NO4SNa required 422.1397.
5-(4-Fluorophenyl)-1-(octylsulfonyl)indoline-2,3-dione (8b)
The titled compound was synthesised from 5-(4-fluorophenyl)indoline-2,3-dione 7b (0.54 g, 2.23 mmol), triethylamine (0.35 mL, 2.51 mmol) and 1-octanesulfonyl chloride (0.44 mL, 2.25 mmol) following general synthetic procedure B. The product was obtained as a yellow solid (0.65 g, 70%); mp 142.3–142.7 °C; 1H NMR (400 MHz, DMSO-d6): δ 8.06 (dd, J = 8.6, 2.2 Hz, 1H, ArH), 7.98 (d, J = 2.0, 1H, ArH), 7.82–7.74 (m, 3H, ArH), 7.36–7.28 (m, 2H, ArH), 3.67–3.59 (m, 2H, CH2), 1.86–1.75 (m, 2H, CH2), 1.43–1.15 (m, 10H, CH2), 0.83 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 178.7 (CO), 162.2 (ArC), 156.6 (CO), 146.1 (ArC), 136.0 (ArC), 135.8 (ArCH), 134.5 (ArC), 128.7 (ArCH), 122.5 (ArCH), 120.1 (ArC), 115.9 (ArCH), 114.6 (ArCH), 53.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.2 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3675, 2970, 2900, 1766, 1739, 1615, 1573, 1519, 1475, 1406, 1393, 1373, 1308, 1292, 1232, 1175, 1139, 1066, 1057, 1027, 944, 891, 856, 831, 784, 763, 726, 714, 701, 619, 605, 590, 567, 532, 497, 484, 466, 447 cm−1; HRMS (+ ESI): Found m/z 440.1303 [M + Na]+, C22H24FNO4SNa required 440.1302.
5-(4-Chlorophenyl)-1-(octylsulfonyl)indoline-2,3-dione (8c)
The titled compound was synthesised from 5-(4-chlorophenyl)indoline-2,3-dione 7c (0.36 g, 1.41 mmol), triethylamine (0.22 mL, 1.58 mmol) and 1-octanesulfonyl chloride (0.28 mL, 1.43 mmol) following general synthetic procedure B. The product was obtained as a yellow solid (0.32 g, 52%); mp 171.7–172.1 °C; 1H NMR (400 MHz, DMSO-d6): δ 8.08 (dd, J = 8.6, 2.2 Hz, 1H, ArH), 8.01 (d, J = 2.0 Hz, 1H, ArH), 7.82–7.75 (m, 3H, ArH), 7.54 (d, J = 8.6 Hz, 2H, ArH), 3.67–3.59 (m, 2H, CH2), 1.86–1.75 (m, 2H, CH2), 1.43–1.15 (m, 10H, CH2), 0.83 (t, J = 7.0 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 178.6 (CO), 156.6 (CO), 146.3 (ArC), 136.8 (ArC), 135.8 (ArCH), 135.6 (ArC), 132.9 (ArC), 129.0 (ArCH), 128.4 (ArCH), 122.5 (ArCH), 120.2 (ArC), 114.6 (ArCH), 53.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.2 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3675, 2970, 2900, 1772, 1746, 1614, 1587, 1568, 1468, 1405, 1394, 1369, 1306, 1287, 1241, 1183, 1168, 1139, 1076, 1066, 1057, 1011, 945, 892, 851, 818, 804, 765, 727, 707, 688, 601, 533, 509, 463, 454, 437 cm−1; HRMS (+ ESI): Found m/z 456.1007 [M + Na]+, C22H24ClNO4SNa required 456.1007.
5-(Naphthalen-2-yl)-1-(octylsulfonyl)indoline-2,3-dione (8d)
The titled compound was synthesised from 5-(naphthalen-2-yl)indoline-2,3-dione 7d (0.42 g, 1.54 mmol), triethylamine (0.24 mL, 1.72 mmol) and 1-octanesulfonyl chloride (0.30 mL, 1.54 mmol) following general synthetic procedure B. The product was obtained as a yellow solid (0.50 g, 73%); mp 125.0-125.4 °C; 1H NMR (400 MHz, DMSO-d6): δ 8.33 (s, 1H, ArH), 8.23 (dd, J = 8.6, 2.1 Hz, 1H, ArH), 8.16 (d, J = 2.0 Hz, 1H, ArH), 8.05–8.00 (m, 2H, ArH), 7.98–7.94 (m, 1H, ArH), 7.93–7.88 (m, 1H, ArH), 7.85 (d, J = 8.6 Hz, 1H, ArH), 7.59–7.52 (m, 2H, ArH), 3.69–3.61 (m, 2H, CH2), 1.88-1.77 (m, 2H, CH2), 1.44–1.15 (m, 10H, CH2), 0.83 (t, J = 7.0 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 178.7 (CO), 156.7 (CO), 146.2 (ArC), 136.8 (ArC), 136.0 (ArCH), 135.2 (ArC), 133.3 (ArC), 132.4 (ArC), 128.7 (ArCH), 128.3 (ArCH), 127.5 (ArCH), 126.6 (ArCH), 126.5 (ArCH), 125.3 (ArCH), 124.6 (ArCH), 122.7 (ArCH), 120.2 (ArC), 114.7 (ArCH), 53.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.4 (CH2), 27.3 (CH2), 22.2 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 2923, 2852, 1766, 1737, 1615, 1578, 1488, 1460, 1424, 1374, 1306, 1293, 1270, 1259, 1235, 1198, 1173, 1136, 1110, 1094, 1038, 1017, 998, 953, 937, 922, 893, 872, 852, 824, 783, 763, 751, 744, 721, 700, 659, 632, 613, 589, 560, 532, 497, 474, 446 cm−1; HRMS (+ ESI): Found m/z 472.1554 [M + Na]+, C26H27NO4SNa required 472.1553.
5-Butyl-1-(octylsulfonyl)indoline-2,3-dione (8e)
The titled compound was synthesised from 5-butylindoline-2,3-dione 7e (0.40 g, 1.95 mmol), triethylamine (0.30 mL, 2.15 mmol) and 1-octanesulfonyl chloride (0.39 mL, 1.99 mmol) following general synthetic procedure B. The product was obtained as a yellow solid (0.41 g, 55%); mp 113.2–113.6 °C; 1H NMR (400 MHz, DMSO-d6): δ 7.64–7.53 (m, 3H, ArH), 3.62–3.56 (m, 2H, CH2), 2.62 (t, J = 7.7 Hz, 2H, CH2), 1.82–1.72 (m, 2H, CH2), 1.59–1.49 (m, 2H, CH2), 1.41–1.16 (m, 12H, CH2), 0.89 (t, J = 7.5 Hz, 3H, CH3), 0.84 (t, J = 7.0 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 178.9 (CO), 156.7 (CO), 145.1 (ArC), 139.5 (ArC), 137.8 (ArCH), 124.4 (ArCH), 119.4 (ArC), 114.0 (ArCH), 53.6 (CH2), 33.7 (CH2), 32.9 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.2 (CH2), 22.0 (CH2), 21.6 (CH2), 13.9 (CH3), 13.7 (CH3); IR (ATR): νmax 3854, 3675, 2968, 2911, 2362, 1762, 1737, 1615, 1584, 1483, 1467, 1406, 1393, 1372, 1293, 1250, 1241, 1223, 1177, 1152, 1134, 1116, 1077, 1066, 1057, 1027, 954, 868, 851, 782, 724, 706, 653, 605, 565, 532, 484, 462, 446 cm−1; HRMS (+ ESI): Found m/z 402.1711 [M + Na]+, C20H29NO4SNa required 402.1710.
1-(Butylsulfonyl)-5-phenylindoline-2,3-dione (9a)
The titled compound was synthesised from 5-phenylindoline-2,3-dione 7a (0.45 g, 1.92 mmol), triethylamine (0.30 mL, 2.15 mmol) and 1-butanesulfonyl chloride (0.25 mL, 1.93 mmol) following general synthetic procedure B. The product was obtained as a yellow sticky solid (0.23 g, 34%); 1H NMR (400 MHz, DMSO-d6): δ 8.08 (dd, J = 8.6, 2.2 Hz, 1H, ArH), 7.98 (d, J = 2.1 Hz, 1H, ArH), 7.80 (d, J = 8.6 Hz, 1H, ArH), 7.75–7.71 (m, 2H, ArH), 7.50 (t, J = 7.8 Hz, 2H, ArH), 7.41 (t, J = 7.3 Hz, 1H, ArH), 3.67–3.60 (m, 2H, CH2), 1.85–1.76 (m, 2H, CH2), 1.47–1.36 (m, 2H, CH2), 0.88 (t, J = 7.4 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 178.7 (CO), 156.6 (CO), 146.1 (ArC), 138.0 (ArC), 137.0 (ArC), 135.9 (ArCH), 129.1 (ArCH), 128.0 (ArCH), 126.5 (ArCH), 122.4 (ArCH), 120.1 (ArC), 114.6 (ArCH), 53.5 (CH2), 24.1 (CH2), 20.7 (CH2), 13.3 (CH3); IR (ATR): νmax 3196, 2961, 2873, 1777, 1736, 1648, 1615, 1588, 1508, 1485, 1472, 1455, 1399, 1368, 1339, 1310, 1293, 1270, 1241, 1183, 1171, 1139, 1117, 1040, 1000, 982, 949, 917, 842, 758, 734, 694, 674, 651, 620, 602, 579, 556, 532, 516, 464, 426 cm−1; HRMS (+ ESI): Found m/z 366.0771 [M + Na]+, C18H17NO4SNa required 366.0770.
General Synthetic Procedure C for Glyoxamide Derivatives
To a solution of N-sulfonylisatin (1.0 equivalent) in dichloromethane (5 mL), 3-dimethylaminopropylamine (1.0 equivalent) was added at 0 °C. The reaction mixture was stirred at room temperature for 6 h. After completion of the reaction, water was added to the reaction mixture and the product was extracted into dichloromethane (3 × 30 mL), washed with brine, dried over anhydrous sodium sulphate and concentrated in vacuo to afford the product.
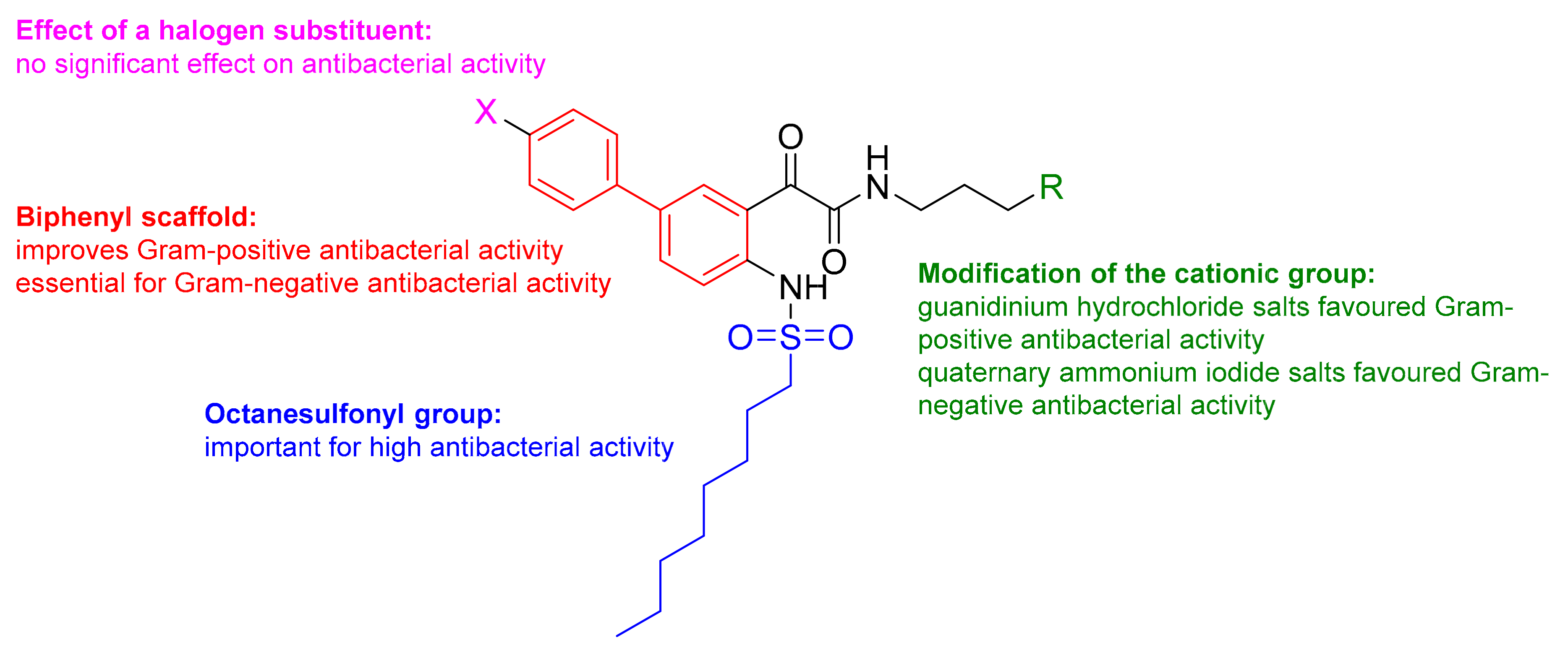
N-(3-(Dimethylamino)propyl)-2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide (11a)
The titled compound was synthesised from 1-(octylsulfonyl)-5-phenylindoline-2,3-dione 8a (0.11 g, 0.28 mmol) and 3-dimethylaminopropylamine (35 μL, 0.28 mmol) following general synthetic procedure C. The product was obtained as a yellow oil (0.13 g, 96%); 1H NMR (400 MHz, CDCl3): δ 8.78 (bs, 1H, NH), 8.72 (d, J = 1.9 Hz, 1H, ArH), 7.87–7.80 (m, 2H, ArH), 7.60–7.55 (m, 2H, ArH), 7.47–7.41 (m, 2H, ArH), 7.39–7.34 (m, 1H, ArH), 3.53 (t, J = 6.0 Hz, 2H, CH2), 3.21–3.15 (m, 2H, CH2), 2.55 (t, J = 6.2 Hz, 2H, CH2), 2.33 (s, 6H, CH3), 1.86–1.76 (m, 4H, CH2), 1.43–1.15 (m, 10H, CH2), 0.85 (t, J = 6.6 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 192.0 (CO), 162.8 (CO), 140.9 (ArC), 139.1 (ArC), 135.8 (ArC), 134.9 (ArCH), 133.6 (ArCH), 129.1 (ArCH), 127.9 (ArCH), 127.0 (ArCH), 120.0 (ArC), 118.6 (ArCH), 58.6 (CH2), 52.7 (CH2), 45.2 (CH3), 39.8 (CH2), 31.8 (CH2), 29.1 (CH2), 29.0 (CH2), 28.2 (CH2), 25.3 (CH2), 23.5 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3327, 3274, 2924, 2854, 1765, 1738, 1616, 1508, 1476, 1395, 1367, 1308, 1261, 1225, 1195, 1143, 1098, 1013, 967, 935, 821, 760, 698, 655, 585, 566, 514, 497, 460, 448 cm−1; HRMS (+ ESI): Found m/z 502.2734 [M + H]+, C27H40N3O4S required 502.2734.
N-(3-(Dimethylamino)propyl)-2-(4’-fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide (11b)
The titled compound was synthesised from 5-(4-fluorophenyl)-1-(octylsulfonyl)indoline-2,3-dione 8b (0.13 g, 0.32 mmol) and 3-dimethylaminopropylamine (40 μL, 0.32 mmol) following general synthetic procedure C. The product was obtained as a yellow oil (0.16 g, 99%); 1H NMR (400 MHz, CDCl3): δ 8.86 (bs, 1H, NH), 8.71 (d, J = 2.2 Hz, 1H, ArH), 7.84 (d, J = 8.7 Hz, 1H, ArH), 7.77 (dd, J = 8.7, 2.2 Hz, 1H, ArH), 7.56–7.50 (m, 2H, ArH), 7.16–7.09 (m, 2H, ArH), 3.53 (t, J = 6.0 Hz, 2H, CH2), 3.20–3.14 (m, 2H, CH2), 2.52 (t, J = 6.1 Hz, 2H, CH2), 2.30 (s, 6H, CH3), 1.86–1.74 (m, 4H, CH2), 1.43–1.16 (m, 10H, CH2), 0.85 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 191.8 (CO), 162.8 (ArC), 162.6 (CO), 140.9 (ArC), 135.3 (ArC), 134.8 (ArC), 134.7 (ArCH), 133.5 (ArCH), 128.6 (ArCH), 119.6 (ArC), 118.6 (ArCH), 116.1 (ArCH), 58.8 (CH2), 52.7 (CH2), 45.3 (CH3), 40.0 (CH2), 31.8 (CH2), 29.1 (CH2), 29.0 (CH2), 28.2 (CH2), 25.3 (CH2), 23.5 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3675, 2970, 2923, 1659, 1635, 1571, 1491, 1393, 1338, 1261, 1222, 1200, 1139, 1066, 1057, 921, 821, 770, 724, 677, 595, 563, 520, 488, 419 cm−1; HRMS (+ ESI): Found m/z 520.2641 [M + H]+, C27H39FN3O4S required 520.2640.
2-(4’-Chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-N-(3-(dimethylamino)propyl)-2-oxoacetamide (11c)
The titled compound was synthesised from 5-(4-chlorophenyl)-1-(octylsulfonyl)indoline-2,3-dione 8c (0.10 g, 0.24 mmol) and 3-dimethylaminopropylamine (30 μL, 0.24 mmol) following general synthetic procedure C. The product was obtained as a yellow oil (0.12 g, 94%); 1H NMR (400 MHz, CDCl3): δ 8.88 (bs, 1H, NH), 8.75 (d, J = 2.2 Hz, 1H, ArH), 7.85 (d, J = 8.8 Hz, 1H, ArH), 7.78 (dd, J = 8.8, 2.3 Hz, 1H, ArH), 7.54–7.47 (m, 2H, ArH), 7.43-7.38 (m, 2H, ArH), 3.52 (t, J = 6.0 Hz, 2H, CH2), 3.21–3.14 (m, 2H, CH2), 2.53–2.48 (m, 2H, CH2), 2.29 (s, 6H, CH3), 1.86–1.74 (m, 4H, CH2), 1.42–1.16 (m, 10H, CH2), 0.85 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 191.7 (CO), 162.5 (CO), 141.2 (ArC), 137.6 (ArC), 134.6 (ArCH), 134.4 (ArC), 134.0 (ArC), 133.5 (ArCH), 129.3 (ArCH), 128.2 (ArCH), 119.6 (ArC), 118.6 (ArCH), 58.9 (CH2), 52.8 (CH2), 45.4 (CH3), 40.1 (CH2), 31.8 (CH2), 29.1 (CH2), 29.0 (CH2), 28.2 (CH2), 25.3 (CH2), 23.5 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3675, 2969, 2922, 1773, 1746, 1636, 1615, 1507, 1482, 1467, 1394, 1338, 1260, 1198, 1140, 1076, 1066, 1012, 919, 852, 817, 773, 697, 600, 564, 539, 509, 492, 436, 426 cm−1; HRMS (+ ESI): Found m/z 536.2344 [M + H]+, C27H39ClN3O4S required 536.2344.
N-(3-(Dimethylamino)propyl)-2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamide (11d)
The titled compound was synthesised from 5-(naphthalen-2-yl)-1-(octylsulfonyl)indoline-2,3-dione 8d (0.15 g, 0.33 mmol) and 3-dimethylaminopropylamine (42 μL, 0.33 mmol) following general synthetic procedure C. The product was obtained as a yellow oil (0.18 g, 96%); 1H NMR (400 MHz, CDCl3): δ 8.87 (d, J = 2.1 Hz, 1H, ArH), 8.85 (bs, 1H, NH), 8.02 (s, 1H, ArH), 7.98–7.84 (m, 5H, ArH), 7.71 (dd, J = 8.5, 1.8 Hz, 1H, ArH), 7.55–7.47 (m, 2H, ArH), 3.55 (t, J = 5.9 Hz, 2H, CH2), 3.23–3.16 (m, 2H, CH2), 2.52 (t, J = 6.2 Hz, 2H, CH2), 2.30 (s, 6H, CH3), 1.87–1.75 (m, 4H, CH2), 1.43–1.17 (m, 10H, CH2), 0.85 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 191.9 (CO), 162.7 (CO), 141.0 (ArC), 136.4 (ArC), 135.7 (ArC), 135.2 (ArCH), 133.9 (ArCH), 133.7 (ArC), 132.9 (ArC), 128.9 (ArCH), 128.4 (ArCH), 127.8 (ArCH), 126.7 (ArCH), 126.4 (ArCH), 125.7 (ArCH), 125.1 (ArCH), 119.7 (ArC), 118.7 (ArCH), 58.9 (CH2), 52.7 (CH2), 45.4 (CH3), 40.1 (CH2), 31.8 (CH2), 29.1 (CH2), 29.0 (CH2), 28.2 (CH2), 25.3 (CH2), 23.6 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3675, 2924, 2855, 1767, 1739, 1641, 1616, 1577, 1492, 1462, 1394, 1333, 1262, 1234, 1201, 1142, 1098, 918, 892, 815, 747, 720, 670, 593, 561, 516, 475 cm−1; HRMS (+ ESI): Found m/z 552.2895 [M + H]+, C31H42N3O4S required 552.2891.
2-(5-Butyl-2-(octylsulfonamido)phenyl)-N-(3-(dimethylamino)propyl)-2-oxoacetamide (11e)
The titled compound was synthesised from 5-butyl-1-(octylsulfonyl)indoline-2,3-dione 8e (0.15 g, 0.40 mmol) and 3-dimethylaminopropylamine (50 μL, 0.40 mmol) following general synthetic procedure C. The product was obtained as a yellow oil (0.18 g, 95%); 1H NMR (400 MHz, CDCl3): δ 8.65 (bs, 1H, NH), 8.21 (d, J = 1.8 Hz, 1H, ArH), 7.67 (d, J = 8.5 Hz, 1H, ArH), 7.41 (dd, J = 8.6, 2.0 Hz, 1H, ArH), 3.56–3.48 (m, 2H, CH2), 3.14–3.08 (m, 2H, CH2), 2.63–2.52 (m, 4H, CH2), 2.34 (s, 6H, CH3), 1.86–1.71 (m, 4H, CH2), 1.63–1.52 (m, 2H, CH2), 1.41–1.17 (m, 12H, CH2), 0.92 (t, J = 7.3 Hz, 3H, CH3), 0.85 (t, J = 7.0 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 192.1 (CO), 163.0 (CO), 139.5 (ArC), 137.6 (ArC), 136.7 (ArCH), 134.6 (ArCH), 119.6 (ArC), 118.5 (ArCH), 58.6 (CH2), 52.4 (CH2), 45.2 (CH3), 39.6 (CH2), 34.9 (CH2), 33.6 (CH2), 31.8 (CH2), 29.1 (CH2), 29.0 (CH2), 28.2 (CH2), 25.3 (CH2), 23.5 (CH2), 22.7 (CH2), 22.4 (CH2), 14.2 (CH3), 14.0 (CH3); IR (ATR): νmax 3674, 2968, 2922, 1609, 1497, 1458, 1405, 1393, 1331, 1256, 1141, 1066, 1057, 934, 834, 781, 732, 668, 562, 520 cm−1; HRMS (+ ESI): Found m/z 482.3047 [M + H]+, C25H44N3O4S required 482.3047.
2-(4-(Butylsulfonamido)-[1,1’-biphenyl]-3-yl)-N-(3-(dimethylamino)propyl)-2-oxoacetamide (12a)
The titled compound was synthesised from 1-(butylsulfonyl)-5-phenylindoline-2,3-dione 9a (0.15 g, 0.44 mmol) and 3-dimethylaminopropylamine (55 μL, 0.44 mmol) following general synthetic procedure C. The product was obtained as a yellow oil (0.18 g, 93%); 1H NMR (400 MHz, CDCl3): δ 8.81 (bs, 1H, NH), 8.73 (d, J = 2.0 Hz, 1H, ArH), 7.87–7.80 (m, 2H, ArH), 7.60–7.55 (m, 2H, ArH), 7.44 (t, J = 8.0 Hz, 2H, ArH), 7.39–7.33 (m, 1H, ArH), 3.53 (t, J = 6.2 Hz, 2H, CH2), 3.22–3.15 (m, 2H, CH2), 2.51 (t, J = 6.2 Hz, 2H, CH2), 2.29 (s, 6H, CH3), 1.84–1.74 (m, 4H, CH2), 1.47–1.36 (m, 2H, CH2), 0.90 (t, J = 7.3 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 192.0 (CO), 162.7 (CO), 140.9 (ArC), 139.1 (ArC), 135.8 (ArC), 134.9 (ArCH), 133.6 (ArCH), 129.1 (ArCH), 127.9 (ArCH), 127.0 (ArCH), 119.6 (ArC), 118.6 (ArCH), 58.8 (CH2), 52.4 (CH2), 45.3 (CH3), 40.0 (CH2), 25.5 (CH2), 25.3 (CH2), 21.5 (CH2), 13.6 (CH3); IR (ATR): νmax 2960, 2871, 2363, 2345, 2183, 2160, 2049, 1978, 1870, 1773, 1734, 1710, 1701, 1685, 1670, 1663, 1654, 1647, 1636, 1617, 1578, 1570, 1560, 1541, 1534, 1522, 1508, 1482, 1466, 1459, 1449, 1395, 1330, 1259, 1194, 1142, 1096, 1025, 921, 794, 761, 733, 697, 681, 669, 616, 583, 555, 535, 477, 428 cm−1; HRMS (+ ESI): Found m/z 466.2109 [M + H]+, C23H32N3O4S required 466.2108.
General Synthetic Procedure D for Tertiary Ammonium Chloride Salts
To a solution of glyoxamide derivative (1.0 equivalent) in diethyl ether (5 mL), 4 M HCl/dioxane (5.0 equivalents) was added. The reaction mixture was stirred at room temperature for 20 min. After completion of reaction, the reaction mixture was concentrated in vacuo, washed thrice with diethyl ether and freeze-dried to afford the product.
N,N-Dimethyl-3-(2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propan-1-aminium chloride (13a)
The titled compound was synthesised from N-(3-(dimethylamino)propyl)-2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide 11a (33 mg, 0.066 mmol) and 4 M HCl/dioxane (0.10 mL, 0.40 mmol) following general synthetic procedure D. The product was obtained as a yellow sticky solid (33 mg, 93%); 1H NMR (600 MHz, DMSO-d6): δ 10.25 (bs, 2H, NH), 9.03 (t, J = 5.8 Hz, 1H, NH), 8.01–7.98 (m, 2H, ArH), 7.67–7.60 (m, 3H, ArH), 7.52–7.48 (m, 2H, ArH), 7.40 (t, J = 7.4 Hz, 1H, ArH), 3.32 (t, J = 6.5 Hz, 2H, CH2), 3.24–3.20 (m, 2H, CH2), 3.12–3.07 (m, 2H, CH2), 2.73 (s, 6H, CH3), 1.97–1.90 (m, 2H, CH2), 1.70–1.64 (m, 2H, CH2), 1.37–1.16 (m, 10H, CH2), 0.82 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 192.1 (CO), 163.8 (CO), 138.3 (ArC), 137.9 (ArC), 135.9 (ArC), 132.9 (ArCH), 130.3 (ArCH), 129.2 (ArCH), 127.9 (ArCH), 126.5 (ArCH), 125.8 (ArC), 122.3 (ArCH), 54.4 (CH2), 51.3 (CH2), 42.0 (CH3), 36.0 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 23.9 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3382, 2924, 2854, 2703, 1647, 1581, 1508, 1483, 1395, 1331, 1267, 1197, 1144, 1075, 974, 919, 842, 761, 697, 681, 616, 586, 509 cm−1; HRMS (+ ESI): Found m/z 502.2731 [M + H]+, C27H40N3O4S required 502.2734.
3-(2-(4’-Fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)-N,N-dimethylpropan-1-aminium chloride (13b)
The titled compound was synthesised from N-(3-(dimethylamino)propyl)-2-(4’-fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide 11b (30 mg, 0.058 mmol) and 4 M HCl/dioxane (0.10 mL, 0.40 mmol) following general synthetic procedure D. The product was obtained as a yellow sticky solid (32 mg, 99%); 1H NMR (600 MHz, DMSO-d6): δ 10.43 (bs, 1H, NH), 10.18 (bs, 1H, NH), 9.02 (t, J = 6.0 Hz, NH), 8.00–7.95 (m, 2H, ArH), 7.73–7.68 (m, 2H, ArH), 7.60 (d, J = 9.0 Hz, 1H, ArH), 7.36–7.31 (m, 2H, ArH), 3.33-3.29 (m, 2H, CH2), 3.21 (t, J = 7.9 Hz, 2H, CH2), 3.12–3.07 (m, 2H, CH2), 2.73 (s, 6H, CH3), 1.98–1.90 (m, 2H, CH2), 1.70–1.63 (m, 2H, CH2), 1.37–1.15 (m, 10H, CH2), 0.82 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 192.0 (CO), 163.7 (CO), 162.1 (ArC), 137.8 (ArC), 135.0 (ArC), 134.8 (ArC), 132.8 (ArCH), 130.1 (ArCH), 128.6 (ArCH), 126.1 (ArC), 122.5 (ArCH), 116.0 (ArCH), 54.4 (CH2), 51.3 (CH2), 42.0 (CH3), 42.0 (CH3), 36.0 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 23.9 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3358, 2925, 2855, 2690, 2360, 1647, 1603, 1515, 1488, 1400, 1332, 1222, 1196, 1143, 1099, 1012, 975, 919, 828, 725, 670, 597, 559, 520, 418 cm−1; HRMS (+ESI): Found m/z 520.2641 [M + H]+, C27H39FN3O4S required 520.2640.
3-(2-(4’-Chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)-N,N-dimethylpropan-1-aminium chloride (13c)
The titled compound was synthesised from 2-(4’-chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-N-(3-(dimethylamino)propyl)-2-oxoacetamide 11c (30 mg, 0.056 mmol) and 4 M HCl/dioxane (0.10 mL, 0.40 mmol) following general synthetic procedure D. The product was obtained as a yellow sticky solid (29 mg, 92%); 1H NMR (600 MHz, DMSO-d6): δ 10.37 (bs, 1H, NH), 10.25 (bs, 1H, NH), 9.02 (t, J = 6.0 Hz, 1H, NH), 8.02–7.98 (m, 2H, ArH), 7.71–7.68 (m, 2H, ArH), 7.63–7.60 (m, 1H, ArH), 7.57–7.54 (m, 2H, ArH), 3.34–3.30 (m, 2H, CH2), 3.21 (t, J = 7.8 Hz, 2H, CH2), 3.12–3.07 (m, 2H, CH2), 2.73 (s, 6H, CH3), 1.98-1.90 (m, 2H, CH2), 1.70–1.63 (m, 2H, CH2), 1.36–1.15 (m, 10H, CH2), 0.82 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 191.9 (CO), 163.6 (CO), 138.1 (ArC), 137.1 (ArC), 134.6 (ArC), 132.8 (ArC), 132.7 (ArCH), 130.2 (ArCH), 129.2 (ArCH), 128.3 (ArCH), 126.1 (ArC), 122.5 (ArCH), 54.4 (CH2), 51.4 (CH2), 42.1 (CH3), 36.0 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 23.9 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3372, 2924, 2854, 2704, 2359, 1643, 1578, 1507, 1481, 1400, 1333, 1273, 1196, 1143, 1092, 1012, 974, 918, 818, 758, 697, 668, 593, 511, 488 cm−1; HRMS (+ ESI): Found m/z 536.2346 [M + H]+, C27H39ClN3O4S required 536.2344.
N,N-Dimethyl-3-(2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamido)propan-1-aminium chloride (13d)
The titled compound was synthesised from N-(3-(dimethylamino)propyl)-2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamide 11d (32 mg, 0.058 mmol) and 4 M HCl/dioxane (0.10 mL, 0.40 mmol) following general synthetic procedure D. The product was obtained as a yellow sticky solid (33 mg, 96%); 1H NMR (600 MHz, DMSO-d6): δ 10.26 (bs, 2H, NH), 9.03 (t, J = 5.9 Hz, 1H, NH), 8.23 (s, 1H, ArH), 8.16–8.13 (m, 2H, ArH), 8.06–8.01 (m, 2H, ArH), 7.96 (d, J = 7.6 Hz, 1H, ArH), 7.83 (dd, J = 8.5, 1.4 Hz, 1H, ArH), 7.65 (d, J = 9.1 Hz, 1H, ArH), 7.60–7.52 (m, 2H, ArH), 3.35–3.29 (m, 2H, CH2), 3.22 (t, J = 7.7 Hz, 2H, CH2), 3.14–3.09 (m, 2H, CH2), 2.74 (s, 6H, CH3), 1.99–1.92 (m, 2H, CH2), 1.72–1.64 (m, 2H, CH2), 1.39–1.14 (m, 10H, CH2), 0.82 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 192.0 (CO), 163.7 (CO), 137.8 (ArC), 135.9 (ArC), 135.6 (ArC), 133.2 (ArC), 133.0 (ArCH), 132.4 (ArC), 130.3 (ArCH), 128.7 (ArCH), 128.2 (ArCH), 127.5 (ArCH), 126.6 (ArCH), 126.6 (ArC), 126.4 (ArCH), 125.2 (ArCH), 124.6 (ArCH), 122.8 (ArCH), 54.5 (CH2), 51.3 (CH2), 42.0 (CH3), 36.0 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 23.9 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3675, 2924, 2854, 2698, 1641, 1494, 1466, 1397, 1331, 1269, 1236, 1201, 1143, 1086, 919, 892, 861, 815, 747, 720, 670, 557, 516, 476 cm−1; HRMS (+ ESI): Found m/z 552.2893 [M + H]+, C31H42N3O4S required 552.2891.
3-(2-(5-Butyl-2-(octylsulfonamido)phenyl)-2-oxoacetamido)-N,N-dimethylpropan-1-aminium chloride (13e)
The titled compound was synthesised from 2-(5-butyl-2-(octylsulfonamido)phenyl)-N-(3-(dimethylamino)propyl)-2-oxoacetamide 11e (33 mg, 0.069 mmol) and 4 M HCl/dioxane (0.10 mL, 0.40 mmol) following general synthetic procedure D. The product was obtained as a yellow sticky solid (35 mg, 99%); 1H NMR (600 MHz, DMSO-d6): δ 10.29 (bs, 1H, NH), 10.04 (bs, 1H, NH), 8.92 (t, J = 6.0 Hz, 1H, NH), 7.54–7.50 (m, 2H, ArH), 7.41 (dd, J = 7.7, 0.9 Hz, 1H, ArH), 3.32–3.27 (m, 2H, CH2), 3.15–3.06 (m, 4H, CH2), 2.74 (s, 6H, CH3), 2.61 (t, J = 7.8 Hz, 2H, CH2), 1.96–1.89 (m, 2H, CH2), 1.66–1.50 (m, 4H, CH2), 1.34–1.15 (m, 12H, CH2), 0.89 (t, J = 7.3 Hz, 3H, CH3), 0.84 (t, J = 7.3 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 192.6 (CO), 164.2 (CO), 138.6 (ArC), 136.4 (ArC), 134.8 (ArCH), 131.7 (ArCH), 125.6 (ArCH), 122.0 (ArCH), 54.5 (CH2), 51.0 (CH2), 42.1 (CH3), 35.9 (CH2), 33.8 (CH2), 32.9 (CH2), 31.1 (CH2), 28.3 (CH2), 28.3 (CH2), 27.3 (CH2), 23.9 (CH2), 22.9 (CH2), 22.0 (CH2), 32.7 (CH2), 13.9 (CH3), 13.7 (CH3); IR (ATR): νmax 3379, 2953, 2922, 2855, 2674, 2360, 1670, 1578, 1524, 1496, 1465, 1443, 1400, 1334, 1253, 1179, 1153, 1086, 978, 918, 906, 874, 836, 799, 759, 677, 604, 570, 544, 516, 475, 432 cm−1; HRMS (+ ESI): Found m/z 482.3046 [M + H]+, C25H44N3O4S required 482.3047.
3-(2-(4-(Butylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)-N,N-dimethylpropan-1-aminium chloride (14a)
The titled compound was synthesised from 2-(4-(butylsulfonamido)-[1,1’-biphenyl]-3-yl)-N-(3-(dimethylamino)propyl)-2-oxoacetamide 12a (35 mg, 0.079 mmol) and 4 M HCl/dioxane (0.10 mL, 0.40 mmol) following general synthetic procedure D. The product was obtained as a yellow sticky solid (34 mg, 91%); 1H NMR (600 MHz, DMSO-d6): δ 10.30 (bs, 1H, NH), 10.18 (bs, 1h, NH), 9.03 (t, J = 6.0 Hz, 1H, NH), 8.01–7.98 (m, 2H, ArH), 7.67–7.64 (m, 2H, ArH), 7.63–7.60 (m, 1H, ArH), 7.52–7.48 (m, 2H, ArH), 7.42–7.38 (m, 1H, ArH), 3.34–3.30 (m, 2H, CH2), 3.24–3.20 (m, 2H, CH2), 3.13–3.07 (m, 2H, CH2), 2.74 (s, 6H, CH3), 1.97–1.91 (m, 2H, CH2), 1.70–1.63 (m, 2H, CH2), 1.41–1.33 (m, 2H, CH2), 0.85 (t, J = 7.5 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 192.1 (CO), 163.8 (CO), 138.3 (ArC), 137.8 (ArC), 136.0 (ArC), 132.9 (ArCH), 130.2 (ArCH), 129.2 (ArCH), 127.9 (ArCH), 126.5 (ArCH), 125.9 (ArC), 122.4 (ArCH), 54.5 (CH2), 51.1 (CH2), 42.1 (CH3), 36.0 (CH2), 24.9 (CH2), 23.9 (CH2), 20.7 (CH2), 13.4 (CH3); IR (ATR): νmax 3363, 2960, 2872, 2690, 2361, 1643, 1581, 1508, 1483, 1394, 1330, 1266, 1239, 1196, 1144, 1076, 974, 921, 843, 800, 761, 698, 681, 616, 585, 539 cm−1; HRMS (+ ESI): Found m/z 446.2109 [M + H] +, C23H32N3O4S required 446.2108.
General Synthetic Procedure E for Quaternary Ammonium Iodide Salts
To a solution of glyoxamide derivative (1.0 equivalent) in THF (5 mL), iodomethane (2.5 equivalents) was added. The reaction mixture was stirred at room temperature for 24 h. After completion of reaction, the reaction mixture was concentrated in vacuo, washed thrice with diethyl ether and freeze-dried to afford the product.
N,N,N-Trimethyl-3-(2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propan-1-aminium iodide (15a)
The titled compound was synthesised from N-(3-(Dimethylamino)propyl)-2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide 11a (33 mg, 0.066 mmol) and iodomethane (10 μL, 0.17 mmol) following general synthetic procedure E. The product was obtained as a yellow sticky solid (40 mg, 94%); 1H NMR (600 MHz, DMSO-d6): δ 10.10 (bs, 1H, NH), 8.98 (t, J = 5.9 Hz, 1H, NH), 8.03–7.97 (m, 2H, ArH), 7.68–7.64 (m, 2H, ArH), 7.59–7.56 (m, 1H, ArH), 7.52–7.48 (m, 2H, ArH), 7.43–7.39 (m, 1H, ArH), 3.39–3.30 (m, 4H, CH2), 3.21–3.16 (m, 2H, CH2), 3.06 (s, 9H, CH2), 2.02–1.95 (m, 2H, CH2), 1.70–1.63 (m, 2H, CH2), 1.39–1.15 (m, 10H, CH3), 0.83 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 191.8 (CO), 163.7 (CO), 138.3 (ArC), 132.7 (ArCH), 130.1 (ArCH), 129.2 (ArC), 129.2 (ArCH), 128.0 (ArCH), 126.8 (ArC), 126.5 (ArCH), 126.2 (ArC), 122.9 (ArCH), 63.5 (CH2), 52.3 (CH3), 51.2 (CH2), 35.9 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.9 (CH2), 22.6 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3396, 3032, 2925, 2854, 1647, 1582, 1508, 1483, 1394, 1330, 1265, 1195, 1140, 1076, 915, 841, 761, 698, 681, 617, 586, 564, 505 cm−1; HRMS (+ ESI): Found m/z 516.2892 [M] +, C28H42N3O4S required 516.2891.
3-(2-(4’-Fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)-N,N,N-trimethylpropan-1-aminium iodide (15b)
The titled compound was synthesised from N-(3-(dimethylamino)propyl)-2-(4’-fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide 11b (32 mg, 0.062 mmol) and iodomethane (10 μL, 0.16 mmol) following general synthetic procedure E. The product was obtained as a yellow sticky solid (39 mg, 95%); 1H NMR (600 MHz, DMSO-d6): δ 10.07 (bs, 1H, NH), 8.97 (t, J = 6.0 Hz, 1H, NH), 7.99–7.94 (m, 2H, ArH), 7.73–7.69 (m, 2H, ArH), 7.56 (d, J = 8.1 Hz, 1H, ArH), 7.35–7.30 (m, 2H, ArH), 3.39–3.30 (m, 4H, CH2), 3.17 (t, J = 7.8 Hz, 2H, CH2), 3.07 (s, 9H, CH3), 2.02–1.95 (m, 2H, CH2), 1.71–1.62 (m, 2H, CH2), 1.39–1.14 (m, 10H, CH2), 0.82 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 191.7 (CO), 163.6 (CO), 162.1 (ArC), 137.4 (ArC), 135.3 (ArC), 134.8 (ArC), 132.6 (ArCH), 130.0 (ArCH), 128.6 (ArCH), 127.2 (ArC), 123.1 (ArCH), 116.0 (ArCH), 63.5 (CH2), 52.3 (CH3), 51.2 (CH2), 35.9 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.9 (CH2), 22.6 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3410, 2925, 2855, 2359, 1647, 1603, 1516, 1488, 1400, 1331, 1262, 1221, 1196, 1141, 1100, 1013, 915, 882, 828, 725, 670, 559, 520 cm−1; HRMS (+ ESI): Found m/z 534.2798 [M] +, C28H41FN3O4S required 534.2796.
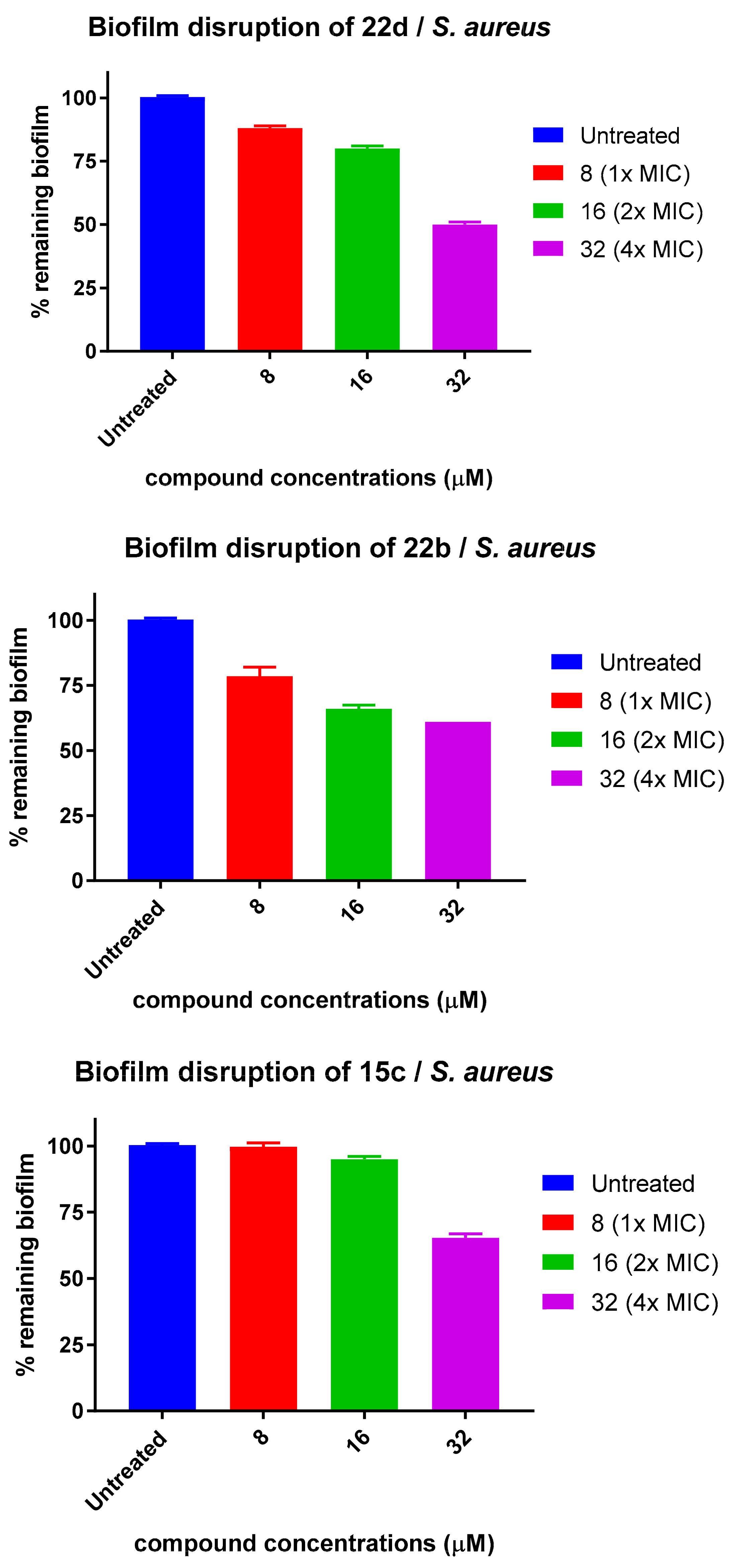
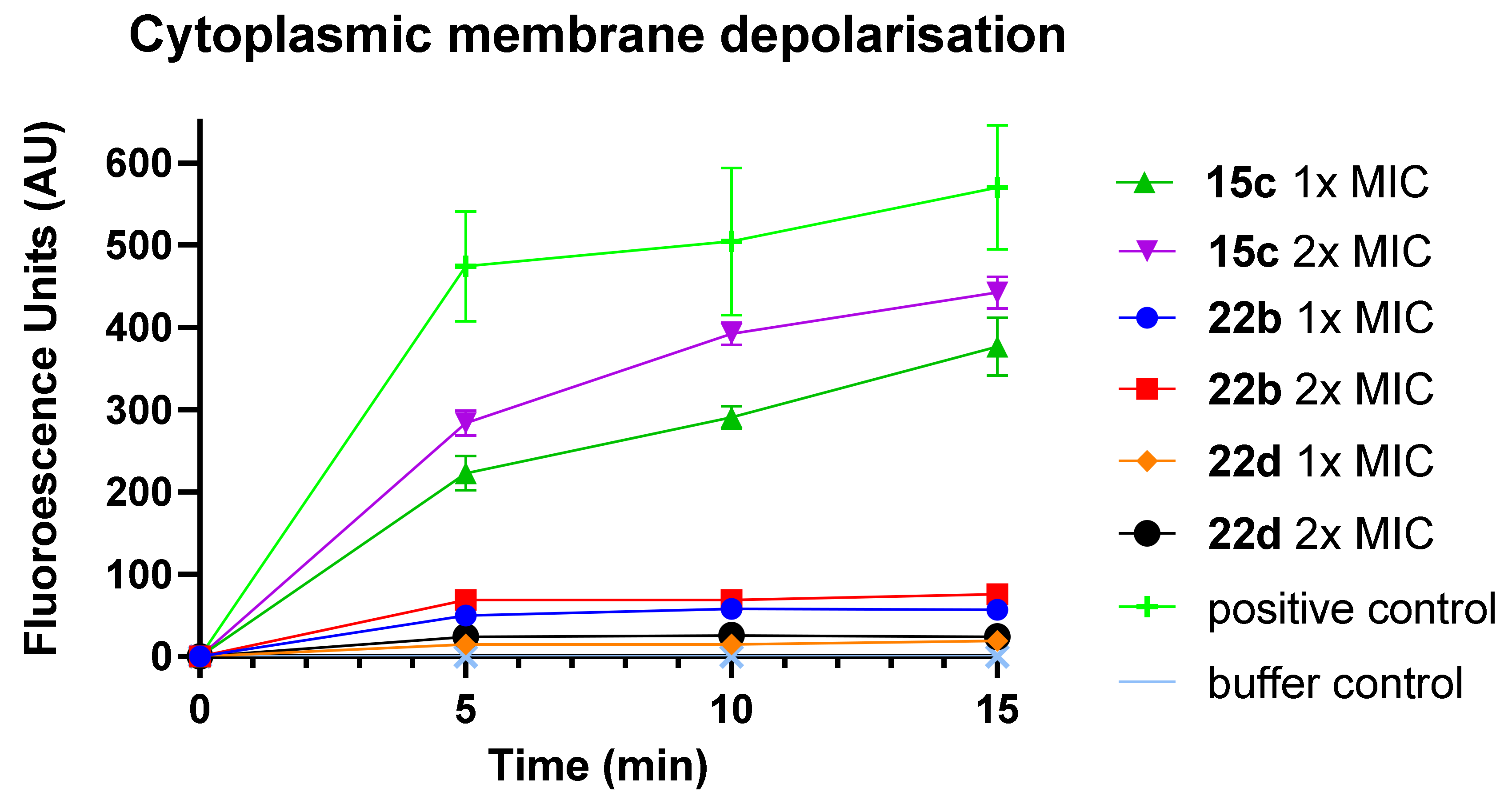
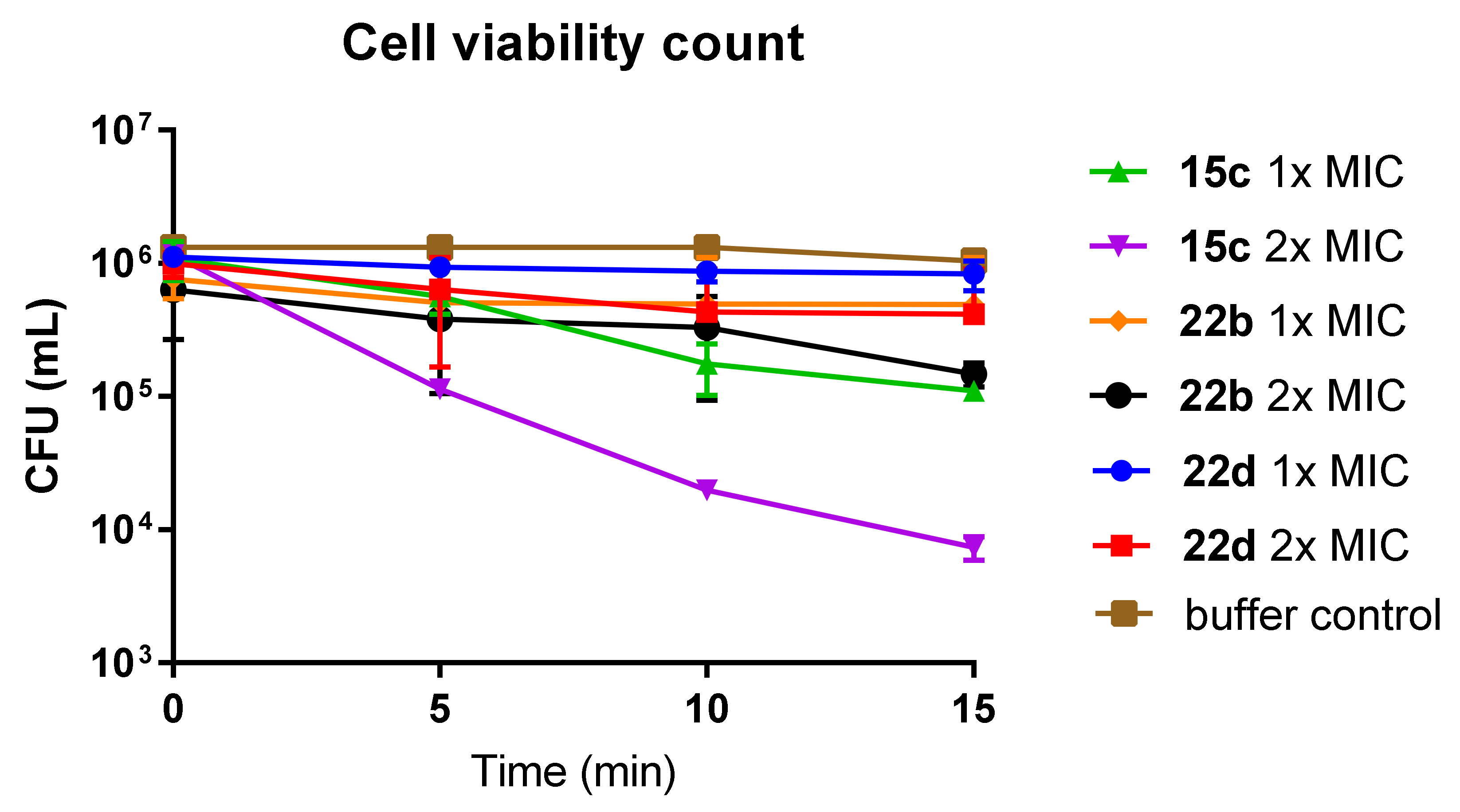
3-(2-(4’-Chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)-N,N,N-trimethylpropan-1-aminium iodide (15c)
The titled compound was synthesised from 2-(4’-chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-N-(3-(dimethylamino)propyl)-2-oxoacetamide 11c (35 mg, 0.065 mmol) and iodomethane (10 μL, 0.16 mmol) following general synthetic procedure E. The product was obtained as a yellow sticky solid (41 mg, 93%); 1H NMR (600 MHz, DMSO-d6): δ 10.09 (bs, 1H, NH), 8.97 (bs, 1H, NH), 8.03–7.96 (m, 2H, ArH), 7.72–7.67 (m, 2H, ArH), 7.60–7.53 (m, 3H, ArH), 3.39–3.30 (m, 4H, CH2), 3.17 (t, J = 7.7 Hz, 2H, CH2), 3.07 (s, 9H, CH3), 2.02–1.95 (m, 2H, CH2), 1.70–1.63 (m, 2H, CH2), 1.38–1.14 (m, 10H, CH2), 0.82 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 191.6 (CO), 163.6 (CO), 137.1 (ArC), 132.8 (ArC), 132.6 (ArCH), 130.0 (ArCH), 129.2 (ArC), 129.1 (ArCH), 128.3 (ArCH), 128.0 (ArC), 127.1 (ArC), 123.1 (ArCH), 63.5 (CH2), 52.3 (CH3), 51.2 (CH2), 35.9 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.9 (CH2), 22.6 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3854, 3807, 3691, 3650, 3629, 2924, 2854, 2360, 1978, 1654, 1648, 1578, 1560, 1508, 1481, 1400, 1330, 1274, 1195, 1141, 1092, 1012, 915, 818, 762, 719, 697, 669, 542, 512, 460, 452, 444, 435, 428, 420 cm−1; HRMS (+ ESI): Found m/z 550.2503 [M] +, C28H41ClN3O4S required 550.2501.
N,N,N-Trimethyl-3-(2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamido)propan-1-aminium iodide (15d)
The titled compound was synthesised from N-(3-(dimethylamino)propyl)-2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamide 11d (35 mg, 0.063 mmol) and iodomethane (10 μL, 0.16 mmol) following general synthetic procedure E. The product was obtained as a yellow sticky solid (40 mg, 90%); 1H NMR (600 MHz, DMSO-d6): δ 10.12 (bs, 1H, NH), 9.00 (t, J = 6.1 Hz, 1H, NH), 8.24 (d, J = 1.5 Hz, 1H, ArH), 8.17–8.13 (m, 2H, ArH), 8.06–8.00 (m, 2H, ArH), 7.98–7.95 (m, 1H, ArH), 7.84 (dd, J = 8.5, 1.9 Hz, 1H, ArH), 7.62 (d, J = 8.4 Hz, 1H, ArH), 7.59–7.54 (m, 2H, ArH), 3.41–3.33 (m, 4H, CH2), 3.19 (t, J = 7.7 Hz, 2H, CH2), 3.07 (s, 9H, CH3), 2.03–1.97 (m, 2H, CH2), 1.72–1.65 (m, 2H, CH2), 1.38–1.30 (m, 2H, CH2), 1.26–1.15 (m, 8H, CH2), 0.82 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 191.7 (CO), 163.7 (CO), 137.4 (ArC), 136.3 (ArC), 135.6 (ArC), 133.2 (ArC), 132.9 (ArCH), 132.4 (ArC), 130.2 (ArCH), 128.8 (ArCH), 128.2 (ArCH), 127.6 (ArCH), 127.5 (ArC), 126.7 (ArCH), 126.5 (ArCH), 125.3 (ArCH), 124.7 (ArCH), 123.3 (ArCH), 63.5 (CH2), 52.3 (CH3), 51.2 (CH2), 35.9 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.9 (CH2), 22.6 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3422, 3051, 2924, 2853, 2360, 1642, 1492, 1466, 1396, 1329, 1263, 1234, 1202, 1189, 1142, 1088, 915, 892, 860, 816, 749, 720, 669, 593, 560, 516, 476, 428 cm−1; HRMS ( + ESI): Found m/z 566.3048 [M] +, C32H44N3O4S required 566.3047.
3-(2-(5-Butyl-2-(octylsulfonamido)phenyl)-2-oxoacetamido)-N,N,N-trimethylpropan-1-aminium iodide (15e)
The titled compound was synthesised from 2-(5-butyl-2-(octylsulfonamido)phenyl)-N-(3-(dimethylamino)propyl)-2-oxoacetamide 11e (32 mg, 0.066 mmol) and iodomethane (10 μL, 0.17 mmol) following general synthetic procedure E. The product was obtained as a yellow sticky solid (35 mg, 85%); 1H NMR (600 MHz, DMSO-d6): δ 9.95 (bs, 1H, NH), 8.88 (t, J = 6.0 Hz, 1H, NH), 7.55–7.51 (m, 2H, ArH), 7.40–7.36 (m, 1H, ArH), 3.40–3.27 (m, 4H, CH2), 3.12–3.05 (m, 11H, CH2, CH3), 2.61 (t, J = 7.6 Hz, 2H, CH2), 2.01–1.94 (m, 2H, CH2), 1.66–1.59 (m, 2H, CH2), 1.58–1.50 (m, 2H, CH2), 1.35–1.15 (m, 12H, CH2), 0.89 (t, J = 7.5 Hz, 3H, CH3), 0.84 (t, J = 7.3 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 192.3 (CO), 164.1 (CO), 139.1 (ArC), 135.9 (ArC), 134.6 (ArCH), 131.5 (ArCH), 126.9 (ArC), 122.7 (ArCH), 63.5 (CH2), 52.3 (CH3), 50.9 (CH2), 35.8 (CH2), 33.8 (CH2), 32.9 (CH2), 31.1 (CH2), 28.3 (CH2), 28.3 (CH2), 27.3 (CH2), 22.9 (CH2), 22.6 (CH2), 22.0 (CH2), 21.7 (CH2), 13.9 (CH3), 13.7 (CH3); IR (ATR): νmax 3424, 2954, 2925, 2855, 2358, 1670, 1577, 1529, 1492, 1466, 1397, 1328, 1233, 1178, 1141, 1074, 914, 837, 775, 723, 668, 553, 509, 424 cm−1; HRMS (+ ESI): Found m/z 496.3202 [M] +, C26H44N3O4S required 496.3204.
3-(2-(4-(Butylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)-N,N,N-trimethylpropan-1-aminium iodide (16a)
The titled compound was synthesised from 2-(4-(butylsulfonamido)-[1,1’-biphenyl]-3-yl)-N-(3-(dimethylamino)propyl)-2-oxoacetamide 12a (34 mg, 0.076 mmol) and iodomethane (12 μL, 0.19 mmol) following general synthetic procedure E. The product was obtained as a yellow sticky solid (26 mg, 58%); 1H NMR (600 MHz, DMSO-d6): δ 10.08 (bs, 1H, NH), 8.98 (t, J = 5.8 Hz, 1H, NH), 8.02–7.98 (m, 2H, ArH), 7.68–7.64 (m, 2H, ArH), 7.60–7.56 (m, 1H, ArH), 7.52–7.47 (m, 2H, ArH), 7.43–7.39 (m, 1H, ArH), 3.39–3.30 (m, 4H, CH2), 3.21–3.16 (m, 2H, CH2), 3.07 (s, 9H, CH2), 2.02–1.95 (m, 2H, CH2), 1.70–1.63 (m, 2H, CH2), 1.41–1.33 (m, 2H, CH3), 0.85 (t, J = 7.4 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 191.8 (CO), 163.7 (CO), 138.3 (ArC), 132.7 (ArCH), 130.1 (ArCH), 129.2 (ArC), 129.2 (ArCH), 127.9 (ArCH), 126.9 (ArC), 126.5 (ArCH), 126.3 (ArC), 123.0 (ArCH), 63.5 (CH2), 52.3 (CH3), 51.0 (CH2), 35.9 (CH2), 25.0 (CH2), 22.6 (CH2), 20.7 (CH2), 13.5 (CH3); IR (ATR): νmax 3195, 3032, 2959, 2872, 2359, 1979, 1644, 1582, 1508, 1482, 1452, 1394, 1329, 1268, 1195, 1140, 1076, 920, 842, 762, 699, 681, 616, 584, 536, 428 cm−1; HRMS (+ ESI): Found m/z 460.2264 [M] +, C24H34N3O4S required 460.2265.
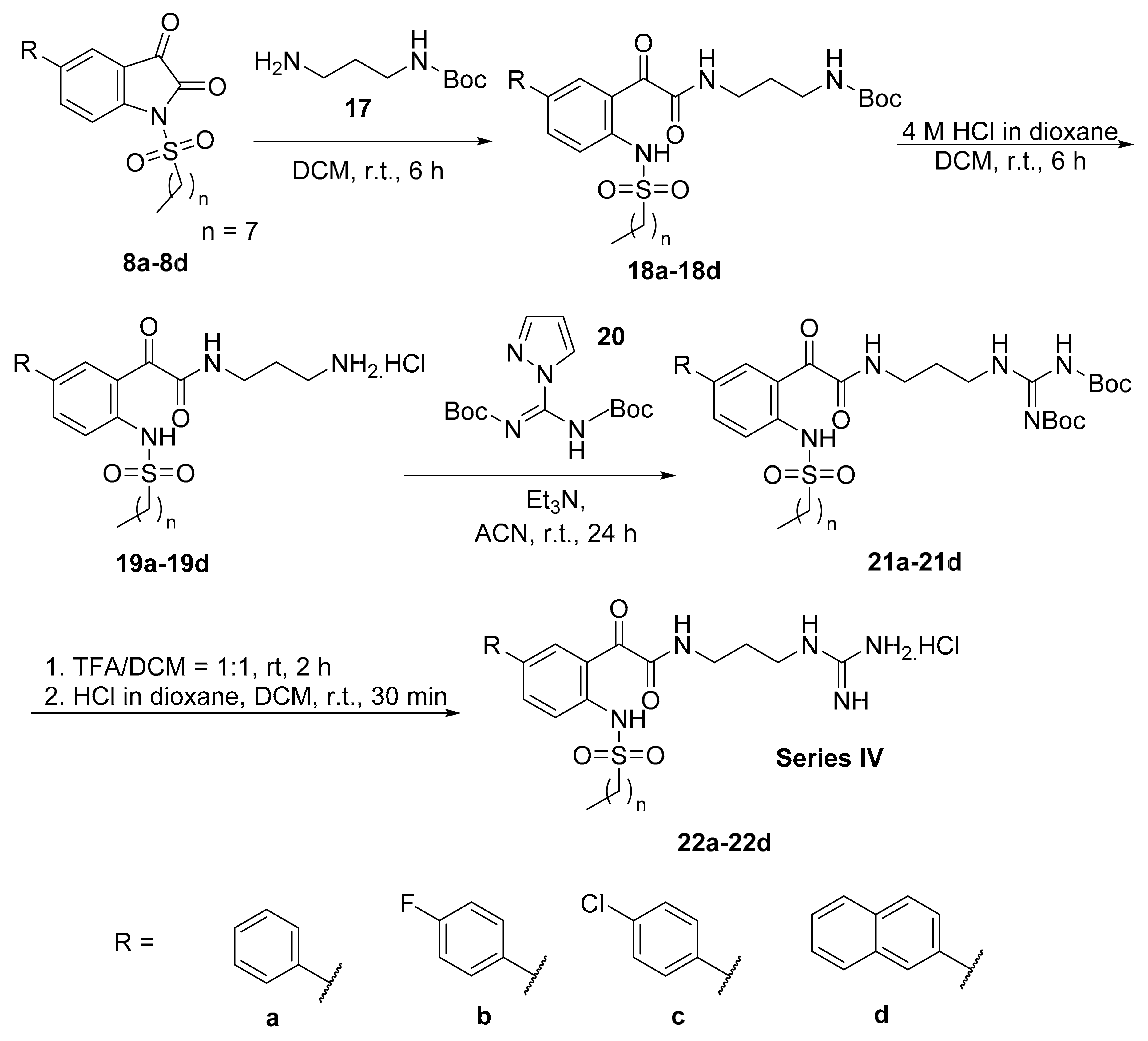
General Synthetic Procedure F for Boc-Protected Glyoxamide Derivatives
To a solution of N-sulfonylisatin (1.0 equivalent) in dichloromethane (10 mL), N-Boc-1,3-propandiamine (1.0 equivalent) in dichloromethane (5 mL) was added dropwise with stirring at 0 °C. The reaction mixture was stirred at room temperature for 6 h. After completion of the reaction, water was added to the reaction mixture and the product was extracted into dichloromethane (3 × 30 mL), washed with brine, dried over anhydrous sodium sulphate and concentrated in vacuo to afford the product.
tert-Butyl (3-(2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl)carbamate (18a)
The titled compound was synthesised from 1-(octylsulfonyl)-5-phenylindoline-2,3-dione 8a (0.32 g, 0.81 mmol) and N-Boc-1,3-propandiamine (0.15 g, 0.82 mmol) following general synthetic procedure F. The product was obtained as a yellow solid (0.45 g, 97%); mp 127.3–127.4 °C; 1H NMR (600 MHz, CDCl3): δ 10.48 (bs, 1H, NH), 8.75 (s, 1H, ArH), 7.88–7.81 (m, 2H, ArH), 7.71 (bs, 1H, NH), 7.59–7.55 (m, 2H, ArH), 7.47–7.43 (m, 2H, ArH), 7.39–7.35 (m, 1H, ArH), 4.81 (bs, 1H, NH), 3.48 (q, J = 6.4 Hz, 2H, CH2), 3.27–3.21 (m, 2H, CH2), 3.21–3.16 (m, 2H, CH2), 1.85–1.78 (m, 2H, CH2), 1.78–1.73 (m, 2H, CH2), 1.44 (s, 9H, CH3), 1.42–1.34 (m, 2H, CH2), 1.30–1.16 (m, 8H, CH2), 0.85 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3): δ 191.5 (CO), 162.9 (CO), 156.9 (CO), 141.1 (ArC), 139.1 (ArC), 135.7 (ArC), 135.1 (ArCH), 133.7 (ArCH), 129.2 (ArCH), 127.9 (ArCH), 126.9 (ArCH), 119.3 (ArC), 118.5 (ArCH), 79.9 (C), 52.8 (CH2), 37.3 (CH2), 36.4 (CH2), 31.8 (CH2), 30.2 (CH2), 29.1 (CH2), 29.0 (CH2), 28.5 (CH3), 28.2 (CH2), 23.5 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3352, 3310, 2919, 2359, 1682, 1666, 1582, 1518, 1483, 1443, 1386, 1364, 1346, 1325, 1294, 1246, 1197, 1171, 1155, 1124, 1069, 1041, 1011, 977, 968, 906, 894, 851, 830, 804, 760, 741, 714, 684, 639, 609, 554, 537, 491, 453, 423 cm−1; HRMS (+ ESI): Found m/z 596.2764 [M + Na] +, C30H43N3O6SNa required 596.2764.
tert-Butyl (3-(2-(4’-fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl) carbamate (18b)
The titled compound was synthesised from 5-(4-fluorophenyl)-1-(octylsulfonyl)indoline-2,3-dione 8b (0.28 g, 0.67 mmol) and N-Boc-1,3-propandiamine (0.12 g, 0.67 mmol) following general synthetic procedure F. The product was obtained as a yellow solid (0.38 g, 96%); mp 138.0–140.3 °C; 1H NMR (400 MHz, CDCl3): δ 10.46 (bs, 1H, NH), 8.72 (s, 1H, ArH), 7.85 (d, J = 8.7 Hz, 1H, ArH), 7.81–7.69 (m, 2H, NH, ArH), 7.56–7.50 (m, 2H, ArH), 7.17–7.09 (m, 2H, ArH), 4.81 (t, J = 5.9 Hz, 1H, NH), 3.47 (q, J = 6.4 Hz, 2H, CH2), 3.29–3.14 (m, 4H, CH2), 1.86–1.71 (m, 4H, CH2), 1.44 (s, 9H, CH3), 1.42–1.33 (m, 2H, CH2), 1.31–1.18 (m, 8H, CH2), 0.85 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 191.3 (CO), 162.8 (CO), 162.8 (ArC), 156.9 (CO), 141.1 (ArC), 135.2 (ArC), 134.9 (ArCH), 134.7 (ArC), 133.5 (ArCH), 128.6 (ArCH), 119.3 (ArC), 118.6 (ArCH), 116.1 (ArCH), 79.9 (C), 52.8 (CH2), 37.3 (CH2), 36.4 (CH2), 31.8 (CH2), 30.2 (CH2), 29.1 (CH2), 29.0 (CH2), 28.5 (CH3), 28.2 (CH2), 23.5 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3357, 3313, 2923, 2856, 2303, 1887, 1680, 1645, 1519, 1487, 1388, 1344, 1247, 1155, 1070, 1011, 976, 906, 854, 826, 771 cm−1; HRMS (+ ESI): Found m/z 614.2670 [M + Na] +, C30H42FN3O6SNa required 614.2671.
tert-Butyl (3-(2-(4’-chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl) carbamate (18c)
The titled compound was synthesised from 5-(4-chlorophenyl)-1-(octylsulfonyl)indoline-2,3-dione 8c (0.30 g, 0.69 mmol) and N-Boc-1,3-propandiamine (0.13 g, 0.71 mmol) following general synthetic procedure F. The product was obtained as a yellow solid (0.41 g, 97%); mp 134.7–135.1 °C; 1H NMR (600 MHz, CDCl3): δ 10.49 (bs, 1H, NH), 8.74 (s, 1H, ArH), 7.85 (d, J = 8.7 Hz, 1H, ArH), 7.78 (dd, J = 8.7, 2.3 Hz, 1H, ArH), 7.77 (bs, 1H, NH), 7.51–7.48 (m, 2H, ArH), 7.43–7.40 (m, 2H, ArH), 4.81 (bs, 1H, NH), 3.47 (q, J = 6.4 Hz, 2H, CH2), 3.27–3.22 (m, 2H, CH2), 3.20–3.16 (m, 2H, CH2), 1.84–1.72 (m, 4H, CH2), 1.44 (s, 9H, CH3), 1.41–1.34 (m, 2H, CH2), 1.29–1.18 (m, 8H, CH2), 0.85 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3): δ 191.3 (CO), 162.7 (CO), 156.9 (CO), 141.3 (ArC), 137.5 (ArC), 134.8 (ArCH), 134.4 (ArC), 134.1 (ArC), 133.5 (ArCH), 129.3 (ArCH), 128.2 (ArCH), 119.3 (ArC), 118.6 (ArCH), 79.9 (C), 52.8 (CH2), 37.3 (CH2), 36.4 (CH2), 31.8 (CH2), 30.2 (CH2), 29.1 (CH2), 29.0 (CH2), 28.5 (CH3), 28.2 (CH2), 23.5 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3352, 3311, 2922, 2855, 2359, 1979, 1682, 1665, 1643, 1581, 1519, 1483, 1443, 1388, 1364, 1345, 1246, 1197, 1155, 1138, 1093, 1070, 1041, 1010, 975, 907, 895, 882, 864, 817, 759, 741, 714, 685, 643, 609, 555, 537, 492, 471, 418 cm−1; HRMS (+ ESI): Found m/z 630.2379 [M + Na] +, C30H42ClN3O6SNa required 630.2375.
tert-Butyl (3-(2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamido)propyl) carbamate (18d)
The titled compound was synthesised from 5-(naphthalen-2-yl)-1-(octylsulfonyl)indoline-2,3-dione 8d (0.31 g, 0.70 mmol) and N-Boc-1,3-propandiamine (0.13 g, 0.70 mmol) following general synthetic procedure F. The product was obtained as a yellow solid (0.41 g, 95%); mp 78.9–80.0 °C; 1H NMR (600 MHz, CDCl3): δ 10.51 (bs, 1H, NH), 8.87 (s, 1H, ArH), 8.01 (s, 1H, ArH), 7.99–7.83 (m, 5H, ArH), 7.79–7.67 (m, 2H, NH, ArH), 7.55–7.46 (m, 2H, ArH), 4.84 (bs, 1H, NH), 3.49 (q, J = 6.5 Hz, 2H, CH2), 3.30–3.16 (m, 4H, CH2), 1.88–1.72 (m, 4H, CH2), 1.44 (s, 9H, CH3), 1.46–1.34 (m, 2H, CH2), 1.31–1.16 (m, 8H, CH2), 0.85 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3): δ 191.5 (CO), 162.9 (CO), 156.9 (CO), 141.1 (ArC), 136.3 (ArC), 135.6 (ArC), 135.4 (ArCH), 133.8 (ArCH), 133.7 (ArC), 132.9 (ArC), 128.9 (ArCH), 128.4 (ArCH), 127.8 (ArCH), 126.7 (ArCH), 126.4 (ArCH), 125.7 (ArCH), 125.0 (ArCH), 119.4 (ArC), 118.6 (ArCH), 79.9 (C), 52.8 (CH2), 37.3 (CH2), 36.5 (CH2), 31.8 (CH2), 30.2 (CH2), 29.1 (CH2), 29.0 (CH2), 28.5 (CH3), 28.2 (CH2), 23.6 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3346, 3055, 2925, 2855, 2285, 2081, 1911, 1683, 1636, 1572, 1497, 1466, 1393, 1365, 1336, 1247, 1140, 1012, 917, 890, 813, 747 cm−1; HRMS (+ ESI): Found m/z 646.2919 [M + Na] +, C34H45N3O6SNa required 646.2921.
General Synthetic Procedure G for Aminoglyoxamides
To a solution of Boc-protected glyoxamide (1.0 equivalent) in dichloromethane (10 mL), 4 M HCl/dioxane (3 mL) was added. The reaction mixture was stirred at room temperature for 6 h. After completion of reaction, the reaction mixture was concentrated in vacuo, washed thrice with diethyl ether and dried under high vacuum to afford the product.
N-(3-Aminopropyl)-2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide hydrochloride (19a)
The titled compound was synthesised from tert-butyl (3-(2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl)carbamate 18a (0.35 g, 0.61 mmol) following general synthetic procedure G. The product was obtained as a yellow solid (0.20 g, 64%); mp 126.2–128.7 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.20 (bs, 1H, NH), 9.05 (t, J = 6.0 Hz, 1H, NH), 8.19–7.91 (m, 5H, NH, ArH), 7.66–7.61 (m, 3H, ArH), 7.51 (t, J = 7.8 Hz, 2H, ArH), 7.41 (t, J = 7.2 Hz, 1H, ArH), 3.38–3.29 (m, 2H, CH2), 3.29–3.22 (m, 2H, CH2), 2.91–2.82 (m, 2H, CH2), 1.89–1.79 (m, 2H, CH2), 1.72–1.62 (m, 2H, CH2), 1.39–1.14 (m, 10H, CH2), 0.82 (t, J = 7.0 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 192.4 (CO), 163.8 (CO), 138.3 (ArC), 138.2 (ArC), 135.6 (ArC), 133.2 (ArCH), 130.5 (ArCH), 129.2 (ArCH), 127.9 (ArCH), 126.4 (ArCH), 124.5 (ArC), 121.7 (ArCH), 51.4 (CH2), 36.7 (CH2), 35.9 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 26.9 (CH2), 22.9 (CH2), 22.0 (CH2), 14.2 (CH3); IR (ATR): νmax 3031, 2921, 2853, 2045, 1961, 1635, 1509, 1482, 1393, 1335, 1264, 1196, 1138, 1025, 917, 838, 759, 696 cm−1; HRMS (+ ESI): Found m/z 474.2419 [M + H] +, C25H36N3O4S required 474.2421.
N-(3-Aminopropyl)-2-(4’-fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide hydrochloride (19b)
The titled compound was synthesised from tert-butyl (3-(2-(4’-fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl) carbamate 18b (0.34 g, 0.58 mmol) following general synthetic procedure G. The product was obtained as a yellow solid (0.27 g, 90%); mp 155.2–157.8 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.19 (bs, 1H, NH), 9.05 (t, J = 5.9 Hz, 1H, NH), 8.14–7.93 (m, 5H, NH, ArH), 7.72–7.65 (m, 2H, ArH), 7.65–7.59 (m, 1H, ArH), 7.34 (t, J = 8.9 Hz, 2H, ArH), 3.37–3.29 (m, 2H, CH2), 3.28–3.21 (m, 2H, CH2), 2.92–2.80 (m, 2H, CH2), 1.90–1.79 (m, 2H, CH2), 1.72–1.61 (m, 2H, CH2), 1.38–1.14 (m, 10H, CH2), 0.82 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 192.3 (CO), 163.7 (CO), 162.1 (ArC), 138.2 (ArC), 134.7 (ArC), 134.6 (ArC), 133.0 (ArCH), 130.3 (ArCH), 128.5 (ArCH), 124.7 (ArC), 121.8 (ArCH), 116.0 (ArCH), 51.4 (CH2), 36.6 (CH2), 35.9 (CH2), 31.1 (CH2), 28.3 (CH2), 28.3 (CH2), 27.3 (CH2), 26.9 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3365, 3161, 2970, 2925, 2753, 2611, 2494, 2343, 2058, 1919, 1633, 1600, 1528, 1491, 1393, 1334, 1259, 1203, 1140, 1083, 1021, 919, 868, 823, 761 cm−1; HRMS (+ ESI): Found m/z 492.2325 [M + H] +, C25H35FN3O4S required 492.2327.
N-(3-Aminopropyl)-2-(4’-chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide hydrochloride (19c)
The titled compound was synthesised from tert-butyl (3-(2-(4’-chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl) carbamate 18c (0.38 g, 0.62 mmol) following general synthetic procedure G. The product was obtained as a yellow solid (0.28 g, 83%); mp 118.8–119.1 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.19 (bs, 1H, NH), 9.04 (t, J = 6.0 Hz, 1H, NH), 8.12–7.88 (m, 5H, NH, ArH), 7.71–7.60 (m, 3H, ArH), 7.57 (d, J = 8.5 Hz, 2H, ArH), 3.38–3.29 (m, 2H, CH2), 3.28–3.21 (m, 2H, CH2), 2.92–2.81 (m, 2H, CH2), 1.89–1.79 (m, 2H, CH2), 1.72–1.61 (m, 2H, CH2), 1.39–1.13 (m, 10H, CH2), 0.82 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 192.2 (CO), 163.7 (CO), 138.4 (ArC), 137.0 (ArC), 134.3 (ArC), 133.0 (ArCH), 132.8 (ArC), 130.4 (ArCH), 129.2 (ArCH), 128.3 (ArCH), 124.8 (ArC), 121.9 (ArCH), 51.5 (CH2), 36.7 (CH2), 35.9 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 26.9 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3808, 2924, 2855, 2360, 2038, 1657, 1636, 1578, 1527, 1509, 1483, 1397, 1340, 1270, 1201, 1139, 1095, 1046, 1010, 919, 875, 815, 772, 700, 668, 596, 562, 494 cm−1; HRMS (+ ESI): Found m/z 508.2033 [M + H] +, C25H35ClN3O4S required 508.2031.
N-(3-Aminopropyl)-2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamide hydrochloride (19d)
The titled compound was synthesised from tert-Butyl (3-(2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamido)propyl) carbamate 18d (0.37 g, 0.59 mmol) following general synthetic procedure G. The product was obtained as a yellow solid (0.22 g, 67%); mp 85.2–87.3 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.23 (bs, 1H, NH), 9.07 (t, J = 5.9 Hz, 1H, NH), 8.22 (s, 1H, ArH), 8.18–8.12 (m, 2H, ArH), 8.10–7.93 (m, 6H, NH, ArH), 7.81 (dd, J = 8.6, 1.8 Hz, 1H, ArH), 7.68 (dd, J = 6.4, 2.8 Hz, 1H, ArH),7.60–7.52 (m, 2H, ArH), 3.40–3.31 (m, 2H, CH2), 3.29–3.22 (m, 2H, CH2), 2.92–2.82 (m, 2H, CH2), 1.92–1.82 (m, 2H, CH2), 1.74–1.63 (m, 2H, CH2), 1.39–1.14 (m, 10H, CH2), 0.81 (t, J = 7.0 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 192.3 (CO), 163.8 (CO), 138.2 (ArC), 135.6 (ArC), 133.2 (ArC), 133.2 (ArCH), 132.3 (ArC), 130.6 (ArCH), 128.9 (ArCH), 128.3 (ArCH), 127.6 (ArC), 127.6 (ArCH), 126.6 (ArCH), 126.4 (ArCH), 125.2 (ArC), 125.2 (ArCH), 124.6 (ArCH), 122.1 (ArCH), 51.4 (CH2), 36.7 (CH2), 35.9 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 26.9 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax3312, 3176, 2921, 2852, 2321, 2050, 1922, 1635, 1494, 1463, 1396, 1333, 1264, 1200, 1138, 1016, 916, 812, 747 cm−1; HRMS ( + ESI): Found m/z 524.2579 [M + H] +, C29H38N3O4S required 524.2578.
General Synthetic Procedure H for Boc-Protected Guanidine Glyoxamides
To a solution of aminoglyoxamides (1.0 equivalent) and N,N’-di-Boc-1H-pyrazole-1- carboxamidine (1.3 equivalents) in acetonitrile (10 mL), triethylamine (2.5 equivalents) in acetonitrile (5 mL) was added dropwise with stirring at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at room temperature for 18 h. After completion of the reaction, the reaction mixture was concentrated in vacuo. The product was purified by flash chromatography on silica using ethyl acetate/n-hexane (1:4) as eluent to afford the product.
(E)-1-tert-Butyl-N-(N’-((tert-butyloxidanyl)carbonyl)-N-(3-(2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl)carbamimidoyl)-1-oxidanecarboxamide (21a)
The titled compound was synthesised from N-(3-aminopropyl)-2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide hydrochloride 19a (0.16 g, 0.30 mmol), N,N’-di-Boc-1H-pyrazole-1-carboxamidine (0.14 g, 0.45 mmol) and triethylamine (0.12 mL, 0.83 mmol) following general synthetic procedure H. The product was obtained as a yellow solid (68 mg, 31%); mp 69.9–70.1 °C; 1H NMR (400 MHz, CDCl3): δ 11.47 (bs, 1H, NH), 10.62 (bs, 1H, NH), 8.68–8.50 (m, 3H, NH, ArH), 7.89–7.80 (m, 2H, ArH), 7.57 (d, J = 7.5 Hz, 2H, ArH), 7.45 (t, J = 7.9 Hz, 2H, ArH), 7.36 (t, J = 7.1 Hz, 1H, ArH), 3.62–3.52 (m, 2H, CH2), 3.46 (q, J = 6.4 Hz, 2H, CH2), 3.21–3.13 (m, 2H, CH2), 1.87–1.76 (m, 4H, CH2), 1.50 (s, 9H, CH3), 1.38 (s, 9H, CH3), 1.43–1.18 (m, 10H, CH2), 0.85 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3): δ 192.6 (CO), 163.7 (CO), 163.0 (CN), 157.4 (CO), 153.3 (CO), 141.2 (ArC), 139.1 (ArC), 135.6 (ArC), 135.0 (ArCH), 133.5 (ArCH), 129.1 (ArCH), 127.9 (ArCH), 127.0 (ArCH), 119.0 (ArC), 118.4 (ArCH), 83.8 (C), 79.9 (C), 52.8 (CH2), 37.3 (CH2), 35.9 (CH2), 31.8 (CH2), 30.1 (CH2), 29.1 (CH2), 29.0 (CH2), 28.4 (CH2), 28.3 (CH3), 28.2 (CH3), 23.6 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3323, 2928, 2360, 1979, 1720, 1638, 1571, 1508, 1483, 1450, 1410, 1366, 1328, 1284, 1228, 1195, 1131, 1051, 1026, 978, 907, 855, 806, 760, 697, 681, 616, 587, 562, 537, 418 cm−1; HRMS (+ ESI): Found m/z 716.3684 [M + H] +, C36H54N5O8S required 716.3688.
(E)-1-tert-Butyl-N-(N’-((tert-butyloxidanyl)carbonyl)-N-(3-(2-(4’-fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl)carbamimidoyl)-1-oxidanecarboxamide (21b)
The titled compound was synthesised from N-(3-aminopropyl)-2-(4’-fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide hydrochloride 19b (0.24 g, 0.45 mmol), N,N’-di-Boc-1H-pyrazole-1-carboxamidine (0.20 g, 0.64 mmol) and triethylamine (0.16 mL, 1.11 mmol) following general synthetic procedure H. The product was obtained as a yellow solid (0.25, 77%); mp 62.8–65.0 °C; 1H NMR (400 MHz, CDCl3): δ 11.47 (bs, 1H, NH), 10.61 (bs, 1H, NH), 8.64 (t, J = 6.0 Hz, 1H, NH), 8.58 (bs, 1H, NH), 8.52 (d, J = 2.1 Hz, 1H, ArH), 7.85 (t, J = 7.9 Hz, 1H, ArH), 7.76 (dd, J = 8.7, 2.2 Hz, 1H, ArH), 7.56–7.49 (m, 2H, ArH), 7.13 (t, J = 8.6 Hz, 2H, ArH), 3.63–3.53 (m, 2H, CH2), 3.46 (q, J = 6.2 Hz, 2H, CH2), 3.20–3.13 (m, 2H, CH2), 1.86–1.76 (m, 4H, CH2), 1.50 (s, 9H, CH3), 1.38 (s, 9H, CH3), 1.43–1.16 (m, 10H, CH2), 0.85 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 192.3 (CO), 163.6 (CO), 162.8 (ArC), 162.7 (CN), 157.3 (CO), 153.3 (CO), 141.1 (ArC), 135.3 (ArC), 134.7 (ArCH), 134.7 (ArC), 133.3 (ArCH), 128.6 (ArCH), 119.1 (ArC), 118.5 (ArCH), 116.1 (ArCH), 83.9 (C), 80.1 (C), 52.8 (CH2), 37.3 (CH2), 35.9 (CH2), 31.8 (CH2), 30.0 (CH2), 29.1 (CH2), 29.0 (CH2), 28.4 (CH2), 28.2 (CH3), 28.2 (CH3), 23.5 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3324, 2926, 2855, 2322, 1890, 1720, 1638, 1570, 1487, 1408, 1326, 1258, 1225, 1128, 1049, 906, 858, 800, 731, 669 cm−1; HRMS ( + ESI): Found m/z 734.3594 [M + H] +, C36H53FN5O8S required 734.3593.
(E)-1-tert-Butyl-N-(N’-((tert-butyloxidanyl)carbonyl)-N-(3-(2-(4’-chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl)carbamimidoyl)-1-oxidanecarboxamide (21c)
The titled compound was synthesised from N-(3-aminopropyl)-2-(4’-chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide hydrochloride 19c (0.16 g, 0.29 mmol), N,N’-di-Boc-1H-pyrazole-1-carboxamidine (0.12 g, 0.38 mmol) and triethylamine (0.10 mL, 0.72 mmol) following general synthetic procedure H. The product was obtained as a yellow solid (0.14 g, 64%); mp 77.1–77.4 °C; 1H NMR (400 MHz, CDCl3): δ 11.47 (bs, 1H, NH), 10.63 (bs, 1H, NH), 8.65 (t, J = 6.3 Hz, 1H, NH), 8.61–8.51 (m, 2H, NH, ArH), 7.86 (d, J = 8.7 Hz, 1H, ArH), 7.77 (dd, J = 8.7, 2.2 Hz, 1H, ArH), 7.52–7.47 (m, 2H, ArH), 7.43–7.39 (m, 2H, ArH), 3.62–3.52 (m, 2H, CH2), 3.46 (q, J = 6.3 Hz, 2H, CH2), 3.20–3.13 (m, 2H, CH2), 1.86–1.75 (m, 4H, CH2), 1.50 (s, 9H, CH3), 1.38 (s, 9H, CH3), 1.43–1.18 (m, 10H, CH2), 0.85 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3): δ 192.3 (CO), 163.5 (CO), 163.1 (CN), 157.5 (CO), 153.4 (CO), 141.5 (ArC), 137.6 (ArC), 134.7 (ArCH), 134.3 (ArC), 134.1 (ArC), 133.4 (ArCH), 129.3 (ArCH), 128.2 (ArCH), 118.9 (ArC), 118.4 (ArCH), 83.8 (C), 79.8 (C), 52.9 (CH2), 37.2 (CH2), 35.9 (CH2), 31.8 (CH2), 30.1 (CH2), 29.1 (CH2), 29.0 (CH2), 28.4 (CH2), 28.3 (CH3), 28.2 (CH3), 23.6 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3322, 2929, 1720, 1637, 1577, 1507, 1482, 1409, 1367, 1328, 1285, 1253, 1228, 1195, 1131, 1094, 1050, 1026, 1012, 979, 907, 856, 817, 770, 698, 657, 592, 562, 489, 419 cm−1; HRMS (+ ESI): Found m/z 772.3120 [M + Na] +, C36H52ClN5O8SNa required 772.3117.
(E)-1-tert-Butyl-N-(N’-((tert-butyloxidanyl)carbonyl)-N-(3-(2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamido)propyl)carbamimidoyl)-1-oxidanecarboxamide (21d)
The titled compound was synthesised from N-(3-Aminopropyl)-2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamide hydrochloride 19d (0.18 g, 0.33 mmol), N,N’-di-Boc-1H-pyrazole-1-carboxamidine (0.13 g, 0.40 mmol) and triethylamine (0.12 mL, 0.83 mmol) following general synthetic procedure H. The product was obtained as a yellow solid (0.13 g, 52%); mp 71.6–73.5 °C; 1H NMR (400 MHz, CDCl3): δ 11.47 (bs, 1H, NH), 10.66 (bs, 1H, NH), 8.71–8.53 (m, 3H, NH, ArH), 8.01 (s, 1H, ArH), 7.98–7.84 (m, 5H, ArH), 7.71 (dd, J = 8.5, 1.7 Hz, 1H, ArH), 7.55–7.46 (m, 2H, ArH), 3.64–3.54 (m, 2H, CH2), 3.48 (q, J = 6.2 Hz, 2H, CH2), 3.23–3.15 (m, 2H, CH2), 1.89–1.76 (m, 4H, CH2), 1.50 (s, 9H, CH3), 1.39 (s, 9H, CH3), 1.44–1.17 (m, 10H, CH2), 0.85 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 192.6 (CO), 163.8 (CO), 162.8 (CN), 157.3 (CO), 153.3 (CO), 141.2 (ArC), 136.4 (ArC), 135.6 (ArC), 135.2 (ArC), 133.7 (ArCH), 133.7 (ArCH), 132.9 (ArC), 128.9 (ArCH), 128.3 (ArCH), 127.8 (ArCH), 126.7 (ArCH), 126.4 (ArCH), 125.7 (ArCH), 125.1 (ArCH), 119.2 (ArC), 118.5 (ArCH), 83.9 (C), 80.2 (C), 52.8 (CH2), 37.5 (CH2), 35.9 (CH2), 31.8 (CH2), 30.0 (CH2), 29.1 (CH2), 29.0 (CH2), 28.3 (CH2), 28.3 (CH3), 28.2 (CH3), 23.6 (CH2), 22.7 (CH2), 14.2 (CH3); IR (ATR): νmax 3323, 2928, 2360, 1979, 1720, 1638, 1571, 1508, 1483, 1450, 1410, 1366, 1328, 1284, 1228, 1195, 1131, 1051, 1026, 978, 907, 855, 806, 760, 697, 681, 616, 587, 562, 537, 418 cm−1; HRMS (+ ESI): Found m/z 766.3845 [M + H] +, C40H56N5O8S required 766.3844.
General Synthetic Procedure I for Guanidinium Hydrochloride Salts
To a solution of Boc-protected guanidine glyoxamide (1.0 equivalent) in dichloromethane (1 mL), trifluoroacetic acid (1 mL) was added. The reaction mixture was stirred at room temperature for 3 h. After completion of the reaction, the reaction mixture was concentrated in vacuo and washed thrice with diethyl ether. To the residue in dichloromethane (1 mL), 4 M HCl/dioxane (1 mL) was added. The reaction mixture was stirred at room temperature for 30 min. After completion of reaction, the reaction mixture was concentrated in vacuo, washed thrice with diethyl ether and freeze-dried to afford the product.
N-(3-Guanidinopropyl)-2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamide hydrochloride (22a)
The titled compound was synthesised from (E)-1-tert-Butyl-N-(N’-((tert-butyloxidanyl)carbonyl)-N-(3-(2-(4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl)carbamimidoyl)-1-oxidanecarboxamide 21a (40 mg, 0.056 mmol) following general synthetic procedure I. The product was obtained as a yellow sticky solid (0.15 g, 50%); 1H NMR (600 MHz, DMSO-d6): δ 10.20 (bs, 1H, NH), 9.01 (t, J = 5.7 Hz, 1H, NH), 8.03–7.99 (m, 2H, ArH), 7.69–7.61 (m, 4H, NH, ArH), 7.57–6.78 (m, 7H, NH, ArH), 3.30 (q, J = 6.5 Hz, 2H, CH2), 3.28–3.23 (m, 2H, CH2), 3.19 (q, J = 6.5 Hz, 2H, CH2), 1.79–1.63 (m, 4H, CH2), 1.37–1.29 (m, 2H, CH2), 1.26–1.15 (m, 8H, CH2), 0.82 (t, J = 7.1 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 192.6 (CO), 163.8 (CO), 156.9 (CN), 138.3 (ArC), 138.2 (ArC), 135.6 (ArC), 133.2 (ArCH), 130.5 (ArCH), 129.2 (ArCH), 127.9 (ArCH), 126.4 (ArCH), 124.5 (ArC), 121.6 (ArCH), 51.4 (CH2), 38.4 (CH2), 36.1 (CH2), 31.1 (CH2), 28.4 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3364, 3163, 2924, 2654, 2360, 1626, 1581, 1528, 1510, 1485, 1459, 1395, 1345, 1265, 1198, 1139, 1067, 924, 909, 849, 759, 683, 621, 587, 560, 530, 481, 424 cm−1; HRMS (+ ESI): Found m/z 516.2636 [M + H] +, C26H38N5O4S required 516.2639.
2-(4’-Fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-N-(3-guanidinopropyl)-2-oxoacetamide hydrochloride (22b)
The titled compound was synthesised from (E)-1-tert-Butyl-N-(N’-((tert-butyloxidanyl)carbonyl)-N-(3-(2-(4’-fluoro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl)carbamimidoyl)-1-oxidanecarboxamide 21b (0.10 g, 0.13 mmol) following general synthetic procedure I. The product was obtained as a yellow sticky solid (49 mg, 61%); 1H NMR (600 MHz, DMSO-d6): δ 10.19 (bs, 1H, NH), 9.01 (t, J = 5.9 Hz, 1H, NH), 8.00–7.96 (m, 2H, ArH), 7.74 (t, J = 5.8 Hz, 1H, NH), 7.71–7.66 (m, 2H, ArH), 7.64–7.60 (m, 1H, ArH), 7.58–6.80 (m, 6H, NH, ArH), 3.30 (q, J = 6.7 Hz, 2H, CH2), 3.26–3.22 (m, 2H, CH2), 3.20 (q, J = 6.7 Hz, 2H, CH2), 1.77–1.71 (m, 2H, CH2), 1.70–1.63 (m, 2H, CH2), 1.36–1.30 (m, 2H, CH2), 1.26–1.15 (m, 8H, CH2), 0.82 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 192.4 (CO), 163.7 (CO), 162.1 (ArC), 157.0 (CN), 138.2 (ArC), 134.7 (ArC), 134.6 (ArC), 133.0 (ArCH), 130.4 (ArCH), 128.5 (ArCH), 124.6 (ArC), 121.8 (ArCH), 116.0 (ArCH), 51.4 (CH2), 38.4 (CH2), 36.1 (CH2), 31.1 (CH2), 28.4 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3313, 3155, 2923, 2853, 2292, 1910, 1639, 1487, 1396, 1330, 1195, 1137, 913, 826, 722 cm−1; HRMS (+ ESI): Found m/z 534.2544 [M + H] +, C26H37FN5O4S required 534.2545.
2-(4’-Chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-N-(3-guanidinopropyl)-2-oxoacetamide hydrochloride (22c)
The titled compound was synthesised from (E)-1-tert-butyl-N-(N’-((tert-butyloxidanyl)carbonyl)-N-(3-(2-(4’-chloro-4-(octylsulfonamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)propyl)carbamimidoyl)-1-oxidanecarboxamide 21c (91 mg, 0.12 mmol) following general synthetic procedure I. The product was obtained as a yellow sticky solid (43 mg, 60%); 1H NMR (600 MHz, DMSO-d6): δ 10.20 (bs, 1H, NH), 9.00 (t, J = 5.8 Hz, 1H, NH), 8.03–7.98 (m, 2H, ArH), 7.71–7.66 (m, 3H, NH, ArH), 7.63 (dd, J = 7.6, 1.5 Hz, 1H, ArH), 7.58–7.54 (m, 2H, ArH), 7.52–6.84 (bs, 4H, NH), 3.30 (q, J = 6.7 Hz, 2H, CH2), 3.27–3.22 (m, 2H, CH2), 3.19 (q, J = 6.6 Hz, 2H, CH2), 1.77–1.71 (m, 2H, CH2), 1.70–1.63 (m, 2H, CH2), 1.36–1.30 (m, 2H, CH2), 1.26–1.15 (m, 8H, CH2), 0.82 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 192.3 (CO), 163.7 (CO), 156.9 (CN), 138.5 (ArC), 137.1 (ArC), 134.2 (ArC), 133.0 (ArCH), 132.8 (ArC), 130.4 (ArCH), 129.2 (ArCH), 128.2 (ArCH), 124.7 (ArC), 121.8 (ArCH), 51.5 (CH2), 38.4 (CH2), 36.1 (CH2), 31.1 (CH2), 28.4 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3164, 2925, 2854, 2360, 1640, 1534, 1507, 1481, 1397, 1332, 1273, 1197, 1139, 1093, 1012, 915, 818, 758, 697, 657, 508 cm−1; HRMS (+ ESI): Found m/z 550.2252 [M + H] +, C26H37ClN5O4S required 550.2249.
N-(3-Guanidinopropyl)-2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamide hydrochloride (22d)
The titled compound was synthesised from (E)-1-tert-butyl-N-(N’-((tert-butyloxidanyl)carbonyl)-N-(3-(2-(5-(naphthalen-2-yl)-2-(octylsulfonamido)phenyl)-2-oxoacetamido)propyl)carbamimidoyl)-1-oxidanecarboxamide 21d (0.10 g, 0.13 mmol) following general synthetic procedure I. The product was obtained as a yellow sticky solid (49 mg, 61%); 1H NMR (600 MHz, DMSO-d6): δ 10.22 (bs, 1H, NH), 9.03 (t, J = 5.8 Hz, 1H, NH), 8.21 (s, 1H, ArH), 8.17–8.14 (m, 2H, ArH), 8.07–7.94 (m, 3H, ArH), 7.81 (dd, J = 8.5, 1.9 Hz, 1H, ArH), 7.73 (t, J = 6.0 Hz, 1H, NH), 7.69–7.66 (m, 1H, ArH), 7.62–6.80 (m, 6H, NH, ArH), 3.32 (q, J = 6.8 Hz, 2H, CH2), 3.28–3.24 (m, 2H, CH2), 3.21 (q, J = 6.8 Hz, 2H, CH2), 1.79–1.72 (m, 2H, CH2), 1.72–1.65 (m, 2H, CH2), 1.38–1.31 (m, 2H, CH2), 1.27–1.15 (m, 8H, CH2), 0.81 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 192.6 (CO), 163.8 (CO), 157.0 (CN), 138.2 (ArC), 135.6 (ArC), 135.5 (ArC), 133.3 (ArCH), 133.2 (ArC), 132.4 (ArC), 130.6 (ArCH), 128.8 (ArCH), 128.2 (ArCH), 127.6 (ArCH), 126.7 (ArCH), 126.4 (ArCH), 125.2 (ArCH), 125.0 (ArC), 124.6 (ArCH), 122.0 (ArCH), 51.4 (CH2), 38.4 (CH2), 36.1 (CH2), 31.1 (CH2), 28.4 (CH2), 28.4 (CH2), 28.3 (CH2), 27.3 (CH2), 22.9 (CH2), 22.0 (CH2), 13.9 (CH3); IR (ATR): νmax 3324, 3159, 3055, 2923, 2853, 2321, 2112, 1924, 1747, 1639, 1493, 1463, 1396, 1328, 1267, 1139, 1189, 913, 814 cm−1; HRMS (+ ESI): Found m/z 566.2793 [M + H] +, C30H40N5O4S required 566.2796.
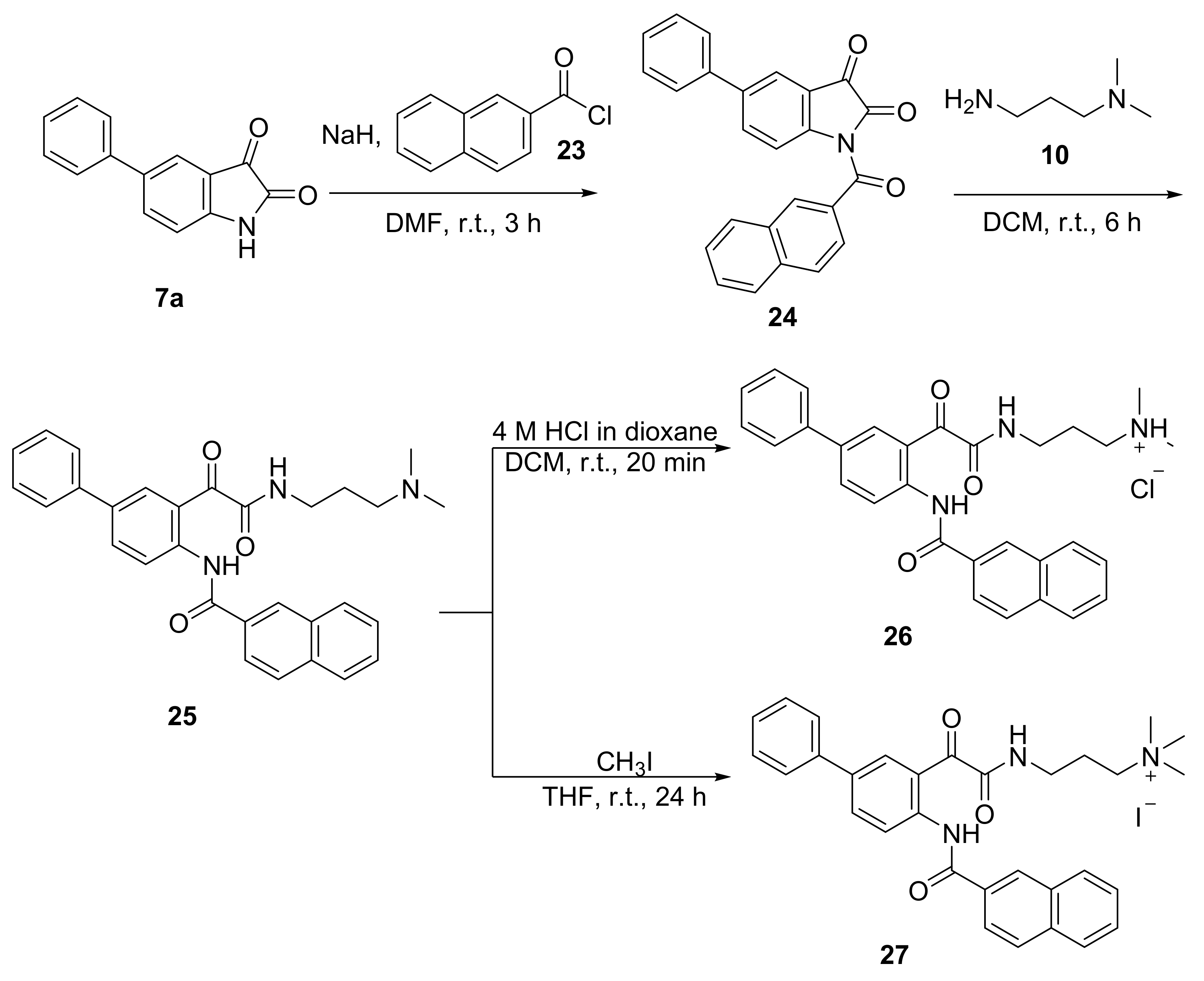
1-(2-Naphthoyl)-5-phenylindoline-2,3-dione (24)
To a suspension of sodium hydride (90 mg, 2.25 mmol) in dimethylformamide (5 mL), slowly dropwise, a solution of 5-phenylindoline-2,3-dione 7a (0.43 g, 1.92 mmol) in dimethylformamide (5 mL) was added at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at 0 °C for 15 min. A solution of 2-naphthoyl chloride (0.38 g, 2.01 mmol) in dimethylformamide (7 mL) was then added slowly dropwise to the reaction mixture at 0 °C with stirring. The reaction mixture was then stirred at room temperature for 3 h. After completion of the reaction, the resulting mixture was poured into 1:1 ice-water mixture. The yellow precipitate was then collected via vacuum filtration and washed with methanol to afford the product as yellow solid (0.25 g, 35%); mp 127.4–127.5 °C; 1H NMR (600 MHz, DMSO-d6): δ 8.60 (d, J = 1.2 Hz, 1H, ArH), 8.16 (dd, J = 8.5, 2.2 Hz, 1H, ArH), 8.09–8.03 (m, 5H, ArH), 7.95 (dd, J = 8.5, 1.7 Hz, 1H, ArH), 7.80–7.77 (m, 2H, ArH), 7.73–7.69 (m, 1H, ArH), 7.67–7.63 (m, 1H, ArH), 7.54–7.50 (m, 2H, ArH), 7.45–7.41 (m, 1H, ArH); 13C NMR (150 MHz, DMSO-d6): δ 180.2 (CO), 168.0 (CO), 157.4 (CO), 147.3 (ArC), 138.2 (ArC), 137.2 (ArC), 135.6 (ArCH), 135.0 (ArC), 131.8 (ArC), 131.2 (ArCH), 131.1 (ArC), 129.2 (ArCH), 129.2 (ArCH), 128.7 (ArCH), 128.1 (ArCH), 127.8 (ArCH), 127.6 (ArCH), 127.0 (ArCH), 126.6 (ArCH), 125.5 (ArCH), 121.9 (ArCH), 120.7 (ArC), 116.7 (ArCH); IR (ATR): νmax 3854, 3675, 2987, 2900, 1948, 1762, 1739, 1692, 1615, 1589, 1508, 1473, 1458, 1406, 1393, 1357, 1306, 1285, 1223, 1193, 1161, 1122, 1066, 1057, 1027, 990, 967, 953, 928, 904, 890, 868, 856, 828, 793, 776, 760, 717, 704, 691, 622, 584, 541, 516, 481, 463 cm−1; HRMS (+ ESI): Found m/z 400.0945 [M + Na] +, C25H15NO3Na required 400.0944.
N-(3-(2-((3-(Dimethylamino)propyl)amino)-2-oxoacetyl)-[1,1’-biphenyl]-4-yl)-2-naphthamide (25)
To a solution of 1-(2-naphthoyl)-5-phenylindoline-2,3-dione 24 (0.11 g, 0.30 mmol) in dichloromethane (5 mL), 3-dimethylaminopropylamine (38 μL, 0.30 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature for 6 h. After completion of the reaction, water was added to the reaction mixture and the product was extracted into dichloromethane (3 × 30 mL), washed with brine, dried over anhydrous sodium sulphate and concentrated in vacuo to afford the product as a yellow solid (0.13 g, 93%); mp 151.6–151.8 °C; 1H NMR (600 MHz, CDCl3): δ 12.21 (bs, 1H, NH), 9.05 (d, J = 9.0 Hz, 1H, ArH), 8.71 (d, J = 2.2 Hz, 1H, ArH), 8.60 (bs, 1H, NH), 8.58 (d, J = 1.3 Hz, 1H, ArH), 8.10 (dd, J = 8.6, 2.0 Hz, 1H, ArH), 8.02 (dd, J = 7.2, 0.6 Hz, 1H, ArH), 7.98 (d, J = 8.6 Hz, 1H, ArH), 7.95–7.90 (m, 2H, ArH), 7.64–7.56 (m, 4H, ArH), 7.48–7.44 (m, 2H, ArH), 7.39–7.35 (m, 1H, ArH), 3.57 (q, J = 5.7 Hz, 2H, CH2), 2.54 (t, J = 6.4 Hz, 2H, CH2), 2.32 (s, 6H, CH3), 1.87–1.81 (m, 2H, CH2); 13C NMR (150 MHz, CDCl3): δ 193.1 (CO), 166.1 (CO), 163.4 (CO), 141.8 (ArC), 139.5 (ArC), 135.7 (ArC), 135.2 (ArC), 135.2 (ArCH), 133.1 (ArCH), 132.9 (ArC), 131.9 (ArC), 129.5 (ArCH), 129.1 (ArCH), 128.9 (ArCH), 128.6 (ArCH), 128.2 (ArCH), 127.9 (ArCH), 127.8 (ArCH), 127.0 (ArCH), 127.0 (ArCH), 123.8 (ArCH), 121.4 (ArCH), 119.6 (ArC), 58.5 (CH2), 45.3 (CH3), 39.6 (CH2), 25.6 (CH2); IR (ATR): νmax 3854, 3675, 3309, 2972, 2900, 2780, 1762, 1735, 1679, 1644, 1627, 1585, 1522, 1492, 1472, 1449, 1394, 1341, 1301, 1285, 1222, 1191, 1132, 1065, 1027, 965, 910, 890, 858, 818, 775, 758, 697, 679, 609, 572, 539, 512, 483, 466, 446 cm−1; HRMS ( + ESI): Found m/z 480.2283 [M + H] +, C30H30N3O3 required 480.2282.
3-(2-(4-(2-Naphthamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)-N,N-dimethylpropan-1-aminium chloride (26)
To a solution of N-(3-(2-((3-(dimethylamino)propyl)amino)-2-oxoacetyl)-[1,1’-biphenyl]-4-yl)-2-naphthamide 25 (32 mg, 0.067 mmol) in dichloromethane (5 mL), 4 M HCl/dioxane (0.10 mL, 0.40 mmol) was added. The reaction mixture was stirred at room temperature for 20 min. After completion of reaction, the reaction mixture was concentrated in vacuo, washed thrice with diethyl ether and freeze-dried to afford the product as a yellow sticky solid (29 mg, 84%); 1H NMR (600 MHz, DMSO-d6): δ 11.43 (s, 1H, NH), 10.38 (bs, 1H, NH), 9.01 (t, J = 6.1 Hz, 1H, NH), 8.63 (s, 1H, ArH), 8.18–8.10 (m, 3H, ArH), 8.07–8.01 (m, 4H, ArH), 7.73–7.64 (m, 4H, ArH), 7.52 (t, J = 7.9 Hz, 2H, ArH), 7.41 (t, J = 7.3 Hz, 2H, ArH), 3.24 (q, J = 6.7 Hz, 2H, CH2), 3.02–2.97 (m, 2H, CH2), 2.63 (s, 6H, CH3), 1.87–1.79 (m, 2H, CH2); 13C NMR (150 MHz, DMSO-d6): δ 190.6 (CO), 165.5 (CO), 163.2 (CO), 138.6 (ArC), 137.6 (ArC), 135.6 (ArC), 134.5 (ArC), 132.1 (ArC), 131.9 (ArCH), 131.2 (ArC), 129.3 (ArCH), 129.2 (ArCH), 129.1 (ArCH), 128.5 (ArCH), 128.2 (ArCH), 128.2 (ArCH), 127.8 (ArCH), 127.8 (ArCH), 127.1 (ArCH), 126.5 (ArCH), 125.5 (ArC), 123.9 (ArCH), 123.0 (ArCH), 54.3 (CH2), 41.9 (CH3), 36.0 (CH2), 23.8 (CH2); IR (ATR): νmax 3331, 3056, 2963, 2681, 2361, 1676, 1643, 1626, 1585, 1523, 1493, 1448, 1396, 1368, 1341, 1306, 1286, 1245, 1219, 1189, 1133, 1068, 967, 912, 891, 849, 819, 761, 699, 681, 572, 512, 487 cm−1; HRMS ( + ESI): Found m/z 480.2281 [M + H] +, C30H30N3O3 required 480.2282.
3-(2-(4-(2-Naphthamido)-[1,1’-biphenyl]-3-yl)-2-oxoacetamido)-N,N,N-trimethylpropan-1-aminium iodide (27)
To a solution of N-(3-(2-((3-(dimethylamino)propyl)amino)-2-oxoacetyl)-[1,1’-biphenyl]-4-yl)-2-naphthamide 25 (34 mg, 0.071 mmol) in THF (5 mL), iodomethane (11 μL, 0.18 mmol) was added. The reaction mixture was stirred at room temperature for 24 h. After completion of reaction, the reaction mixture was concentrated in vacuo, washed thrice with diethyl ether and freeze-dried to afford the product as a yellow sticky solid (35 mg, 79%); 1H NMR (600 MHz, DMSO-d6): δ 10.37 (bs, 1H, NH), 8.98 (t, J = 6.0 Hz, 1H, NH), 8.61 (d, J = 1.1 Hz, 1H, ArH), 8.14–8.08 (m, 3H, ArH), 8.07–8.01 (m, 4H, ArH), 7.73–7.64 (m, 4H, ArH), 7.54–7.49 (m, 2H, ArH), 7.44–7.40 (m, 1H, ArH), 3.30–3.25 (m, 2H, CH2), 3.23 (q, J = 6.6 Hz, 2H, CH2), 2.95 (s, 9H, CH2), 1.90–1.82 (m, 2H, CH2); 13C NMR (150 MHz, DMSO-d6): δ 190.3 (CO), 165.6 (CO), 163.0 (CO), 138.7 (ArC), 137.4 (ArC), 135.6 (ArC),134.5 (ArC), 132.1 (ArC), 131.8 (ArCH), 131.3 (ArC), 129.3 (ArCH), 129.2 (ArCH), 129.0 (ArCH), 128.5 (ArCH), 128.3 (ArCH), 128.2 (ArCH), 127.8 (ArCH), 127.8 (ArCH), 127.2 (ArCH), 126.5 (ArCH), 125.8 (ArC), 123.9 (ArCH), 123.1 (ArCH), 63.3 (CH2), 52.2 (CH3), 35.9 (CH2), 22.5 (CH2); IR (ATR): νmax 3319, 2999, 2360, 2160, 1978, 1677, 1644, 1625, 1586, 1525, 1494, 1447, 1399, 1342, 1306, 1286, 1198, 1068, 966, 912, 868, 850, 825, 763, 698, 681, 609, 573, 517, 488, 472, 426 cm−1; HRMS (+ ESI): Found m/z 494.2439 [M] +, C31H32N3O3 required 494.2438.

 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}