On the Way to Understanding the Interplay between the RNA Structure and Functions in Cells: A Genome-Wide Perspective


Abstract
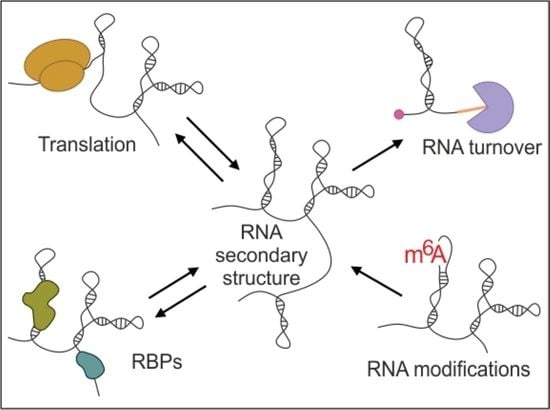
1. Introduction
2. Approaches for RNA Structure Determination
3. The Correlation between mRNA Structure and Translation
3.1. The 5′ and 3′ UTRs
3.2. The Coding Sequence (CDS)
4. RNA Structure in Relation to RNA Stability and Degradation
5. The Relationship between RNA Structure and Proteins Binding
6. Impact of RNA Modifications on RNA Structure In Vivo
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hendrix, D.K.; Brenner, S.E.; Holbrook, S.R. RNA structural motifs: Building blocks of a modular biomolecule. Q. Rev. Biophys. 2005, 38, 221–243. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.K. Dawn of the in vivo RNA structurome and interactome. Biochem. Soc. Trans. 2016, 44, 1395–1410. [Google Scholar] [CrossRef] [PubMed]
- Piao, M.; Sun, L.; Zhang, Q.C. RNA Regulations and Functions Decoded by Transcriptome-wide RNA Structure Probing. Genom. Proteom. Bioinform. 2017, 15, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, S.A.; Kidwell, M.A.; Doudna, J.A. Insights into RNA structure and function from genome-wide studies. Nat. Rev. Genet. 2014, 15, 469–479. [Google Scholar] [CrossRef]
- Ignatova, Z.; Narberhaus, F. Systematic probing of the bacterial RNA structurome to reveal new functions. Curr. Opin. Microbiol. 2017, 36, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Faure, G.; Ogurtsov, A.Y.; Shabalina, S.A.; Koonin, E.V. Role of mRNA structure in the control of protein folding. Nucleic Acids Res. 2016, 44, 10898–10911. [Google Scholar] [CrossRef]
- Wan, Y.; Qu, K.; Zhang, Q.C.; Flynn, R.A.; Manor, O.; Ouyang, Z.; Zhang, J.; Spitale, R.C.; Snyder, M.P.; Segal, E.; et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 2014, 505, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2013, 505, 696–700. [Google Scholar] [CrossRef]
- Rouskin, S.; Zubradt, M.; Washietl, S.; Kellis, M.; Weissman, J.S. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 2013, 505, 701–705. [Google Scholar] [CrossRef]
- Spitale, R.C.; Flynn, R.A.; Zhang, Q.C.; Crisalli, P.; Lee, B.; Jung, J.-W.; Kuchelmeister, H.Y.; Batista, P.J.; Torre, E.A.; Kool, E.T.; et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 2015, 519, 486–490. [Google Scholar] [CrossRef]
- Smola, M.J.; Christy, T.W.; Inoue, K.; Nicholson, C.O.; Friedersdorf, M.; Keene, J.D.; Lee, D.M.; Calabrese, J.M.; Weeks, K.M. SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc. Natl. Acad. Sci. USA 2016, 113, 10322–10327. [Google Scholar] [CrossRef]
- Bevilacqua, P.C.; Ritchey, L.E.; Su, Z.; Assmann, S.M. Genome-Wide Analysis of RNA Secondary Structure. Annu. Rev. Genet. 2016, 50, 235–266. [Google Scholar] [CrossRef]
- Strobel, E.J.; Yu, A.M.; Lucks, J.B. High-throughput determination of RNA structures. Nat. Rev. Genet. 2018, 19, 615–634. [Google Scholar] [CrossRef]
- Kubota, M.; Tran, C.; Spitale, R. Progress and challenges for chemical probing of RNA structure inside living cells. Nat. Methods 2015, 11, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.M. Review toward all RNA structures, concisely. Biopolymers 2015, 103, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.M. Rediscovering RNA. RNA 2015, 21, 714–715. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol 2011, 6, 26. [Google Scholar] [CrossRef]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef]
- Xu, Z.; Mathews, D.H. Prediction of Secondary Structures Conserved in Multiple RNA Sequences. Adv. Struct. Saf. Stud. 2016, 1490, 35–50. [Google Scholar] [CrossRef]
- Mailler, E.; Paillart, J.-C.; Marquet, R.; Smyth, R.P.; Vivet-Boudou, V. The evolution of RNA structural probing methods: From gels to next-generation sequencing. Wiley Interdiscip. Rev. RNA 2018, 10, e1518. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef]
- Deigan, K.E.; Li, T.W.; Mathews, D.H.; Weeks, K.M. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. USA 2008, 106, 97–102. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, B.; Ding, X.; Liu, T.; Hu, X.; Yip, K.; Yang, Z.R.; Mathews, D.H.; Lu, Z.J. Improved prediction of RNA secondary structure by integrating the free energy model with restraints derived from experimental probing data. Nucleic Acids Res. 2015, 43, 7247–7259. [Google Scholar] [CrossRef] [PubMed]
- Busan, S.; Weidmann, C.A.; Sengupta, A.; Weeks, K.M. Guidelines for SHAPE Reagent Choice and Detection Strategy for RNA Structure Probing Studies. Biochemistry 2019, 58, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Ritchey, L.E.; Su, Z.; Tang, Y.; Tack, D.C.; Assmann, S.M.; Bevilacqua, P.C. Structure-seq2: Sensitive and accurate genome-wide profiling of RNA structure in vivo. Nucleic Acids Res. 2017, 45, e135. [Google Scholar] [CrossRef]
- Zubradt, M.; Gupta, P.; Persad, S.; Lambowitz, A.M.; Weissman, J.S.; Rouskin, S. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 2016, 14, 75–82. [Google Scholar] [CrossRef]
- Siegfried, N.A.; Busan, S.; Rice, G.M.; Nelson, J.A.; Weeks, K.M. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 2014, 11, 959–965. [Google Scholar] [CrossRef]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010, 467, 103–107. [Google Scholar] [CrossRef]
- Bevilacqua, P.C.; Assmann, S.M. Technique Development for Probing RNA Structure In Vivo and Genome-Wide. Cold Spring Harb. Perspect. Biol. 2018, 10, a032250. [Google Scholar] [CrossRef]
- Lawley, P.D.; Brookes, P.; Swann, P.F.; Magee, P.N.; Orr, D.J.; Jarman, M. Further studies on the alkylation of nucleic acids and their constituent nucleotides. Biochem. J. 1963, 89, 127–138. [Google Scholar] [CrossRef]
- Wells, S.E.; Hughes, J.M.; Igel, A.H.; Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Enzym. Eng. Evol. Gen. Methods 2000, 318, 479–493. [Google Scholar] [CrossRef]
- Merino, E.J.; Wilkinson, K.A.; Coughlan, J.L.; Weeks, K.M. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 2005, 127, 4223–4231. [Google Scholar] [CrossRef]
- Spitale, R.C.; Flynn, R.A.; Torre, E.A.; Kool, E.T.; Chang, H.Y. RNA structural analysis by evolving SHAPE chemistry. Wiley Interdiscip. Rev. RNA 2014, 5, 867–881. [Google Scholar] [CrossRef]
- Lorenz, R.; Wolfinger, M.T.; Tanzer, A.; Hofacker, I.L. Predicting RNA secondary structures from sequence and probing data. Methods 2016, 103, 86–98. [Google Scholar] [CrossRef]
- Sloma, M.F.; Mathews, D.H. Improving RNA Secondary Structure Prediction with Structure Mapping Data. Enzym. Eng. Evol. Gen. Methods 2015, 553, 91–114. [Google Scholar] [CrossRef]
- Sun, L.; Fazal, F.M.; Li, P.; Broughton, J.P.; Lee, B.; Tang, L.; Huang, W.; Kool, E.T.; Chang, H.Y.; Zhang, Q.C. RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol. 2019, 26, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Zhang, J.; Heng, J.; Gong, J.; Zhang, T.; Li, P.; Sun, B.-F.; Yang, Y.; Zhao, Y.-L.; Wang, H.-L.; et al. RNA structural dynamics regulate early embryogenesis through controlling transcriptome fate and function. Genome Biol. 2020, 21, 1–27. [Google Scholar] [CrossRef]
- Beaudoin, J.-D.; Novoa, E.M.; Vejnar, C.E.; Yartseva, V.; Takacs, C.M.; Kellis, M.; Giraldez, A.J. Analyses of mRNA structure dynamics identify embryonic gene regulatory programs. Nat. Struct. Mol. Biol. 2018, 25, 677–686. [Google Scholar] [CrossRef]
- Burkhardt, D.H.; Rouskin, S.; Zhang, Y.; Li, G.-W.; Weissman, J.S.; Gross, C.A. Operon mRNAs are organized into ORF-centric structures that predict translation efficiency. eLife 2017, 6, 811. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Q.; Yang, X.; Zhang, Y.; Norris, M.; Chen, X.; Cheema, J.; Ding, Y. In vivo nuclear RNA structurome reveals RNA-structure regulation of mRNA processing in plants. bioRxiv 2019, 839506. preprint. [Google Scholar] [CrossRef]
- Deng, H.; Cheema, J.; Zhang, H.; Woolfenden, H.C.; Norris, M.; Liu, Z.; Liu, Q.; Yang, X.; Yang, M.; Deng, X.; et al. Rice In Vivo RNA Structurome Reveals RNA Secondary Structure Conservation and Divergence in Plants. Mol. Plant 2018, 11, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Mustoe, A.M.; Busan, S.; Rice, G.M.; Hajdin, C.E.; Peterson, B.K.; Ruda, V.M.; Kubica, N.; Nutiu, R.; Baryza, J.L.; Weeks, K.M. Pervasive Regulatory Functions of mRNA Structure Revealed by High-Resolution SHAPE Probing. Cell 2018, 173, 181–195.e18. [Google Scholar] [CrossRef] [PubMed]
- Aw, J.G.A.; Shen, Y.; Wilm, A.; Sun, M.; Ni Lim, X.; Boon, K.-L.; Tapsin, S.; Chan, Y.-S.; Tan, C.-P.; Sim, A.Y.; et al. In Vivo Mapping of Eukaryotic RNA Interactomes Reveals Principles of Higher-Order Organization and Regulation. Mol. Cell 2016, 62, 603–617. [Google Scholar] [CrossRef]
- Del Campo, C.; Bartholomäus, A.; Fedyunin, I.; Ignatova, Z. Secondary Structure across the Bacterial Transcriptome Reveals Versatile Roles in mRNA Regulation and Function. PLoS Genet. 2015, 11, e1005613. [Google Scholar] [CrossRef]
- Incarnato, D.; Neri, F.; Anselmi, F.; Oliviero, S. Genome-wide profiling of mouse RNA secondary structures reveals key features of the mammalian transcriptome. Genome Biol. 2014, 15, 491. [Google Scholar] [CrossRef]
- Takyar, S.; Hickerson, R.P.; Noller, H.F. mRNA Helicase Activity of the Ribosome. Cell 2005, 120, 49–58. [Google Scholar] [CrossRef]
- Geisberg, J.V.; Moqtaderi, Z.; Fan, X.; Ozsolak, F.; Struhl, K. Global Analysis of mRNA Isoform Half-Lives Reveals Stabilizing and Destabilizing Elements in Yeast. Cell 2014, 156, 812–824. [Google Scholar] [CrossRef]
- Moqtaderi, Z.; Geisberg, J.V.; Struhl, K. Extensive Structural Differences of Closely Related 3′ mRNA Isoforms: Links to Pab1 Binding and mRNA Stability. Mol. Cell. 2018, 72, 849–861. [Google Scholar] [CrossRef]
- Wu, X.; Bartel, D.P. Widespread Influence of 3′-End Structures on Mammalian mRNA Processing and Stability. Cell 2017, 169, 905–917. [Google Scholar] [CrossRef]
- Roost, C.; Lynch, S.R.; Batista, P.J.; Qu, K.; Chang, H.Y.; Kool, E.T. Structure and thermodynamics of N6-methyladenosine in RNA: A spring-loaded base modification. J. Am. Chem. Soc. 2015, 137, 2107–2115. [Google Scholar] [CrossRef]
- Su, Z.; Tang, Y.; Ritchey, L.E.; Tack, D.C.; Zhu, M.; Bevilacqua, P.C.; Assmann, S.M. Genome-wide RNA structurome reprogramming by acute heat shock globally regulates mRNA abundance. Proc. Natl. Acad. Sci. USA 2018, 115, 12170–12175. [Google Scholar] [CrossRef] [PubMed]
- De Groot, N.S.; Armaos, A.; Graña-Montes, R.; Alriquet, M.; Calloni, G.; Vabulas, R.M.; Tartaglia, G.G. RNA structure drives interaction with proteins. Nat. Commun. 2019, 10, 3246. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell. Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Chiaruttini, C.; Guillier, M. On the role of mRNA secondary structure in bacterial translation. Wiley Interdiscip. Rev. RNA 2020, 11, e1579. [Google Scholar] [CrossRef]
- Bolduc, F.; Garant, J.-M.; Allard, F.; Perreault, J.-P. Irregular G-quadruplexes Found in the Untranslated Regions of Human mRNAs Influence Translation. J. Biol. Chem. 2016, 291, 21751–21760. [Google Scholar] [CrossRef]
- Cohen-Chalamish, S.; Hasson, A.; Weinberg, D.; Namer, L.S.; Banai, Y.; Osman, F.; Kaempfer, R. Dynamic refolding of IFN-gamma mRNA enables it to function as PKR activator and translation template. Nat. Chem. Biol. 2009, 5, 896–903. [Google Scholar] [CrossRef]
- Baird, S.D.; Lewis, S.M.; Turcotte, M.; Holcik, M. A search for structurally similar cellular internal ribosome entry sites. Nucleic Acids Res. 2007, 35, 4664–4677. [Google Scholar] [CrossRef]
- Lee, A.S.Y.; Kranzusch, P.J.; Cate, J.H. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 2015, 522, 111–114. [Google Scholar] [CrossRef]
- Weingarten-Gabbay, S.; Elias-Kirma, S.; Nir, R.; Gritsenko, A.A.; Stern-Ginossar, N.; Yakhini, Z.; Weinberger, A.; Segal, E. Comparative genetics. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 2016, 351. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Green, R. Connections Underlying Translation and mRNA Stability. J. Mol. Biol. 2016, 428, 3558–3564. [Google Scholar] [CrossRef] [PubMed]
- Tuck, A.C.; Rankova, A.; Arpat, A.B.; Liechti, L.A.; Hess, D.; Iesmantavicius, V.; Castelo-Szekely, V.; Gatfield, D.; Bühler, M. Mammalian RNA Decay Pathways Are Highly Specialized and Widely Linked to Translation. Mol. Cell 2020, 77, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, H.; Fukao, A.; Funakami, Y.; Duncan, K.E.; Fujiwara, T. Emerging Evidence of Translational Control by AU-Rich Element-Binding Proteins. Front. Genet. 2019, 10, 332. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Brand, M.D. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995, 312, 163–167. [Google Scholar] [CrossRef]
- Ashe, M.P.; De Long, S.K.; Sachs, A.B. Glucose Depletion Rapidly Inhibits Translation Initiation in Yeast. Mol. Biol. Cell 2000, 11, 833–848. [Google Scholar] [CrossRef]
- Li, F.; Zheng, Q.; Vandivier, L.E.; Willmann, M.R.; Chen, Y.; Gregory, B.D. Regulatory Impact of RNA Secondary Structure across the Arabidopsis Transcriptome. Plant. Cell 2012, 24, 4346–4359. [Google Scholar] [CrossRef]
- Gagliardi, D.; Dziembowski, A. 5′ and 3′ modifications controlling RNA degradation: From safeguards to executioners. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Ringner, M.; Krogh, M. Folding free energies of 5′-UTRs impact post-transcriptional regulation on a genomic scale in yeast. PLoS Comput. Biol. 2005, 1, e72. [Google Scholar]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA Translation and Stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castellóo, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Mayya, V.K.; Duchaine, T.F. Ciphers and Executioners: How 3′-Untranslated Regions Determine the Fate of Messenger RNAs. Front. Genet. 2019, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C. Regulation by 3′-Untranslated Regions. Annu. Rev. Genet. 2017, 51, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Niu, D.-K. Relationship Between mRNA Stability and Length: An Old Question with a New Twist. Biochem. Genet. 2007, 45, 131–137. [Google Scholar] [CrossRef]
- Decker, C.J.; Parker, R. mRNA decay enzymes: Decappers conserved between yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 12512–12514. [Google Scholar] [CrossRef]
- Vejnar, C.E.; Abdelmessih, M.; Takacs, C.; Yartseva, V.; Oikonomou, P.; Christiano, R.; Stoeckius, M.; Lau, S.; Lee, M.; Beaudoin, J.-D. Genome wide analysis of 3′ UTR sequence elements and proteins regulating mRNA stability during maternal-to-zygotic transition in zebrafish. Genome Res. 2019, 29, 1100–1114. [Google Scholar] [CrossRef]
- Boel, G.; Letso, R.; Neely, H.; Price, W.N.; Wong, K.-H.; Su, M.; Luff, J.D.; Valecha, M.; Everett, J.K.; Acton, T.B.; et al. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 2016, 529, 358–363. [Google Scholar] [CrossRef]
- Presnyak, V.; Alhusaini, N.; Chen, Y.-H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef]
- Bazzini, A.A.; Viso, F.; Moreno-Mateos, M.A.; Johnstone, T.; Vejnar, C.E.; Qin, Y.; Yao, J.; Khokha, M.K.; Giraldez, A.J. Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. EMBO J. 2016, 35, 2087–2103. [Google Scholar] [CrossRef]
- Burrow, D.A.; Martin, S.; Quail, J.F.; Alhusaini, N.; Coller, J.; Cleary, M.D. Attenuated Codon Optimality Contributes to Neural-Specific mRNA Decay in Drosophila. Cell Rep. 2018, 24, 1704–1712. [Google Scholar] [CrossRef]
- Wu, Q.; Medina, S.G.; Kushawah, G.; Devore, M.L.; Castellano, L.; Hand, J.M.; Wright, M.; Bazzini, A.A. Translation affects mRNA stability in a codon-dependent manner in human cells. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Wheeler, E.C.; Van Nostrand, E.L.; Yeo, G.W. Advances and challenges in the detection of transcriptome-wide protein-RNA interactions. Wiley Interdiscip Rev. RNA 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 2019, 176, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, D.; Freese, P.; Alexis, M.S.; Su, A.; Hochman, M.; Palden, T.; Bazile, C.; Lambert, N.J.; Van Nostrand, E.L.; Pratt, G.A.; et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 2018, 70, 854–867. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, B.M.; Horos, R.; Fischer, B.; Castelló, A.; Eichelbaum, K.; Alleaume, A.-M.; Schwarzl, T.; Curk, T.; Foehr, S.; Huber, W.; et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 2015, 6, 10127. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, E.; Harris, M.E. Specificity and nonspecificity in RNA–protein interactions. Nat. Rev. Mol. Cell Biol. 2015, 16, 533–544. [Google Scholar] [CrossRef]
- Ray, D.; Kazan, H.; Cook, K.; Weirauch, M.T.; Najafabadi, H.S.; Li, X.; Gueroussov, S.; Albu, M.; Zheng, H.; Yang, A.; et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 2013, 499, 172–177. [Google Scholar] [CrossRef]
- Kumari, P.; Aeschimann, F.; Gaidatzis, D.; Keusch, J.J.; Ghosh, P.; Neagu, A.; Pachulska-Wieczorek, K.; Bujnicki, J.; Gut, H.; Großhans, H.; et al. Evolutionary plasticity of the NHL domain underlies distinct solutions to RNA recognition. Nat. Commun. 2018, 9, 1549. [Google Scholar] [CrossRef]
- Pachulska-Wieczorek, K.; Błaszczyk, L.; Biesiada, M.; Adamiak, R.W.; Purzycka, K.J. The matrix domain contributes to the nucleic acid chaperone activity of HIV-2 Gag. Retrovirology 2016, 13, 18. [Google Scholar] [CrossRef]
- Woodson, S.A.; Panja, S.; Santiago-Frangos, A. Proteins That Chaperone RNA Regulation. Regul. RNA Bact. Archaea 2018, 6, 383–397. [Google Scholar] [CrossRef]
- Casas-Vila, N.; Sayols, S.; Pérez-Martínez, L.; Scheibe, M.; Butter, F. The RNA fold interactome of evolutionary conserved RNA structures in S. cerevisiae. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Mukherjee, N.; Wessels, H.-H.; Lebedeva, S.; Sajek, M.; Ghanbari, M.; Garzia, A.; Munteanu, A.; Yusuf, D.; Farazi, T.; Hoell, J.I.; et al. Deciphering human ribonucleoprotein regulatory networks. Nucleic Acids Res. 2018, 47, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Konig, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010, 17, 909–915. [Google Scholar] [CrossRef]
- Furic, L.; Maher-Laporte, M.; DesGroseillers, L. A genome-wide approach identifies distinct but overlapping subsets of cellular mRNAs associated with Staufen1- and Staufen2-containing ribonucleoprotein complexes. RNA 2007, 14, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Incarnato, D.; Oliviero, S. The RNA Epistructurome: Uncovering RNA Function by Studying Structure and Post-Transcriptional Modifications. Trends Biotechnol. 2017, 35, 318–333. [Google Scholar] [CrossRef]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, E.M.; Kietrys, A.M.; Kool, E.T. Chemical and structural effects of base modifications in messenger RNA. Nature 2017, 541, 339–346. [Google Scholar] [CrossRef]
- Delaunay, S.; Frye, M. RNA modifications regulating cell fate in cancer. Nature 2019, 21, 552–559. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S.R.; Qian, S.-B. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; He, C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015, 29, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.S.; Wang, X.; Beadell, A.V.; Lu, Z.; Shi, H.; Kuuspalu, A.; Ho, R.K.; He, C. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 2017, 542, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.-J.; Ringeling, F.R.; Vissers, C.; Jacob, F.; Pokrass, M.; Jimenez-Cyrus, D.; Su, Y.; Kim, N.-S.; Zhu, Y.; Zheng, L.; et al. Temporal Control of Mammalian Cortical Neurogenesis by m6A Methylation. Cell 2017, 171, 877–889. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Zhang, J.; Zhu, J. The role of m6A RNA methylation in human cancer. Mol. Cancer 2019, 18, 103. [Google Scholar] [CrossRef]
- Jaffrey, S.R.; Kharas, M.G. Emerging links between m6A and misregulated mRNA methylation in cancer. Genome Med. 2017, 9, 2. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Roundtree, I.A.; He, C. RNA epigenetics—Chemical messages for posttranscriptional gene regulation. Curr. Opin. Chem. Biol. 2016, 30, 46–51. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Method | Application Described in this Review | Used Probe | Modification Readout | Condition |
|---|---|---|---|---|
| icSHAPE | Mouse [10,37], human [37], zebrafish [38] | NAI-N3 | RT-stop | In vivo and in vitro |
| DMS-seq | Yeast [9], zebrafish [39], E. coli [40] | DMS | RT-stop | In vivo and in vitro |
| SHAPE-Structure-seq | A. thaliana [41] | NAI | RT-stop | In vivo and in vitro |
| Structure-seq | A. thaliana [8], rice [42] | DMS | RT-stop | In vivo |
| SHAPE-MaP | E. coli [43] | 1M7 | RT-mutate | In vivo and in vitro |
| SPLASH | Human and yeast [44] | Biotinylated psoralen | Mapping of ligated junctions | In vivo |
| PARS | E. coli [45] | RNase V1 (dsRNA) and S1 (ssRNA) | Fragments analysis | In vitro |
| CIRS-seq | Mouse [46] | DMS and CMCT | RT-stop | In vitro |
| Work | Organism | Main Conclusions |
|---|---|---|
| Del Campo et al., 2015 [45] | E. coli | The unstructured sequence upstream of the start codon is a general feature of E. coli genes and is positively correlated with gene expression. |
| Mustoe et al., 2018 [43] | E. coli | Translation is the main source of mRNA structural destabilization in cells. The structure in RBS is a strong determinant of TE. CDS structure is not critical for TE. |
| Burkhardt et al., 2017 [40] | E. coli | The structure in RBS does not determine TE. The intrinsic CDS structure plays the critical role in TE tuning. |
| Beaudoin et al., 2018 [39] | Zebrafish | Translation guides RNA structure rather than structure guiding translation. The ribosome is a major remodeler of RNA structure. Structural elements in the 3′ UTR are major regulators of transcript stability during the MZT. |
| Shi et al., 2020 [38] | Zebrafish | TE is correlated with RNA unfolding. |
| Rouskin et al., 2014 [9] | Yeast | ATP-dependent processes strongly contribute to the unfolded state of mRNAs inside cells. |
| Geisberg et al., 2014 [48] | Yeast | The double-stranded structures at the 3′-ends, involving or not involving poly(A) tails, are a critical determinant of mRNA stability. |
| Moqtaderi et al., 2018 [49] | Yeast | The single-strandedness in the proximity of 3′-end, double-strandedness of the poly(A) tail, together with low Pab1 binding, are linked with mRNA stability and are evolutionarily conserved. |
| Aw et al., 2016 [44] | Human and yeast | The structure of 5′-UTRs is negatively correlated with mRNA stability, whereas the secondary structure in 3′ UTRs is associated with longer mRNA half-life. |
| Wu et al., 2017 [50] | Human | In cells, 3′-ends are generally more folded than are other mRNA regions and their structure regulates mRNA metabolic stability. Specific structure of the 3′-end can facilitate cleavage and polyadenylation of mRNAs. |
| Roost et al., 2015 [51] | Human | Ex vivo studies of human transcriptome confirmed the structural RNA changes at the m6A modification sites, with a strong tendency for unwinding RNA secondary structure. |
| Sun et al., 2019 [37] | Mouse and human | The intrinsic RNA structure plays a central role in connecting transcription, translation, and RNA degradation. The majority of the transcripts preserve their structure as they transfer from chromatin to the nucleoplasm and cytoplasm. RBPs and RNA modifications account for local RNA structure changes between cellular compartments. CDS structure and TE are only weakly correlated. More-structured RNAs tended to have shorter half-lives. RNA degradation is not RNA-region specific. |
| Spitale et al., 2015 [10] | Mouse | m6A modifications impact RNA structure in vivo, favoring the transition from paired to unpaired RNA. |
| Deng et al., 2018 [42] | Rice | Higher m6A modification tends to have less RNA structure in the 3′ UTR in plants. |
| Su et al., 2018 [52] | Rice | Transcripts are subjected to degradation by a mechanism involving secondary structure unfolding in 5′ and 3′ UTRs. |
| Ding et al., 2014 [8] | A. thaliana | Less structured regions immediately upstream the start codon region facilitate ribosome binding and increase TE. |
| Liu et al., 2019 [41] | A. thaliana | Nuclear mRNAs fold differently from cytosolic mRNAs. |
| Sanchez De Groot et al., 2019 [53] | Various | Highly structured RNAs bind a large amount of proteins. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrzejewska, A.; Zawadzka, M.; Pachulska-Wieczorek, K. On the Way to Understanding the Interplay between the RNA Structure and Functions in Cells: A Genome-Wide Perspective. Int. J. Mol. Sci. 2020, 21, 6770. https://doi.org/10.3390/ijms21186770
Andrzejewska A, Zawadzka M, Pachulska-Wieczorek K. On the Way to Understanding the Interplay between the RNA Structure and Functions in Cells: A Genome-Wide Perspective. International Journal of Molecular Sciences. 2020; 21(18):6770. https://doi.org/10.3390/ijms21186770
Chicago/Turabian StyleAndrzejewska, Angelika, Małgorzata Zawadzka, and Katarzyna Pachulska-Wieczorek. 2020. "On the Way to Understanding the Interplay between the RNA Structure and Functions in Cells: A Genome-Wide Perspective" International Journal of Molecular Sciences 21, no. 18: 6770. https://doi.org/10.3390/ijms21186770
APA StyleAndrzejewska, A., Zawadzka, M., & Pachulska-Wieczorek, K. (2020). On the Way to Understanding the Interplay between the RNA Structure and Functions in Cells: A Genome-Wide Perspective. International Journal of Molecular Sciences, 21(18), 6770. https://doi.org/10.3390/ijms21186770