Pathogenic Mechanisms of Myeloma Bone Disease and Possible Roles for NRF2




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Bone Metabolism
3. Myeloma Induced Bone Disease
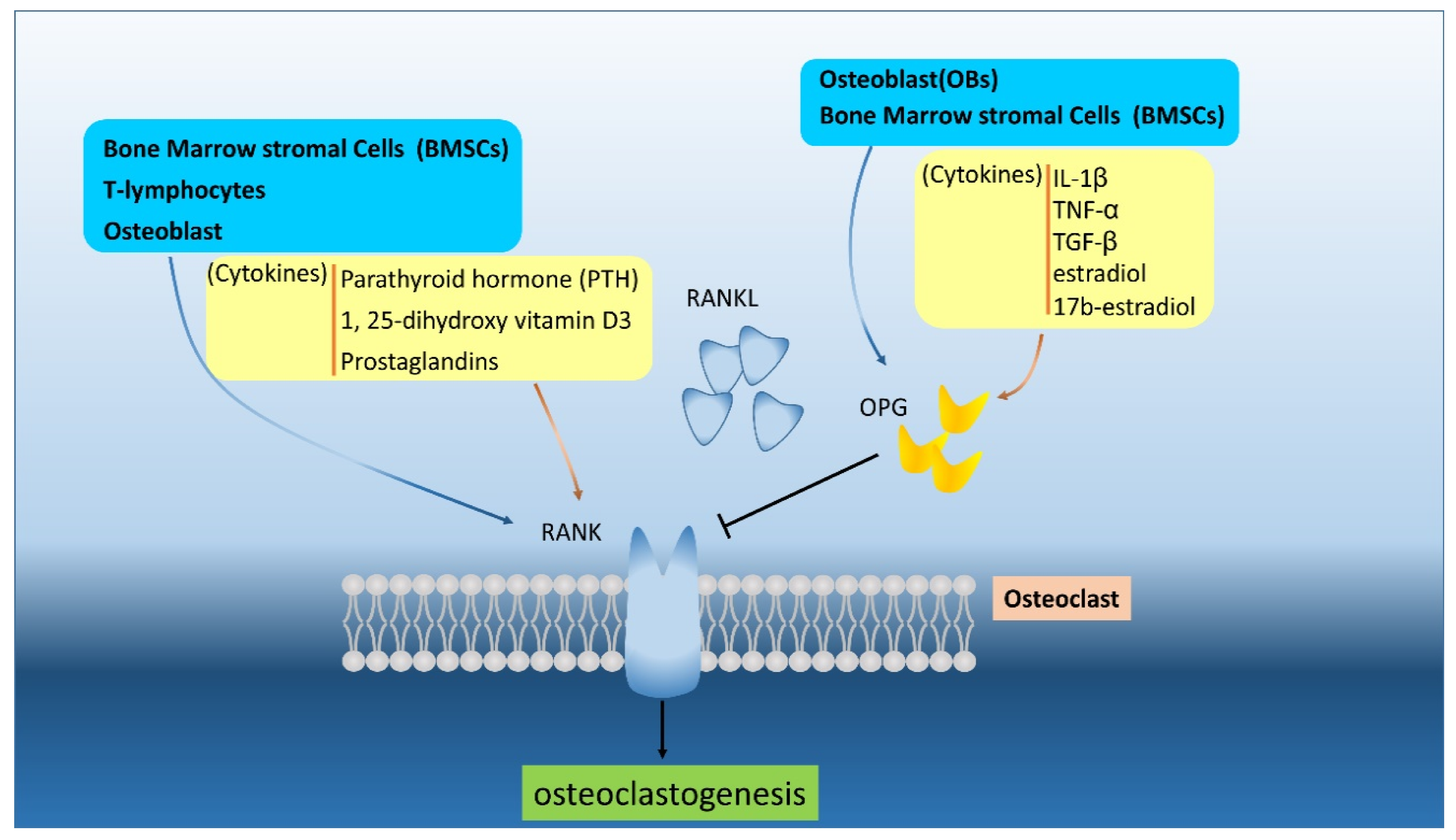
4. The RANK/RANKL Pathway
5. Introduction of NRF2 (or Structure, Function, and Regulation of NRF2)
6. The Role of NRF2 in the Myeloma Microenvironment
7. The Role of NRF2 in Bone Metabolism
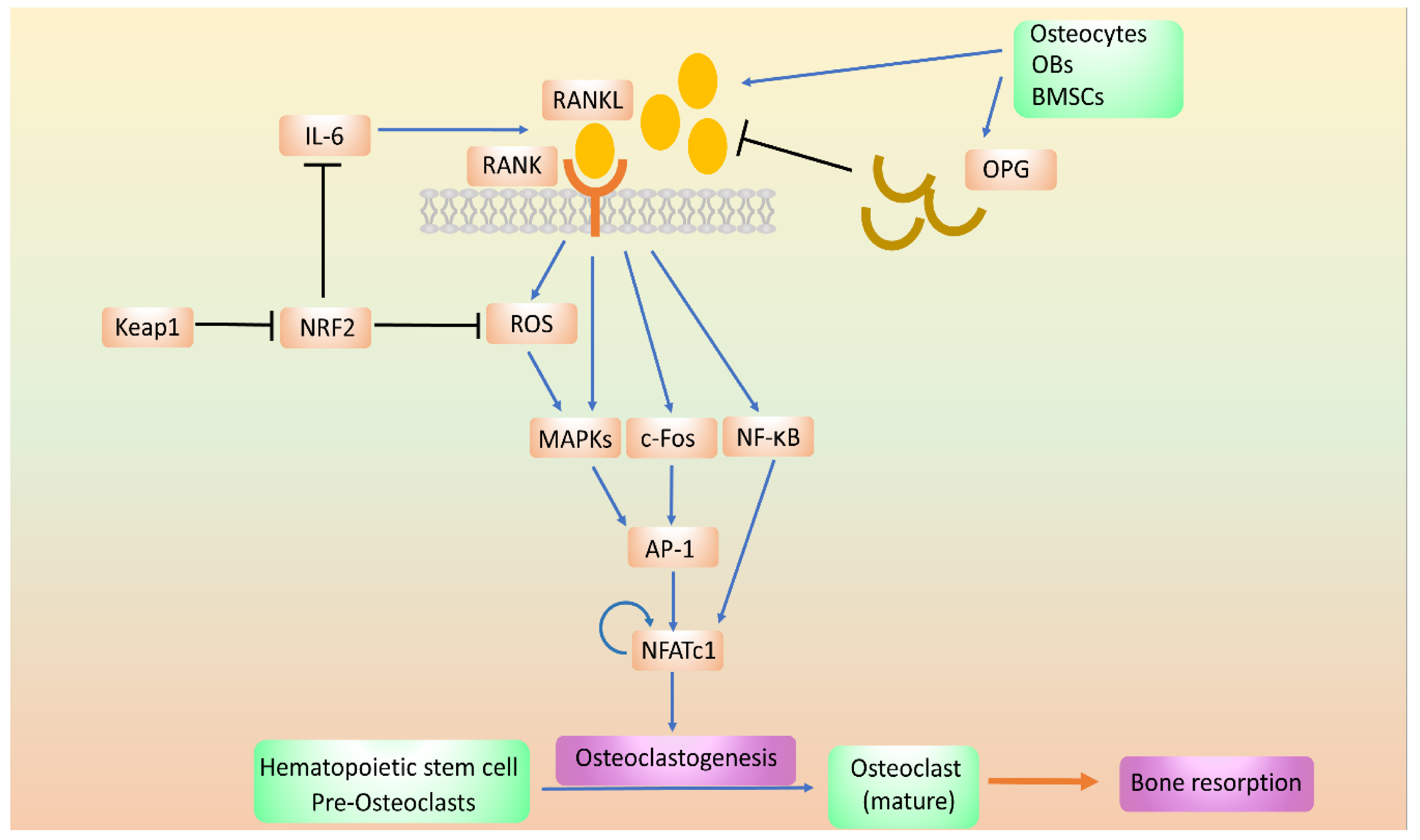
8. The Effects of NRF2 on OC
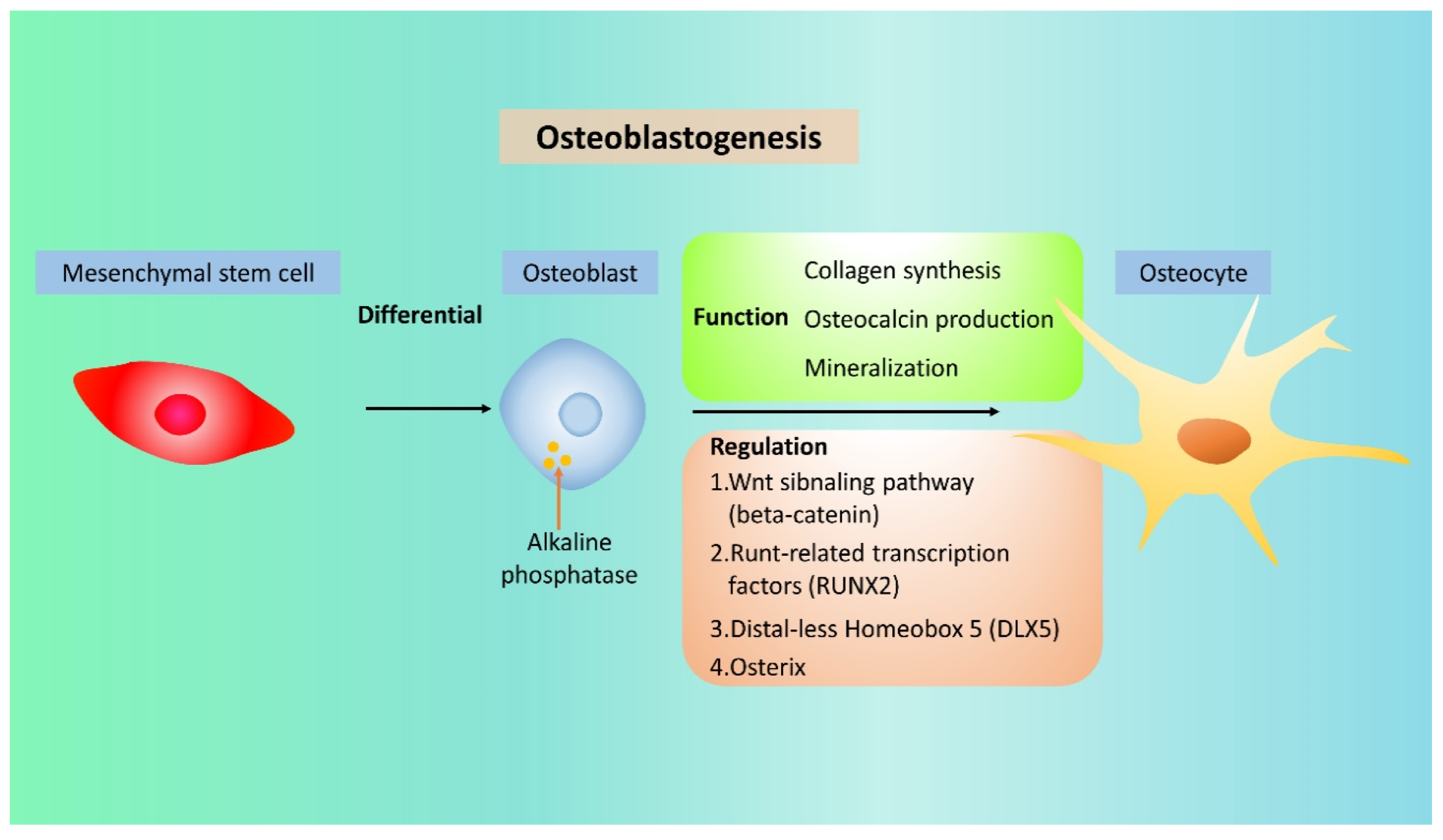
9. The Role of NRF2 in OB
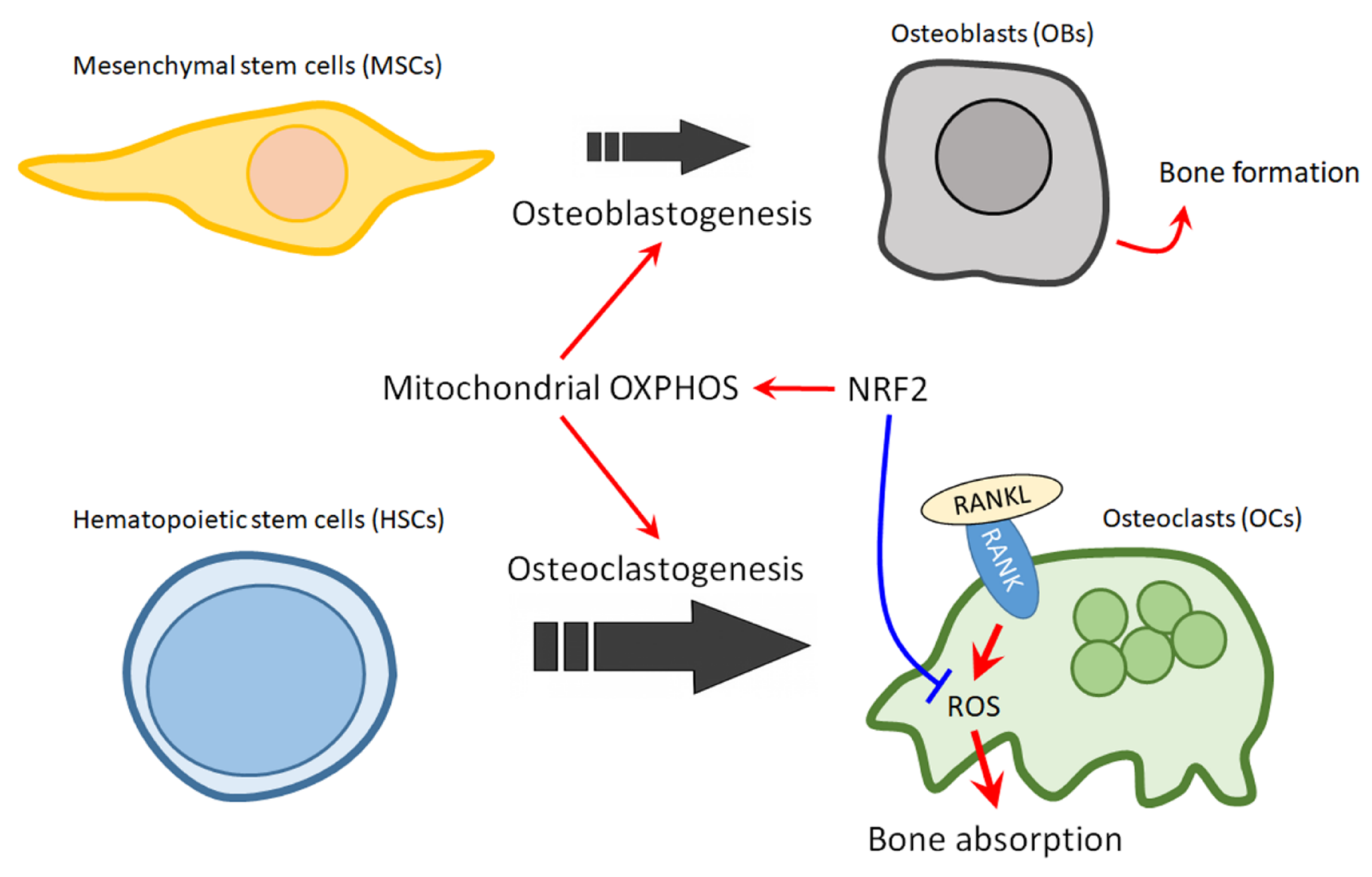
10. The Role of NRF2 in Mitochondrial Regulation of Bone Homeostasis
11. The Role of NRF2 in Mitochondrial Regulation in Myeloma
12. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ARE | antioxidant response elements |
| bZIP | basic leucine zipper protein family |
| β-TrCP | β-transducin repeat-containing protein |
| BMSCs | bone marrow stromal cells |
| CCL3 | chemokine C-C motif 3 |
| CBP | CREB-binding protein |
| CUL1-RBX1 | Cullin 1-RING Box 1 |
| CUL3-RBX1 | Cullin 3-RING Box 1 |
| DDK-1 | dickkopf-1 |
| DLX5 | Distal-Less Homeobox 5 |
| GCLC | glutamate-cysteine ligase catalytic |
| GSH | glutathione |
| GSTs | glutathione S-transferases |
| HSCs | Hematopoietic stem cells |
| IL-1,6 | Interleukin-1,6 |
| MIP-1-alpha | macrophage inflammatory protein 1 |
| M-GCSF | macrophage-colony stimulating factor |
| MAPK | mitogen-activated protein kinase |
| MM | Multiple myeloma |
| Maf | Musculoaponeurotic fibrosarcoma |
| MDSCs | myeloid-derived suppressor cells |
| MBD | Myeloma bone disease |
| MSCs | Mesenchymal stem cells |
| NQO | NADPH-quinone oxidoreductase |
| RANKL | NF-kappa B ligand |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NFATc1 | nuclear-factor-activated T-cell cytoplasmic 1 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| OC-STAMP | osteoclast stimulatory transmembrance protein |
| OB | Osteoblast |
| OC | Osteoclast |
| OPG | Osteoprotegerin |
| OXPHOS | oxidative phosphorylation |
| PTHrP | parathyroid hormone-related protein |
| PTH | Parathyroid hormone |
| NQO1 | NAD(P)H: quinone osidoreductase 1 |
| RANKL | RANK ligand |
| ROS | reactive oxygen species |
| RXRα | retinoid X receptor alpha |
| RUNX2 | Runt-related transcription factor 2 |
| TRAP | tartrate-resistant acid phosphatase |
| TGF-beta | transforming growth factor beta |
| TNF-alpha | tumor necrosis factor-alpha |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VEGF | vascular endothelial growth factor |
References
- Moreau, P.; San Miguel, J.; Sonneveld, P.; Mateos, M.V.; Zamagni, E.; Avet-Loiseau, H.; Hajek, R.; Dimopoulos, M.A.; Ludwig, H. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol 2017, 28, iv52–iv61. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kumar, S. Multiple Myeloma: Diagnosis and Treatment. Mayo Clin. Proc. 2016, 91, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Chappard, D.; Bouvard, B.; Basle, M.F.; Legrand, E.; Audran, M. Bone metastasis: Histological changes and pathophysiological mechanisms in osteolytic or osteosclerotic localizations. A review. Morphol. Bull. L’association Anat. 2011, 95, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Hinge, M.; Andersen, K.T.; Lund, T.; Jorgensen, H.B.; Holdgaard, P.C.; Ormstrup, T.E.; Ostergaard, L.L.; Plesner, T. Bone healing in multiple myeloma: A prospective evaluation of the impact of first-line anti-myeloma treatment. Haematologica 2016, 101, e419–e422. [Google Scholar] [CrossRef]
- Silbermann, R.; Roodman, G.D. Current Controversies in the Management of Myeloma Bone Disease. J. Cell. Physiol. 2016, 231, 2374–2379. [Google Scholar] [CrossRef]
- Roodman, G.D. Pathogenesis of myeloma bone disease. Blood Cells Mol. Dis. 2004, 32, 290–292. [Google Scholar] [CrossRef]
- Aly, A.; Onukwugha, E.; Woods, C.; Mullins, C.D.; Kwok, Y.; Qian, Y.; Arellano, J.; Balakumaran, A.; Hussain, A. Measurement of skeletal related events in SEER-Medicare: A comparison of claims-based methods. BMC Med. Res. Methodol. 2015, 15, 65. [Google Scholar] [CrossRef]
- Kim, C.; Bhatta, S.; Cyprien, L.; Fonseca, R.; Hernandez, R.K. Incidence of skeletal-related events among multiple myeloma patients in the United States at oncology clinics: Observations from real-world data. J. Bone. Oncol. 2019, 14, 100215. [Google Scholar] [CrossRef]
- Sonmez, M.; Akagun, T.; Topbas, M.; Cobanoglu, U.; Sonmez, B.; Yilmaz, M.; Ovali, E.; Omay, S.B. Effect of pathologic fractures on survival in multiple myeloma patients: A case control study. J. Exp. Clin. Cancer Res. CR 2008, 27, 11. [Google Scholar] [CrossRef]
- Ntanasis-Stathopoulos, I.; Fotiou, D.; Terpos, E. CCL3 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1231, 13–21. [Google Scholar] [PubMed]
- Politou, M.; Terpos, E.; Anagnostopoulos, A.; Szydlo, R.; Laffan, M.; Layton, M.; Apperley, J.F.; Dimopoulos, M.A.; Rahemtulla, A. Role of receptor activator of nuclear factor-kappa B ligand (RANKL), osteoprotegerin and macrophage protein 1-alpha (MIP-1a) in monoclonal gammopathy of undetermined significance (MGUS). Br. J. Haematol. 2004, 126, 686–689. [Google Scholar] [CrossRef]
- Terpos, E.; Szydlo, R.; Apperley, J.F.; Hatjiharissi, E.; Politou, M.; Meletis, J.; Viniou, N.; Yataganas, X.; Goldman, J.M.; Rahemtulla, A. Soluble receptor activator of nuclear factor kappaB ligand-osteoprotegerin ratio predicts survival in multiple myeloma: Proposal for a novel prognostic index. Blood 2003, 102, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A. Myeloma bone disease: From biology findings to treatment approaches. Blood 2019, 133, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.H.; Hsiao, H.H. NRF2 Is One of the Players Involved in Bone Marrow Mediated Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 3503. [Google Scholar] [CrossRef] [PubMed]
- Hadjidakis, D.J.; Androulakis, I.I. Bone remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef]
- Raggatt, L.J.; Partridge, N.C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, Y.; Pacios, S.; Li, S.; Graves, D.T. Cellular and Molecular Aspects of Bone Remodeling. Front. Oral Biol. 2016, 18, 9–16. [Google Scholar]
- Janckila, A.J.; Takahashi, K.; Sun, S.Z.; Yam, L.T. Naphthol-ASBI phosphate as a preferred substrate for tartrate-resistant acid phosphatase isoform 5b. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2001, 16, 788–793. [Google Scholar] [CrossRef]
- Janckila, A.J.; Takahashi, K.; Sun, S.Z.; Yam, L.T. Tartrate-resistant acid phosphatase isoform 5b as serum marker for osteoclastic activity. Clin. Chem. 2001, 47, 74–80. [Google Scholar] [CrossRef]
- Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; Yasuda, H.; Yano, K.; Morinaga, T.; Higashio, K. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem. Biophys. Res. Commun. 1998, 253, 395–400. [Google Scholar] [CrossRef]
- Takahashi, N.; Udagawa, N.; Suda, T. A new member of tumor necrosis factor ligand family, ODF/OPGL/TRANCE/RANKL, regulates osteoclast differentiation and function. Biochem. Biophys. Res. Commun. 1999, 256, 449–455. [Google Scholar] [CrossRef]
- Chambers, T.J. Regulation of the differentiation and function of osteoclasts. J. Pathol. 2000, 192, 4–13. [Google Scholar] [CrossRef]
- Kogianni, G.; Mann, V.; Noble, B.S. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2008, 23, 915–927. [Google Scholar] [CrossRef]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Tsukii, K.; Shima, N.; Mochizuki, S.; Yamaguchi, K.; Kinosaki, M.; Yano, K.; Shibata, O.; Udagawa, N.; Yasuda, H.; Suda, T.; et al. Osteoclast differentiation factor mediates an essential signal for bone resorption induced by 1 alpha,25-dihydroxyvitamin D3, prostaglandin E2, or parathyroid hormone in the microenvironment of bone. Biochem. Biophys. Res. Commun. 1998, 246, 337–341. [Google Scholar] [CrossRef]
- Qiang, Y.W.; Chen, Y.; Stephens, O.; Brown, N.; Chen, B.; Epstein, J.; Barlogie, B.; Shaughnessy, J.D.J. Myeloma-derived Dickkopf-1 disrupts Wnt-regulated osteoprotegerin and RANKL production by osteoblasts: A potential mechanism underlying osteolytic bone lesions in multiple myeloma. Blood 2008, 112, 196–207. [Google Scholar] [CrossRef]
- Weitzmann, M.N. Bone and the Immune System. Toxicol. Pathol. 2017, 45, 911–924. [Google Scholar] [CrossRef]
- Zupan, J.; Komadina, R.; Marc, J. The relationship between osteoclastogenic and anti-osteoclastogenic pro-inflammatory cytokines differs in human osteoporotic and osteoarthritic bone tissues. J. Biomed. Sci. 2012, 19, 28. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Khosla, S.; Dunstan, C.R.; Lacey, D.L.; Boyle, W.J.; Riggs, B.L. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2000, 15, 2–12. [Google Scholar] [CrossRef]
- Huang, W.; Yang, S.; Shao, J.; Li, Y.P. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front. Biosci. A J. Virtual Libr. 2007, 12, 3068–3092. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Kuboki, Y.; Miyata, T. Differentiation of mesenchymal stem cells into osteoblasts on honeycomb collagen scaffolds. Biotechnol. Bioeng. 2006, 95, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Harada, S.; Rodan, G.A. Control of osteoblast function and regulation of bone mass. Nature 2003, 423, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Filzmoser, J.M.; Pecherstorfer, M.; Podar, K. Myeloma Bone Disease: Update on Pathogenesis and Novel Treatment Strategies. Pharmaceutics 2018, 10, 202. [Google Scholar] [CrossRef]
- Chen, A.; Hamamura, K.; Zhang, P.; Chen, Y.; Yokota, H. Systems analysis of bone remodelling as a homeostatic regulator. IET Syst. Biol. 2010, 4, 52–63. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Nakashima, T.; Takayanagi, H. Osteocyte control of osteoclastogenesis. Bone 2013, 54, 258–263. [Google Scholar] [CrossRef]
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef]
- Nakahama, K. Cellular communications in bone homeostasis and repair. Cell. Mol. Life Sci. CMLS 2010, 67, 4001–4009. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Tsourdi, E.; Jähn, K.; Rauner, M.; Busse, B.; Bonewald, L.F. Physiological and pathological osteocytic osteolysis. J. Musculoskelet. Neuronal Interact. 2018, 18, 292–303. [Google Scholar] [PubMed]
- Giuliani, N.; Colla, S.; Rizzoli, V. Update on the pathogenesis of osteolysis in multiple myeloma patients. Acta Bio-Med. Atenei Parm. 2004, 75, 143–152. [Google Scholar]
- Giuliani, N.; Rizzoli, V.; Roodman, G.D. Multiple myeloma bone disease: Pathophysiology of osteoblast inhibition. Blood 2006, 108, 3992–3996. [Google Scholar] [CrossRef]
- Roodman, G.D. Pathogenesis of myeloma bone disease. J. Cell. Biochem. 2010, 109, 283–291. [Google Scholar]
- Terpos, E.; Christoulas, D.; Gavriatopoulou, M. Biology and treatment of myeloma related bone disease. Metab. Clin. Exp. 2018, 80, 80–90. [Google Scholar] [CrossRef]
- Christoulas, D.; Terpos, E.; Dimopoulos, M.A. Pathogenesis and management of myeloma bone disease. Expert Rev. Hematol. 2009, 2, 385–398. [Google Scholar] [CrossRef]
- Gregory, C. MM-induced osteolysis: Partners in crime. Blood 2008, 112, 3–4. [Google Scholar] [CrossRef]
- Teitelbaum, S.L.; Ross, F.P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003, 4, 638–649. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Theoleyre, S.; Wittrant, Y.; Tat, S.K.; Fortun, Y.; Redini, F.; Heymann, D. The molecular triad OPG/RANK/RANKL: Involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004, 15, 457–475. [Google Scholar] [CrossRef]
- Graham, J.M.; Ayati, B.P.; Holstein, S.A.; Martin, J.A. The role of osteocytes in targeted bone remodeling: A mathematical model. PLoS ONE 2013, 8, e63884. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Takayanagi, H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol. Metab. TEM 2012, 23, 582–590. [Google Scholar] [CrossRef]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef]
- Walsh, M.C.; Choi, Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front. Immunol. 2014, 5, 511. [Google Scholar] [CrossRef]
- Chaweewannakorn, W.; Ariyoshi, W.; Okinaga, T.; Fujita, Y.; Maki, K.; Nishihara, T. Ameloblastin attenuates RANKL-mediated osteoclastogenesis by suppressing activation of nuclear factor of activated T-cell cytoplasmic 1 (NFATc1). J. Cell. Physiol. 2019, 234, 1745–1757. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, X. Receptor activator of nuclear factor-κB ligand (RANKL)/RANK/osteoprotegerin system in bone and other tissues (review). Mol. Med. Rep. 2015, 11, 3212–3218. [Google Scholar] [CrossRef]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Kostenuik, P.J.; Shalhoub, V. Osteoprotegerin: A physiological and pharmacological inhibitor of bone resorption. Curr. Pharm. Des. 2001, 7, 613–635. [Google Scholar] [CrossRef]
- Giuliani, N.; Colla, S.; Sala, R.; Moroni, M.; Lazzaretti, M.; La Monica, S.; Bonomini, S.; Hojden, M.; Sammarelli, G.; Barillè, S.; et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand (RANKL) in T lymphocytes: A potential role in multiple myeloma bone disease. Blood 2002, 100, 4615–4621. [Google Scholar] [CrossRef]
- Standal, T.; Seidel, C.; Hjertner, Ø.; Plesner, T.; Sanderson, R.D.; Waage, A.; Borset, M.; Sundan, A. Osteoprotegerin is bound, internalized, and degraded by multiple myeloma cells. Blood 2002, 100, 3002–3007. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D.; et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res. 2016, 76, 1089–1100. [Google Scholar] [CrossRef]
- Giuliani, N.; Ferretti, M.; Bolzoni, M.; Storti, P.; Lazzaretti, M.; Dalla Palma, B.; Bonomini, S.; Martella, E.; Agnelli, L.; Neri, A.; et al. Increased osteocyte death in multiple myeloma patients: Role in myeloma-induced osteoclast formation. Leukemia 2012, 26, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell. Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef]
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar] [CrossRef]
- Tian, W.; Rojo de la Vega, M.; Schmidlin, C.J.; Ooi, A.; Zhang, D.D. Kelch-like ECH-associated protein 1 (KEAP1) differentially regulates nuclear factor erythroid-2-related factors 1 and 2 (NRF1 and NRF2). J. Biol. Chem. 2018, 293, 2029–2040. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid. Med. Cell Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar]
- Nioi, P.; McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: Reassessment of the ARE consensus sequence. Biochem. J. 2003, 374, 337–348. [Google Scholar] [CrossRef]
- Pi, J.; Hayes, J.D.; Yamamoto, M. New insights into nuclear factor erythroid 2-related factors in toxicology and pharmacology. Toxicol. Appl. Pharmacol. 2019, 367, 33–35. [Google Scholar] [CrossRef]
- Pi, J.; Freeman, M.L.; Yamamoto, M. Nrf2 in toxicology and pharmacology: The good, the bad and the ugly? Toxicol. Appl. Pharmacol. 2010, 244, 1–3. [Google Scholar] [CrossRef]
- Lacher, S.E.; Lee, J.S.; Wang, X.; Campbell, M.R.; Bell, D.A.; Slattery, M. Beyond antioxidant genes in the ancient Nrf2 regulatory network. Free Radic. Biol. Med. 2015, 88, 452–465. [Google Scholar] [CrossRef]
- Marini, M.G.; Asunis, I.; Chan, K.; Chan, J.Y.; Kan, Y.W.; Porcu, L.; Cao, A.; Moi, P. Cloning MafF by recognition site screening with the NFE2 tandem repeat of HS2: Analysis of its role in globin and GCSl genes regulation. Blood Cells Mol. Dis. 2002, 29, 145–158. [Google Scholar] [CrossRef]
- Marini, M.G.; Chan, K.; Casula, L.; Kan, Y.W.; Cao, A.; Moi, P. hMAF, a small human transcription factor that heterodimerizes specifically with Nrf1 and Nrf2. J. Biol. Chem. 1997, 272, 16490–16497. [Google Scholar] [CrossRef]
- Wang, X.; Tomso, D.J.; Chorley, B.N.; Cho, H.Y.; Cheung, V.G.; Kleeberger, S.R.; Bell, D.A. Identification of polymorphic antioxidant response elements in the human genome. Hum. Mol. Genet. 2007, 16, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef]
- Jeyapaul, J.; Jaiswal, A.K. Nrf2 and c-Jun regulation of antioxidant response element (ARE)-mediated expression and induction of gamma-glutamylcysteine synthetase heavy subunit gene. Biochem. Pharmacol. 2000, 59, 1433–1439. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells Devoted Mol. Cell. Mech. 2003, 8, 379–391. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Kizaki, M.; Tabayashi, T. The Role of Intracellular Signaling Pathways in the Pathogenesis of Multiple Myeloma and Novel Therapeutic Approaches. J. Clin. Exp. Hematop. JCEH 2016, 56, 20–27. [Google Scholar] [CrossRef]
- Manni, S.; Carrino, M.; Semenzato, G.; Piazza, F. Old and Young Actors Playing Novel Roles in the Drama of Multiple Myeloma Bone Marrow Microenvironment Dependent Drug Resistance. Int. J. Mol. Sci. 2018, 19, 19. [Google Scholar] [CrossRef]
- Shay, G.; Hazlehurst, L.; Lynch, C.C. Dissecting the multiple myeloma-bone microenvironment reveals new therapeutic opportunities. J. Mol. Med. 2016, 94, 21–35. [Google Scholar] [CrossRef]
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. Bone marrow micro-environment is a crucial player for myelomagenesis and disease progression. Oncotarget 2017, 8, 20394–20409. [Google Scholar] [CrossRef]
- Andrews, S.W.; Kabrah, S.; May, J.E.; Donaldson, C.; Morse, H.R. Multiple myeloma: The bone marrow microenvironment and its relation to treatment. Br. J. Biomed. Sci. 2013, 70, 110–120. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Kobayashi, Y.; Yamasaki, S.; Kawakami, A.; Eguchi, K.; Sasaki, H.; Sakai, H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: Modulation of the expression by osteotropic factors and cytokines. Biochem. Biophys. Res. Commun. 2000, 275, 768–775. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Buelna-Chontal, M.; Zazueta, C. Redox activation of Nrf2 & NF-κB: A double end sword? Cell. Signal. 2013, 25, 2548–2557. [Google Scholar]
- Zhang, F.; Peng, W.; Zhang, J.; Dong, W.; Yuan, D.; Zheng, Y.; Wang, Z. New strategy of bone marrow mesenchymal stem cells against oxidative stress injury via Nrf2 pathway: Oxidative stress preconditioning. J. Cell. Biochem. 2019, 120, 19902–19914. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, S.; Lee, H.; Yang, Y.; Jeong, W. Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic. Biol. Med. 2013, 65, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Piddock, R.E.; Marlein, C.R.; Abdul-Aziz, A.; Shafat, M.S.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. Myeloma-derived macrophage inhibitory factor regulates bone marrow stromal cell-derived IL-6 via c-MYC. J. Hematol. Oncol. 2018, 11, 66. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef]
- Hengeveld, P.J.; Kersten, M.J. B-cell activating factor in the pathophysiology of multiple myeloma: A target for therapy? Blood Cancer J. 2015, 5, e282. [Google Scholar] [CrossRef]
- Shen, X.; Guo, Y.; Yu, J.; Qi, J.; Shi, W.; Wu, X.; Ni, H.; Ju, S. miRNA-202 in bone marrow stromal cells affects the growth and adhesion of multiple myeloma cells by regulating B cell-activating factor. Clin. Exp. Med. 2016, 16, 307–316. [Google Scholar] [CrossRef]
- Li, W.; Suwanwela, N.C.; Patumraj, S. Curcumin by down-regulating NF-kB and elevating Nrf2, reduces brain edema and neurological dysfunction after cerebral I/R. Microvasc. Res. 2016, 106, 117–127. [Google Scholar] [CrossRef]
- Riz, I.; Hawley, T.S.; Marsal, J.W.; Hawley, R.G. Noncanonical SQSTM1/p62-Nrf2 pathway activation mediates proteasome inhibitor resistance in multiple myeloma cells via redox, metabolic and translational reprogramming. Oncotarget 2016, 7, 66360–66385. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Xu, A.H.; Yang, Y.; Li, J. Role of Nrf2 in bone metabolism. J. Biomed. Sci. 2015, 22, 101. [Google Scholar] [CrossRef]
- Sun, Y.; Abdul Aziz, A.; Bowles, K.; Rushworth, S. High NRF2 expression controls endoplasmic reticulum stress induced apoptosis in multiple myeloma. Cancer Lett 2018, 412, 37–45. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol 2017, 7, 85. [Google Scholar] [CrossRef]
- Sun, Y.X.; Li, L.; Corry, K.A.; Zhang, P.; Yang, Y.; Himes, E.; Mihuti, C.L.; Nelson, C.; Dai, G.; Li, J. Deletion of Nrf2 reduces skeletal mechanical properties and decreases load-driven bone formation. Bone 2015, 74, 1–9. [Google Scholar] [CrossRef]
- Pellegrini, G.G.; Cregor, M.; McAndrews, K.; Morales, C.C.; McCabe, L.D.; McCabe, G.P.; Peacock, M.; Burr, D.; Weaver, C.; Bellido, T. Nrf2 regulates mass accrual and the antioxidant endogenous response in bone differently depending on the sex and age. PLoS ONE 2017, 12, e0171161. [Google Scholar] [CrossRef]
- Zhou, H.; Newnum, A.B.; Martin, J.R.; Li, P.; Nelson, M.T.; Moh, A.; Fu, X.Y.; Yokota, H.; Li, J. Osteoblast/osteocyte-specific inactivation of Stat3 decreases load-driven bone formation and accumulates reactive oxygen species. Bone 2011, 49, 404–411. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Bellido, T.; Roodman, G.D. Role of osteocytes in multiple myeloma bone disease. Curr. Opin. Supportive Palliat. Care 2014, 8, 407–413. [Google Scholar] [CrossRef]
- Romas, E.; Gillespie, M.T.; Martin, T.J. Involvement of receptor activator of NFkappaB ligand and tumor necrosis factor-alpha in bone destruction in rheumatoid arthritis. Bone 2002, 30, 340–346. [Google Scholar] [CrossRef]
- Kanzaki, H.; Shinohara, F.; Kajiya, M.; Kodama, T. The Keap1/Nrf2 protein axis plays a role in osteoclast differentiation by regulating intracellular reactive oxygen species signaling. J. Biol. Chem. 2013, 288, 23009–23020. [Google Scholar] [CrossRef]
- Xu, H.; Liu, T.; Li, J.; Xu, J.; Chen, F.; Hu, L.; Zhang, B.; Zi, C.; Wang, X.; Sheng, J. Oxidation derivative of (-)-epigallocatechin-3-gallate (EGCG) inhibits RANKL-induced osteoclastogenesis by suppressing RANK signaling pathways in RAW 264.7 cells. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 118, 109237. [Google Scholar] [CrossRef]
- Kanzaki, H.; Shinohara, F.; Itohiya, K.; Yamaguchi, Y.; Katsumata, Y.; Matsuzawa, M.; Fukaya, S.; Miyamoto, Y.; Wada, S.; Nakamura, Y. RANKL induces Bach1 nuclear import and attenuates Nrf2-mediated antioxidant enzymes, thereby augmenting intracellular reactive oxygen species signaling and osteoclastogenesis in mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 781–792. [Google Scholar] [CrossRef]
- Kanzaki, H.; Shinohara, F.; Kanako, I.; Yamaguchi, Y.; Fukaya, S.; Miyamoto, Y.; Wada, S.; Nakamura, Y. Molecular regulatory mechanisms of osteoclastogenesis through cytoprotective enzymes. Redox Biol. 2016, 8, 186–191. [Google Scholar] [CrossRef]
- Rana, T.; Schultz, M.A.; Freeman, M.L.; Biswas, S. Loss of Nrf2 accelerates ionizing radiation-induced bone loss by upregulating RANKL. Free Radic. Biol. Med. 2012, 53, 2298–2307. [Google Scholar] [CrossRef] [PubMed]
- Narimiya, T.; Kanzaki, H.; Yamaguchi, Y.; Wada, S.; Katsumata, Y.; Tanaka, K.; Tomonari, H. Nrf2 activation in osteoblasts suppresses osteoclastogenesis via inhibiting IL-6 expression. BONE Rep. 2019, 11, 100228. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Lee, Y.; Kim, K.H.; Lee, Z.H.; Joo, M.; Kim, H.H. Nrf2 is a novel regulator of bone acquisition. BONE 2014, 63, 36–46. [Google Scholar] [CrossRef]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.I.; Lee, G.R.; Lee, J.; Kim, N.; Kwon, M.; Kim, H.J.; Kim, N.Y.; Park, J.H.; Jeong, W. Dehydrocostus lactone inhibits NFATc1 via regulation of IKK, JNK, and Nrf2, thereby attenuating osteoclastogenesis. BMB Rep. 2020, 53, 218–222. [Google Scholar] [CrossRef]
- Lee, H.I.; Lee, J.; Hwang, D.; Lee, G.R.; Kim, N.; Kwon, M.; Lee, H.; Piao, D.; Kim, H.J.; Kim, N.Y.; et al. Dehydrocostus lactone suppresses osteoclast differentiation by regulating NFATc1 and inhibits osteoclast activation through modulating migration and lysosome function. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 9685–9694. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lee, K.S.; Yi, S.H.; Kook, S.H.; Lee, J.C. Acteoside suppresses RANKL-mediated osteoclastogenesis by inhibiting c-Fos induction and NF-κB pathway and attenuating ROS production. PLoS ONE 2013, 8, e80873. [Google Scholar] [CrossRef]
- Takayanagi, H. The role of NFAT in osteoclast formation. Ann. N. Y. Acad. Sci. 2007, 1116, 227–237. [Google Scholar] [CrossRef]
- Gangemi, S.; Allegra, A.; Alonci, A.; Cristani, M.; Russo, S.; Speciale, A.; Penna, G.; Spatari, G.; Cannavo, A.; Bellomo, G.; et al. Increase of novel biomarkers for oxidative stress in patients with plasma cell disorders and in multiple myeloma patients with bone lesions. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2012, 61, 1063–1067. [Google Scholar] [CrossRef]
- Hinoi, E.; Fujimori, S.; Wang, L.; Hojo, H.; Uno, K.; Yoneda, Y. Nrf2 negatively regulates osteoblast differentiation via interfering with Runx2-dependent transcriptional activation. J. Biol. Chem. 2006, 281, 18015–18024. [Google Scholar] [CrossRef]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010, 339, 189–195. [Google Scholar] [CrossRef]
- Ducy, P.; Starbuck, M.; Priemel, M.; Shen, J.; Pinero, G.; Geoffroy, V.; Amling, M.; Karsenty, G. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999, 13, 1025–1036. [Google Scholar] [CrossRef]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef]
- Inada, M.; Yasui, T.; Nomura, S.; Miyake, S.; Deguchi, K.; Himeno, M.; Sato, M.; Yamagiwa, H.; Kimura, T.; Yasui, N.; et al. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 1999, 214, 279–290. [Google Scholar]
- Komori, T. Roles of Runx2 in Skeletal Development. Adv. Exp. Med. Biol. 2017, 962, 83–93. [Google Scholar]
- Zhang, S.; Xiao, Z.; Luo, J.; He, N.; Mahlios, J.; Quarles, L.D. Dose-dependent effects of Runx2 on bone development. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2009, 24, 1889–1904. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 123, 590–600. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef]
- Wu, K.L.H.; Wu, C.W.; Chao, Y.M.; Hung, C.Y.; Chan, J.Y.H. Impaired Nrf2 regulation of mitochondrial biogenesis in rostral ventrolateral medulla on hypertension induced by systemic inflammation. Free Radic. Biol. Med. 2016, 97, 58–74. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef]
- Ludtmann, M.H.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef]
- Pang, S.; Lynn, D.A.; Lo, J.Y.; Paek, J.; Curran, S.P. SKN-1 and Nrf2 couples proline catabolism with lipid metabolism during nutrient deprivation. Nat. Commun. 2014, 5, 5048. [Google Scholar] [CrossRef]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef]
- Kang, T.C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. CB 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.X.; Sui, B.D.; Qiu, X.Y.; Hu, C.H.; Jin, Y. Mitochondrial Regulation of Stem Cells in Bone Homeostasis. Trends Mol. Med. 2020, 26, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.C.; Wisidagama, D.R.; Bensard, C.; Zhao, H.; Wei, P.; Tanner, J.; Flores, A.; Mohlman, J.; Sorensen, L.K.; Earl, C.S.; et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 2017, 19, 1027–1036. [Google Scholar] [CrossRef]
- Snoeck, H.W. Mitochondrial regulation of hematopoietic stem cells. Curr. Opin. Cell Biol. 2017, 49, 91–98. [Google Scholar] [CrossRef]
- Chen, C.T.; Shih, Y.R.; Kuo, T.K.; Lee, O.K.; Wei, Y.H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968. [Google Scholar] [CrossRef]
- Gao, J.; Feng, Z.; Wang, X.; Zeng, M.; Liu, J.; Han, S.; Xu, J.; Chen, L.; Cao, K.; Long, J.; et al. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2018, 25, 229–240. [Google Scholar] [CrossRef]
- Treiber, N.; Maity, P.; Singh, K.; Kohn, M.; Keist, A.F.; Ferchiu, F.; Sante, L.; Frese, S.; Bloch, W.; Kreppel, F.; et al. Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell 2011, 10, 239–254. [Google Scholar] [CrossRef]
- Liu, S.; Yang, L.; Mu, S.; Fu, Q. Epigallocatechin-3-Gallate Ameliorates Glucocorticoid-Induced Osteoporosis of Rats in Vivo and in Vitro. Front. Pharmacol. 2018, 9, 447. [Google Scholar] [CrossRef]
- Tang, X.; Ma, S.; Li, Y.; Sun, Y.; Zhang, K.; Zhou, Q.; Yu, R. Evaluating the Activity of Sodium Butyrate to Prevent Osteoporosis in Rats by Promoting Osteal GSK-3beta/Nrf2 Signaling and Mitochondrial Function. J. Agric. Food Chem. 2020, 68, 6588–6603. [Google Scholar] [CrossRef]
- Zeng, R.; Faccio, R.; Novack, D.V. Alternative NF-kappaB Regulates RANKL-Induced Osteoclast Differentiation and Mitochondrial Biogenesis via Independent Mechanisms. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2015, 30, 2287–2299. [Google Scholar] [CrossRef]
- Jin, Z.; Wei, W.; Yang, M.; Du, Y.; Wan, Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. 2014, 20, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, Y.D.; Kim, H.J.; Lee, Z.H.; Kim, H.H. SOD2 and Sirt3 Control Osteoclastogenesis by Regulating Mitochondrial ROS. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Bartell, S.M.; Kim, H.N.; Ambrogini, E.; Han, L.; Iyer, S.; Serra Ucer, S.; Rabinovitch, P.; Jilka, R.L.; Weinstein, R.S.; Zhao, H.; et al. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat. Commun. 2014, 5, 3773. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.L.Z.; Manar, S.; Shafat, K.; Bowles, M.; Rushworth, S.A. Dual Activation of NRF2 in Multiple Myeloma and Bone Marrow Mesenchymal Stromal Cells Regulates Chemotherapy Resistance. Blood 2016, 128, 3287. [Google Scholar]
- Liu, H.Y.; Tuckett, A.Z.; Fennell, M.; Garippa, R.; Zakrzewski, J.L. Repurposing of the CDK inhibitor PHA-767491 as a NRF2 inhibitor drug candidate for cancer therapy via redox modulation. Invest. New Drugs 2018, 36, 590–600. [Google Scholar] [CrossRef]
- Harder, B.; Tian, W.; La Clair, J.J.; Tan, A.C.; Ooi, A.; Chapman, E.; Zhang, D.D. Brusatol overcomes chemoresistance through inhibition of protein translation. Mol. Carcinog. 2017, 56, 1493–1500. [Google Scholar] [CrossRef]
- Song, I.S.; Kim, H.K.; Lee, S.R.; Jeong, S.H.; Kim, N.; Ko, K.S.; Rhee, B.D.; Han, J. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int. J. Cancer 2013, 133, 1357–1367. [Google Scholar] [CrossRef]
- Zhan, X.; Yu, W.; Franqui-Machin, R.; Bates, M.L.; Nadiminti, K.; Cao, H.; Amendt, B.A.; Jethava, Y.; Frech, I.; Zhan, F.; et al. Alteration of mitochondrial biogenesis promotes disease progression in multiple myeloma. Oncotarget 2017, 8, 111213–111224. [Google Scholar] [CrossRef]
- Ortiz-Ruiz, A.; Ruiz-Heredia, Y.; Samur, M.; Aguilar-Garrido, P.; Morales, M.L.; Valeri, A.; Garcia-Ortiz, A.; Barrio, S.; Bárcena, C.; Garcia-Martin, R.M. Increase of Mitochondrial Activity Contributes to the Bortezomib-Relapsed in Multiple Myeloma, a Novel Therapeutic Opportunity; American Society of Hematology: Washington, DC, USA, 2019. [Google Scholar]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Yang, Y.-X.; Zhe, H.; He, Z.-X.; Zhou, S.-F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Dev. Ther. 2014, 8, 2075–2088. [Google Scholar]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yen, C.-H.; Hsu, C.-M.; Hsiao, S.Y.; Hsiao, H.-H. Pathogenic Mechanisms of Myeloma Bone Disease and Possible Roles for NRF2. Int. J. Mol. Sci. 2020, 21, 6723. https://doi.org/10.3390/ijms21186723
Yen C-H, Hsu C-M, Hsiao SY, Hsiao H-H. Pathogenic Mechanisms of Myeloma Bone Disease and Possible Roles for NRF2. International Journal of Molecular Sciences. 2020; 21(18):6723. https://doi.org/10.3390/ijms21186723
Chicago/Turabian StyleYen, Chia-Hung, Chin-Mu Hsu, Samuel Yien Hsiao, and Hui-Hua Hsiao. 2020. "Pathogenic Mechanisms of Myeloma Bone Disease and Possible Roles for NRF2" International Journal of Molecular Sciences 21, no. 18: 6723. https://doi.org/10.3390/ijms21186723
APA StyleYen, C.-H., Hsu, C.-M., Hsiao, S. Y., & Hsiao, H.-H. (2020). Pathogenic Mechanisms of Myeloma Bone Disease and Possible Roles for NRF2. International Journal of Molecular Sciences, 21(18), 6723. https://doi.org/10.3390/ijms21186723