Disorders of Human Coenzyme Q10 Metabolism: An Overview


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
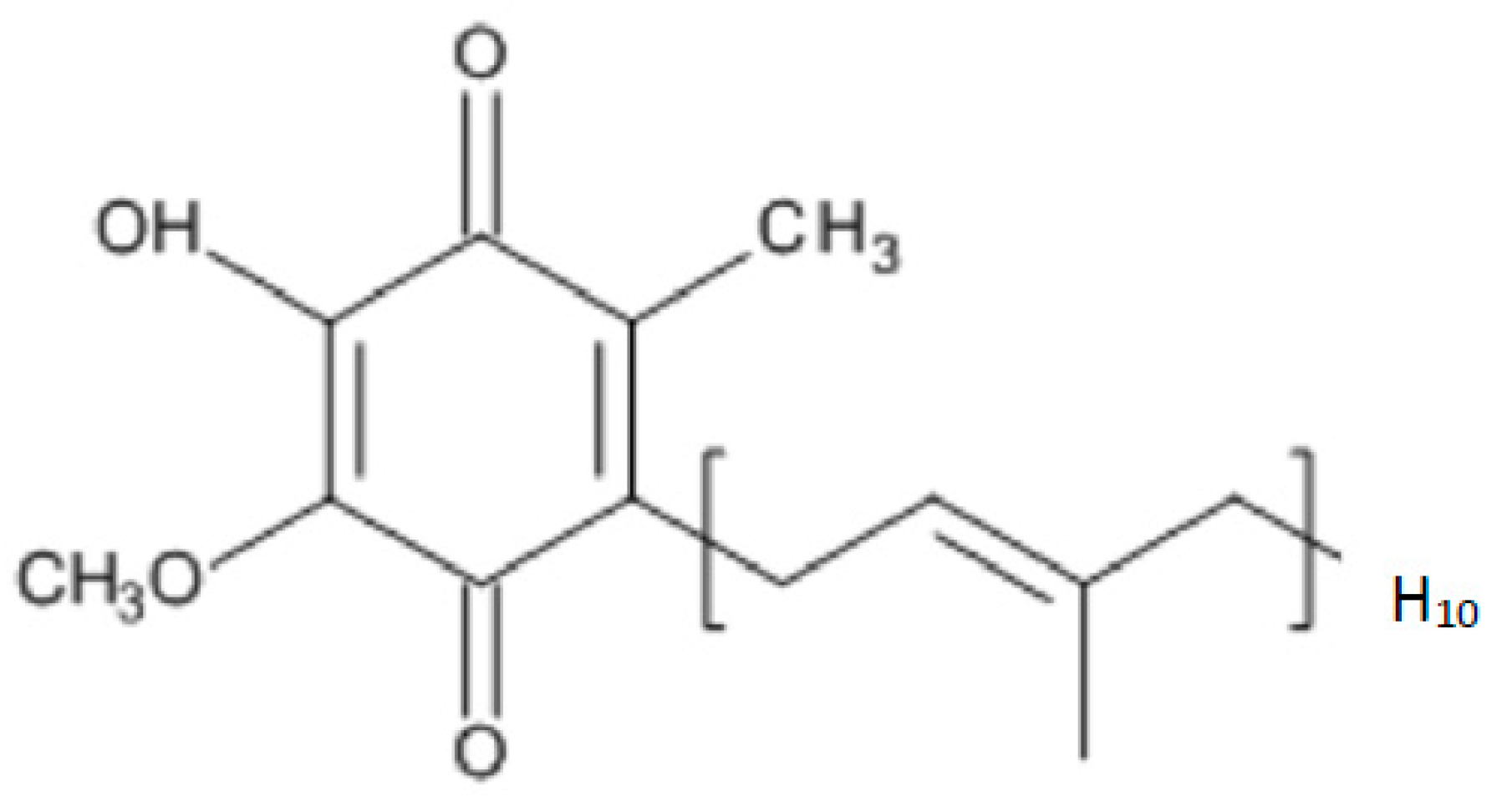
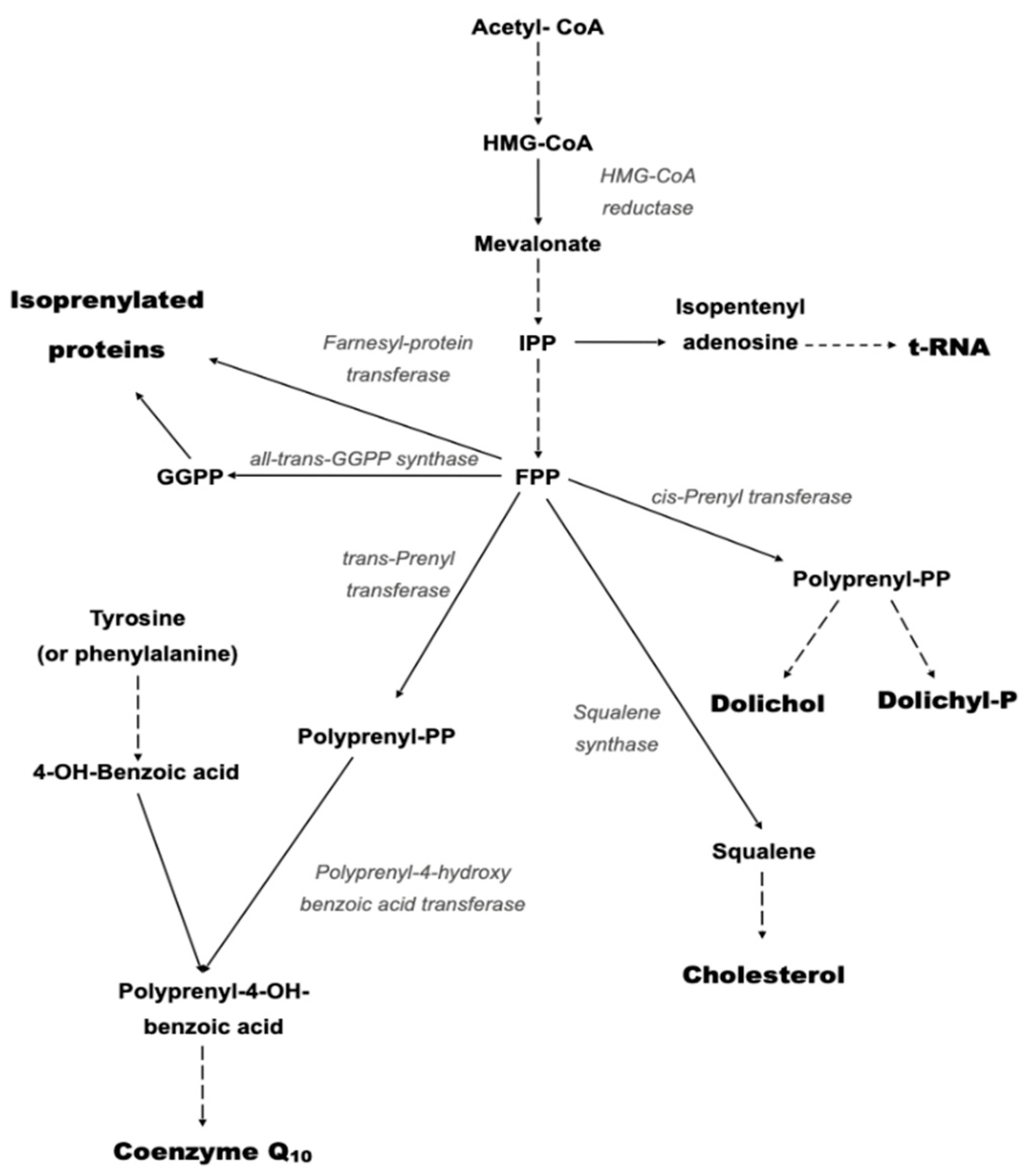
1. Introduction
2. Deficiency of CoQ10
2.1. Primary CoQ10 Deficiency
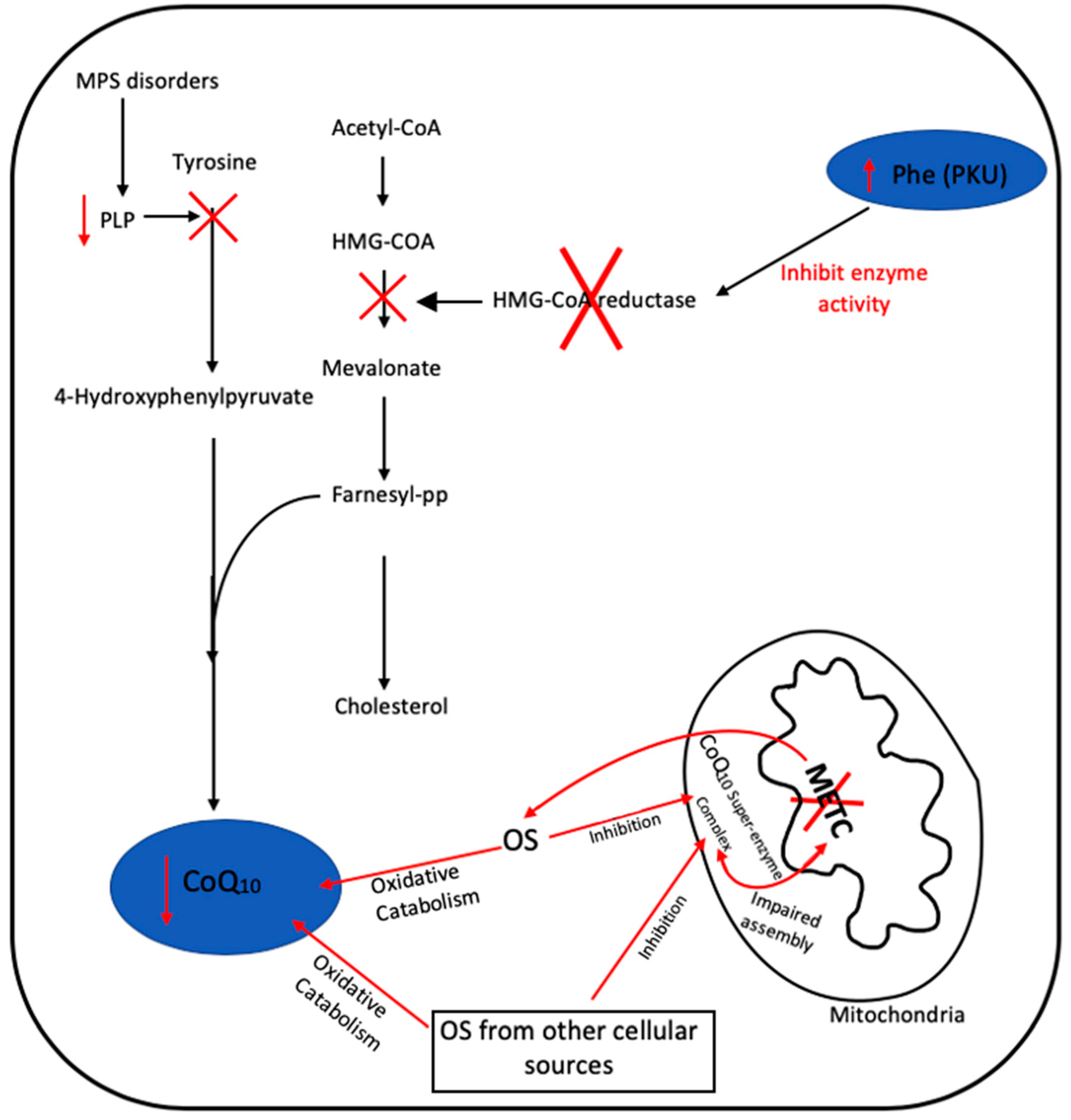
2.2. Secondary CoQ10 Deficiency
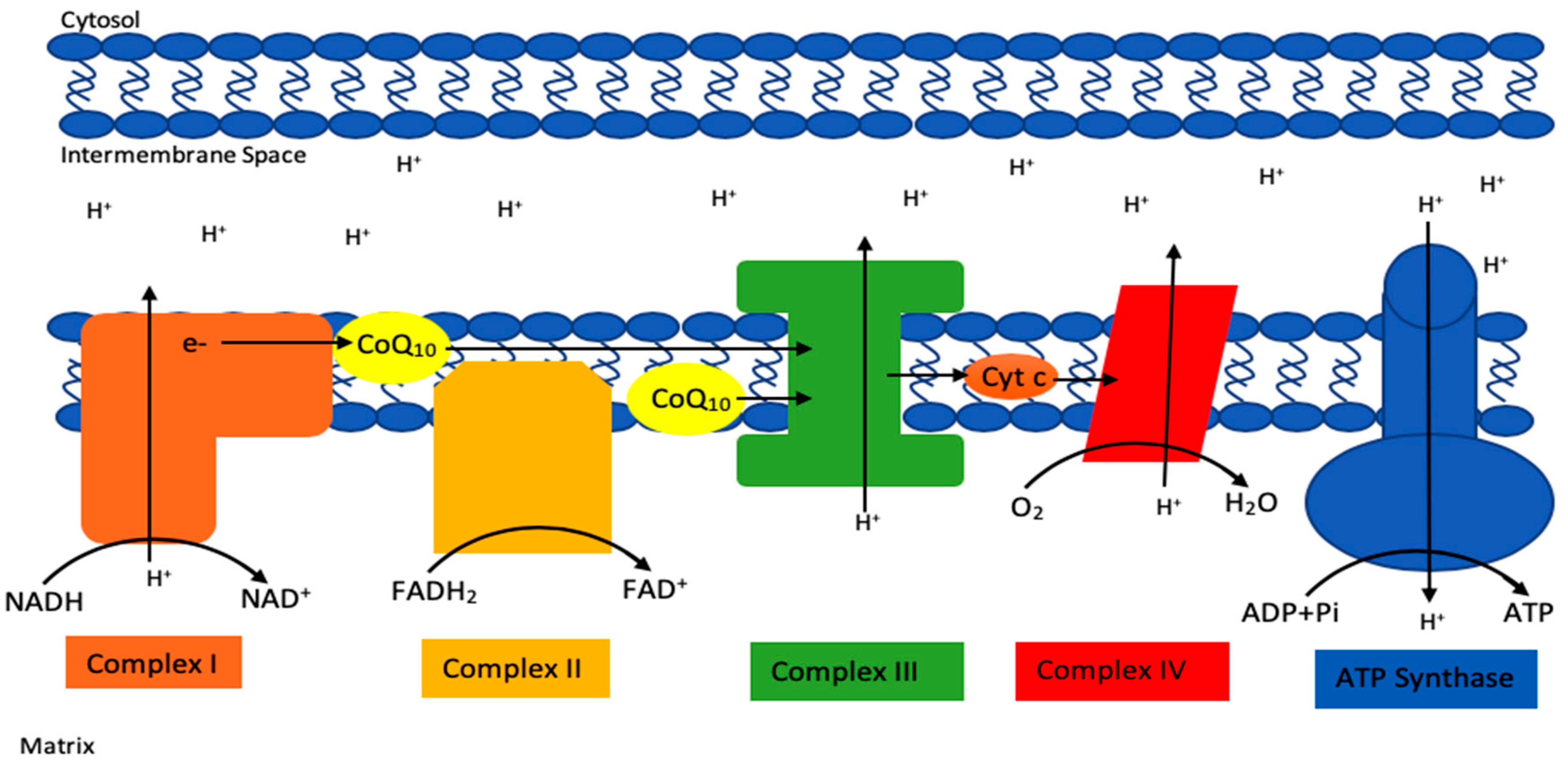
3. Clinical Consequences of a CoQ10 Deficiency
4. Clinical Assessment of CoQ10
5. Supplementation with CoQ10
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| CKD | Chronic kidney disease |
| CoQ10 | Coenzyme Q10 |
| METC | Mitochondrial electron transport chain |
| NAFLD | Non-alcoholic fatty liver disease |
| OS | Oxidative stress |
References
- Crane, F.L. Biochemical functions of coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef]
- Hargreaves, I.P. Ubiquinone: Cholesterol’s reclusive cousin. Ann. Clin. Biochem. 2003, 40, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.J.; Vazquez-Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochim. Biophys. Acta Bioenergetics 2016, 1857, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Rodick, T.C.; Seibels, D.R.; Babu, J.R.; Huggins, K.W.; Ren, G.; Mathews, S.T. Potential role of coenzyme Q10 in health and disease conditions. Nutr. Diet. Suppl. 2018, 10, 1–11. [Google Scholar] [CrossRef]
- Schmelzer, C.; Lindner, I.; Rimbach, G.; Niklowitz, P.; Menke, T.; Döring, F. Functions of coenzyme Q10in inflammation and gene expression. BioFactors 2008, 32, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.A.; Heales, S.J.; Rahman, K.; Sexton, D.W.; Hargreaves, I. The Effect of Cellular Coenzyme Q10 Deficiency on Lysosomal Acidification. J. Clin. Med. 2020, 9, 1923. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Luna-Sanchez, M.; Ziosi, M.; Hidalgo-Gutierrez, A.; Kleiner, G.; López, L.C. The Role of Sulfide Oxidation Impairment in the Pathogenesis of Primary CoQ Deficiency. Front. Physiol. 2017, 8, 525. [Google Scholar] [CrossRef]
- Salvi, F.; Gadda, G. Human choline dehydrogenase: Medical promises and biochemical challenges. Arch. Biochem. Biophys. 2013, 537, 243–252. [Google Scholar] [CrossRef]
- Hancock, C.N.; Liu, W.; Alvord, W.G.; Phang, J.M. Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino Acids 2015, 48, 859–872. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Yubero-Serrano, E.M.; Villalba, J.M.; Miranda, J.L. Coenzyme Q10: From bench to clinic in aging diseases, a translational review. Crit. Rev. Food Sci. Nutr. 2018, 59, 2240–2257. [Google Scholar] [CrossRef]
- Weber, C.; Bysted, A.; Hłlmer, G. The coenzyme Q10 content of the average Danish diet. Int. J. Vitam. Nutr. Res. 1997, 67, 123–129. [Google Scholar] [PubMed]
- Martelli, A.; Testai, L.; Colletti, A.; Cicero, A.F.G. Coenzyme Q10: Clinical Applications in Cardiovascular Diseases. Antioxidants 2020, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Kalén, A.; Appelkvist, E.L.; Dallner, G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 1989, 24, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P.; Duncan, A.J.; Heales, S.J.; Land, J.M.; Hargreaves, I.P. The Effect of HMG-CoA Reductase Inhibitors on Coenzyme Q10. Drug Saf. 2005, 28, 659–676. [Google Scholar] [CrossRef] [PubMed]
- Potgieter, M.; Pretorius, E.; Pepper, M.S. Primary and secondary coenzyme Q10 deficiency: The role of therapeutic supplementation. Nutr. Rev. 2013, 71, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Trevisson, E.; Doimo, M.; Navas, P. Primary Coenzyme Q10 Deficiency; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef] [PubMed]
- Allan, C.M.; Awad, A.M.; Johnson, J.S.; Shirasaki, D.I.; Wang, C.; Blaby-Haas, C.E.; Merchant, S.S.; Loo, J.A.; Clarke, C.F. Identification of Coq11, a New Coenzyme Q Biosynthetic Protein in the CoQ-Synthome in Saccharomyces cerevisiae. J. Boil. Chem. 2015, 290, 7517–7534. [Google Scholar] [CrossRef] [PubMed]
- Forsgren, M.; Attersand, A.; Lake, S.; Grünler, J.; Swiezewska, E.; Dallner, G.; Climent, I. Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem. J. 2004, 382, 519–526. [Google Scholar] [CrossRef]
- Tran, U.C.; Clarke, C.F. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion 2007, 7, S62–S71. [Google Scholar] [CrossRef]
- Ozeir, M.; Mühlenhoff, U.; Webert, H.; Lill, R.; Fontecave, M.; Pierrel, F. Coenzyme Q Biosynthesis: Coq6 Is Required for the C5-Hydroxylation Reaction and Substrate Analogs Rescue Coq6 Deficiency. Chem. Boil. 2011, 18, 1134–1142. [Google Scholar] [CrossRef]
- Cuiwen, H.; Black, D.S.; Nguyen, T.P.T.; Wang, C.; Srinivasan, C.; Clarke, C.F. Yeast Coq9 controls deamination of coenzyme Q intermediates that derive from para-aminobenzoic acid. Biochim. Biophys. Acta Bioenergetics 2015, 1851, 1227–1239. [Google Scholar] [CrossRef][Green Version]
- Ozeir, M.; Pelosi, L.; Ismail, A.; Mellot-Draznieks, C.; Fontecave, M.; Pierrel, F. Coq6 Is Responsible for the C4-deamination Reaction in Coenzyme Q Biosynthesis in Saccharomyces cerevisiae. J. Boil. Chem. 2015, 290, 24140–24151. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.X.; Hsieh, E.J.; Watanabe, S.; Allan, C.M.; Chen, J.Y.; Tran, U.C.; Clarke, C.F. Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of Coq polypeptides in yeast coq8 mutants. Biochim. Biophys. Acta Mol. Cell Boil. Lipids 2011, 1811, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Lohman, D.C.; Forouhar, F.; Beebe, E.T.; Stefely, M.S.; Minogue, C.E.; Ulbrich, A.; Stefely, J.A.; Sukumar, S.; Luna-Sánchez, M.; Jochem, A.; et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, E4697–E4705. [Google Scholar] [CrossRef]
- Barros, M.H.; Johnson, A.; Gin, P.; Marbois, B.N.; Clarke, C.F.; Tzagoloff, A. TheSaccharomyces cerevisiae COQ10Gene Encodes a START Domain Protein Required for Function of Coenzyme Q in Respiration. J. Boil. Chem. 2005, 280, 42627–42635. [Google Scholar] [CrossRef]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chrétien, M.; Delahodde, A.; Bacq, D.; de Lonlay, P.; Munnich, A.; Rötig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef]
- Ivanyi, B.; Rácz, G.Z.; Gál, P.; Brinyiczki, K.; Bodi, I.; Kalmar, T.; Maróti, Z.; Bereczki, C. Diffuse mesangial sclerosis in a PDSS2 mutation-induced coenzyme Q10 deficiency. Pediatr. Nephrol. 2017, 33, 439–446. [Google Scholar] [CrossRef]
- Bosch, A.M.; Kamsteeg, E.-J.; Rodenburg, R.; van Deutekom, A.W.; Buis, D.R.; Engelen, M.; Cobben, J.M. Coenzyme Q10 deficiency due to a COQ4 gene defect causes childhood-onset spinocerebellar ataxia and stroke-like episodes. Mol. Genet. Metab. Rep. 2018, 17, 19–21. [Google Scholar] [CrossRef]
- Malicdan, M.C.V.; Vilboux, T.; Ben-Zeev, B.; Guo, J.; Eliyahu, A.; Pode-Shakked, B.; Dori, A.; Kakani, S.; Chandrasekharappa, S.C.; Ferreira, C.R. A novel inborn error of the coenzyme Q10 biosynthesis pathway: Cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Human Mutat. 2018, 39, 69–79. [Google Scholar] [CrossRef]
- Doimo, M.; Desbats, M.A.; Cerqua, C.; Cassina, M.; Trevisson, E.; Salviati, L. Genetics of Coenzyme Q10 Deficiency. Mol. Syndr. 2014, 5, 156–162. [Google Scholar] [CrossRef]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4–dihydroxybensoic acid. J. Med. Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Ruiz-Lopez, M.; Slow, E.; Tarnopolsky, M.; Lang, A.E.; Munhoz, R.P. ADCK3-related Coenzyme Q10 Deficiency: A Potentially Treatable Genetic Disease. Mov. Disord. Clin. Pr. 2018, 5, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Wang, Q.; Wang, J.; Liu, F.; Shen, H.; Fu, H.; Mao, J. Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal disease and ADCK4 mutation: Case reports and literature review. Medicine 2017, 96. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; López, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.I.; Hardy, J.; et al. A Nonsense Mutation in COQ9 Causes Autosomal-Recessive Neonatal-Onset Primary Coenzyme Q10 Deficiency: A Potentially Treatable Form of Mitochondrial Disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Montero, R.; Santos-Ocaña, C.; Salviati, L.; Navas, P.; Artuch, R. Molecular diagnosis of coenzyme Q10 deficiency: An update. Expert Rev. Mol. Diagn. 2018, 18, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Castellotti, B.; Mariotti, C.; Rimoldi, M.; Fancellu, R.; Plumari, M.; Caimi, S.; Uziel, G.; Nardocci, N.; Moroni, I.; Zorzi, G.; et al. Ataxia with oculomotor apraxia type1 (AOA1): Novel and recurrent aprataxin mutations, coenzyme Q10 analyses, and clinical findings in Italian patients. Neurogenetics 2011, 12, 193–201. [Google Scholar] [CrossRef]
- Aeby, A.; Sznajer, Y.; Cave, H.; Rebuffat, E.; van Coster, R.; Rigal, O.; van Bogaert, P. Cardiofaciocutaneous (CFC) syndrome associated with muscular coenzyme Q10 deficiency. J. Inherit. Metab. Dis. 2007, 30, 827. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef]
- Balreira, A.; Boczonadi, V.; Barca, E.; Pyle, A.; Bánsági, B.; Appleton, M.; Graham, C.; Hargreaves, I.P.; Rasic, V.M.; Lochmüller, H.; et al. ANO10 mutations cause ataxia and coenzyme Q10 deficiency. J. Neurol. 2014, 261, 2192–2198. [Google Scholar] [CrossRef]
- Hargreaves, I.P. Coenzyme Q10 as a therapy for mitochondrial disease. Int. J. Biochem. Cell Boil. 2014, 49, 105–111. [Google Scholar] [CrossRef]
- Montero, R.; Grazina, M.; López-Gallardo, E.; Montoya, J.; Briones, P.; Navarro-Sastre, A.; Land, J.M.; Hargreaves, I.P.; Artuch, R.; O’Callaghan, M.D.M.; et al. Coenzyme Q10 deficiency in mitochondrial DNA depletion syndromes. Mitochondrion 2013, 13, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Hargreaves, I. Coenzyme Q10 and Degenerative Disorders Affecting Longevity: An Overview. Antioxidants 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.; Mantle, D.; Milford, D. Chronic kidney disease and coenzyme Q10 supplementation. J. Kidney Care 2019, 4, 82–90. [Google Scholar] [CrossRef]
- Mantle, D. Coenzyme Q10 supplementation for diabetes and its complications: An overview. Br. J. Diabetes Vasc. Dis. 2017, 17, 145–148. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Chaudhuri, R.; Yang, P.; Maghzal, G.J.; Thomas, K.C.; Krycer, J.R.; Humphrey, S.; Parker, B.L.; Fisher-Wellman, K.H.; Meoli, C.C.; et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. eLife 2018, 7, 32111. [Google Scholar] [CrossRef]
- Montero, R.; Yubero, D.; Salgado, M.C.; González, M.J.; Campistol, J.; O’Callaghan, M.D.M.; Pineda, M.; Delgadillo, V.; Maynou, J.; Fernandez, G.; et al. Plasma coenzyme Q10 status is impaired in selected genetic conditions. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Matalonga, L.; Arias, Á.; Coll, M.J.; García-Villoria, J.; Gort, L.; Ribes, A. Treatment effect of coenzyme Q10 and an antioxidant cocktail in fibroblasts of patients with Sanfilippo disease. J. Inherit. Metab. Dis. 2013, 37, 439–446. [Google Scholar] [CrossRef]
- Yubero, D.; Montero, R.; Martín, M.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Rodríguez-Aguilera, J.C.; Hargreaves, I.; Salviati, L.; et al. Secondary coenzyme Q 10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef]
- Neergheen, V.; Hargreaves, I. Secondary coenzyme Q10 deficiency: Causes and consequence. Coenzyme Q10-Uses, Health Effects and Role in Disease; Nova Science Publishers: New York, NY, USA, 2018; pp. 89–111. [Google Scholar]
- Fernández, A.M.; Cordero, M.D.; Garrido-Maraver, J.; Alcocer-Gómez, E.; Barquero, N.C.; Carmona-López, M.I.; Sánchez-Alcázar, J.A.; de Miguel, M. Oral treatment with amitriptyline induces coenzyme Q deficiency and oxidative stress in psychiatric patients. J. Psychiatr. Res. 2012, 46, 341–345. [Google Scholar] [CrossRef]
- Luna-Sánchez, M.; Hidalgo-Gutiérrez, A.; Hildebrandt, T.M.; Chaves-Serrano, J.; Barriocanal-Casado, E.; Santos-Fandila, Á.; Romero, M.; Sayed, R.K.A.; Duarte, J.; Prokisch, H.; et al. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol. Med. 2016, 9, 78–95. [Google Scholar] [CrossRef]
- Ziosi, M.; di Meo, I.; Kleiner, G.; Gao, X.; Barca, E.; Sanchez-Quintero, M.J.; Tadesse, S.; Jiang, H.; Qiao, C.; Rodenburg, R.; et al. Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol. Med. 2016, 9, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Ogasahara, S.; Engel, A.G.; Frens, D.; Mack, D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc. Natl. Acad. Sci. USA 1989, 86, 2379–2382. [Google Scholar] [CrossRef] [PubMed]
- Emmanuele, V.; López, L.C.; Berardo, A.; Naini, A.; Tadesse, S.; Wen, B.; D’Agostino, E.; Solomon, M.; DiMauro, S.; Quinzii, C.; et al. Heterogeneity of Coenzyme Q10Deficiency. Arch. Neurol. 2012, 69, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P.; Lane, A.; Sleiman, P.M. The coenzyme Q10 status of the brain regions of Parkinson’s disease patients. Neurosci. Lett. 2008, 447, 17–19. [Google Scholar] [CrossRef]
- Mantle, D. Coenzyme Q10 and cardiovascular disease: An overview. Br. J. Cardiol. 2015, 22, 1–7. [Google Scholar]
- Hargreaves, I.P.; Sheena, Y.; Land, J.M.; Heales, S.J.R. Glutathione deficiency in patients with mitochondrial disease: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2005, 28, 81–88. [Google Scholar] [CrossRef]
- Päivä, H.; Thelen, K.M.; Coster, R.; Smet, J.; Paepe, B.; Mattila, K.M.; Laaksonen, R.; Lehtimäki, T.; von Bergmann, K.; Lütjohann, D. High-dose statins and skeletal muscle metabolism in humans: A randomized, controlled trial. Clin. Pharmacol. Ther. 2005, 78, 60–68. [Google Scholar] [CrossRef]
- Yubero, D.; Montero, R.; Artuch, R.; Land, J.M.; Heales, S.J.; Hargreaves, I. Biochemical Diagnosis of Coenzyme Q10 Deficiency. Mol. Syndr. 2014, 5, 147–155. [Google Scholar] [CrossRef]
- Molyneux, S.L.; Young, J.M.; Florkowski, C.M.; Lever, M.; George, P.M. Coenzyme Q10: Is There a Clinical Role and a Case for Measurement? Clin. Biochem. Rev. 2008, 29, 71–82. [Google Scholar]
- Duberley, K.E.C.; Abramov, A.Y.; Chalasani, A.; Heales, S.J.; Rahman, S.; Hargreaves, I.P. Human neuronal coenzyme Q10 deficiency results in global loss of mitochondrial respiratory chain activity, increased mitochondrial oxidative stress and reversal of ATP synthase activity: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2012, 36, 63–73. [Google Scholar] [CrossRef]
- Duncan, A.J.; Heales, S.J.; Mills, K.; Eaton, S.; Land, J.M.; Hargreaves, I.P. Determination of Coenzyme Q10 Status in Blood Mononuclear Cells, Skeletal Muscle, and Plasma by HPLC with Di-Propoxy-Coenzyme Q10 as an Internal Standard. Clin. Chem. 2005, 51, 2380–2382. [Google Scholar] [CrossRef] [PubMed]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta Mol. Basis Dis. 1995, 1271, 195–204. [Google Scholar] [CrossRef]
- Duberley, K.E.C.; Hargreaves, I.P.; Chaiwatanasirikul, K.-A.; Heales, S.J.R.; Land, J.M.; Rahman, S.; Mills, K.; Eaton, S. Coenzyme Q10quantification in muscle, fibroblasts and cerebrospinal fluid by liquid chromatography/tandem mass spectrometry using a novel deuterated internal standard. Rapid Commun. Mass Spectrom. 2013, 27, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Montero, R.; Ramos, M.; Neergheen, V.; Navas, P.; Artuch, R.; Hargreaves, I. Determination of urinary coenzyme Q10by HPLC with electrochemical detection: Reference values for a paediatric population. BioFactors 2015, 41, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Hargreaves, I.P. Ataxia and coenzyme Q10: An overview. Br. J. Neurosci. Nurs. 2018, 14, 108–114. [Google Scholar] [CrossRef]
- Musumeci, O.; Naini, A.; Slonim, A.E.; Skavin, N.; Hadjigeorgiou, G.L.; Krawiecki, N.; Weissman, B.M.; Tsao, C.-Y.; Mendell, J.R.; Shanske, S.; et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurol. 2001, 56, 849–855. [Google Scholar] [CrossRef]
- Lamperti, C.; Naini, A.; Hirano, M.; de Vivo, D.; Bertini, E.; Servidei, S.; Valeriani, M.; Lynch, D.; Banwell, B.; Berg, M.; et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology 2003, 60, 1206–1208. [Google Scholar] [CrossRef]
- Hidalgo-Gutiérrez, A.; Barriocanal-Casado, E.; Bakkali, M.; Díaz-Casado, M.E.; Sánchez-Maldonado, L.; Romero, M.; Sayed, R.K.A.; Prehn, C.; Escames, G.; Duarte, J.; et al. β- RA reduces DMQ /CoQ ratio and rescues the encephalopathic phenotype in Coq9 R239X mice. EMBO Mol. Med. 2018, 11, e9466. [Google Scholar] [CrossRef]
- Diomedi-Camassei, F.; di Giandomenico, S.; Santorelli, F.M.; Caridi, G.; Piemonte, F.; Montini, G.; Ghiggeri, G.M.; Murer, L.; Barisoni, L.; Pastore, A.; et al. COQ2 Nephropathy: A Newly Described Inherited Mitochondriopathy with Primary Renal Involvement. J. Am. Soc. Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef]
- Salviati, L.; Trevisson, E.; Rodríguez-Hernández, Á.; Casarin, A.; Pertegato, V.; Doimo, M.; Cassina, M.; Agosto, C.; Desbats, M.A.; Sartori, G.; et al. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J. Med. Genet. 2012, 49, 187–191. [Google Scholar] [CrossRef]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steurer, G.; Littarru, G.P. The Effect of Coenzyme Q 10 on Morbidity and Mortality in Chronic Heart Failure. JACC Hear. Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef]
- Mortensen, A.L.; Rosenfeldt, F.; Filipiak, K.J. Effect of coenzyme Q10 in Europeans with chronic heart failure: A sub-group analysis of the Q-SYMBIO randomized double-blind trial. Cardiol. J. 2019, 26, 147–156. [Google Scholar] [CrossRef] [PubMed]
- el Ghoroury, E.A.; Raslan, H.M.; Badawy, E.; el Saaid, G.S.; Agybi, M.H.; Siam, I.; I Salem, S. Malondialdehyde and coenzyme Q10 in platelets and serum in type 2 diabetes mellitus: Correlation with glycemic control. Blood Coagul. Fibrinolysis 2009, 20, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, R.K.; Hosseinzadeh-Attar, M.J.; Eshraghian, M.R.; Nakhjavani, M.; Khorami, E.; Esteghamati, A. The effect of coenzyme Q10 supplementation on metabolic status of type 2 diabetic patients. Minerva Gastroenterol. Dietol. 2013, 59, 231–236. [Google Scholar]
- Zahedi, H.; Eghtesadi, S.; Seifirad, S.; Rezaee, N.; Shidfar, F.; Heydari, I.; Golestan, B.; Jazayeri, S. Effects of CoQ10 Supplementation on Lipid Profiles and Glycemic Control in Patients with Type 2 Diabetes: A randomized, double blind, placebo-controlled trial. J. Diabetes Metab. Disord. 2014, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh-Attar, M.; Mohammadi, R.K.; Eshraghian, M.; Nakhjavani, M.; Khorrami, E.; Ebadi, M.; Esteghamati, A. Reduction in asymmetric dimethylarginine plasma levels by coenzyme Q10 supplementation in patients with type 2 diabetes mellitus. Minerva Endocrinol. 2015, 40, 259–266. [Google Scholar] [PubMed]
- Zhang, S.-Y.; Yang, K.-L.; Zeng, L.-T.; Wu, X.-H.; Huang, H.-Y. Effectiveness of Coenzyme Q10 Supplementation for Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Int. J. Endocrinol. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Macunluoglu, B.; Kaya, Y.; Atakan, A.; Ari, E.; Kaspar, C.; Demir, H.; Alp, H.H.; Asicioglu, E.; Kedrah, A.E. Serum coenzyme Q 10 levels are associated with coronary flow reserve in hemodialysis patients. Hemodial. Int. 2012, 17, 339–345. [Google Scholar] [CrossRef]
- Triolo, L.; Lippa, S.; Oradei, A.; de Sole, P.; Mori, R. Serum Coenzyme Q10 in Uremic Patients on Chronic Hemodialysis. Nephron 1994, 66, 153–156. [Google Scholar] [CrossRef]
- Yeung, C.K.; Billings, F.T.; Claessens, A.J.; Roshanravan, B.; Linke, L.; Sundell, M.B.; Ahmad, S.; Shao, B.; Shen, D.D.; Ikizler, T.A.; et al. Coenzyme Q10 dose-escalation study in hemodialysis patients: Safety, tolerability, and effect on oxidative stress. BMC Nephrol. 2015, 16, 183. [Google Scholar] [CrossRef]
- Singh, R.B.; Khanna, H.K.; Niaz, M.A. Randomized, Double-blind Placebo-controlled Trial of Coenzyme Q10 in Chronic Renal Failure: Discovery of a New Role. J. Nutr. Environ. Med. 2000, 10, 281–288. [Google Scholar] [CrossRef]
- Emma, F.; Montini, G.; Parikh, S.M.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic Markers of Lipid Peroxidation and Antioxidants in Patients with Nonalcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef]
- Farhangi, M.A.; Alipour, B.; Jafarvand, E.; Khoshbaten, M. Oral Coenzyme Q10 Supplementation in Patients with Nonalcoholic Fatty Liver Disease: Effects on Serum Vaspin, Chemerin, Pentraxin 3, Insulin Resistance and Oxidative Stress. Arch. Med. Res. 2014, 45, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Farsi, F.; Mohammad-Shahi, M.; Alavinejad, P.; Rezazadeh, A.; Zarei, M.; Engali, K.A. Functions of Coenzyme Q10 Supplementation on Liver Enzymes, Markers of Systemic Inflammation, and Adipokines in Patients Affected by Nonalcoholic Fatty Liver Disease: A Double-Blind, Placebo-Controlled, Randomized Clinical Trial. J. Am. Coll. Nutr. 2015, 35, 1–8. [Google Scholar] [CrossRef]
- López-Lluch, G.; del Pozo-Cruz, J.; Sánchez-Cuesta, A.; Cortés-Rodríguez, A.B.; Navas, P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition 2019, 57, 133–140. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hargreaves, I.; Heaton, R.A.; Mantle, D. Disorders of Human Coenzyme Q10 Metabolism: An Overview. Int. J. Mol. Sci. 2020, 21, 6695. https://doi.org/10.3390/ijms21186695
Hargreaves I, Heaton RA, Mantle D. Disorders of Human Coenzyme Q10 Metabolism: An Overview. International Journal of Molecular Sciences. 2020; 21(18):6695. https://doi.org/10.3390/ijms21186695
Chicago/Turabian StyleHargreaves, Iain, Robert A. Heaton, and David Mantle. 2020. "Disorders of Human Coenzyme Q10 Metabolism: An Overview" International Journal of Molecular Sciences 21, no. 18: 6695. https://doi.org/10.3390/ijms21186695
APA StyleHargreaves, I., Heaton, R. A., & Mantle, D. (2020). Disorders of Human Coenzyme Q10 Metabolism: An Overview. International Journal of Molecular Sciences, 21(18), 6695. https://doi.org/10.3390/ijms21186695