Mouse Ataxin-2 Expansion Downregulates CamKII and Other Calcium Signaling Factors, Impairing Granule—Purkinje Neuron Synaptic Strength

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Transcriptome Profile of Atxn2-CAG100-KIN Mouse Cerebellum before Disease Onset
2.2. Ataxin-2 Pathology Alters the Expression of Ca2+ Channels and Transporters
2.3. Subcellular Ca2+ Imbalance Promotes Ataxin-2 Relocalization into Stress Granules
2.4. Pre-Onset Dysregulation of the Ca2+/Calmodulin-Dependent Protein Kinase Pathway
2.5. Impact of Ataxin-2 Pathology on the CamKIV-Modulated RNA Splicing Factor Khdrbs1/Sam68
2.6. Alternative Splicing Profile of Nrxn1 in Ataxin-2 Pathology
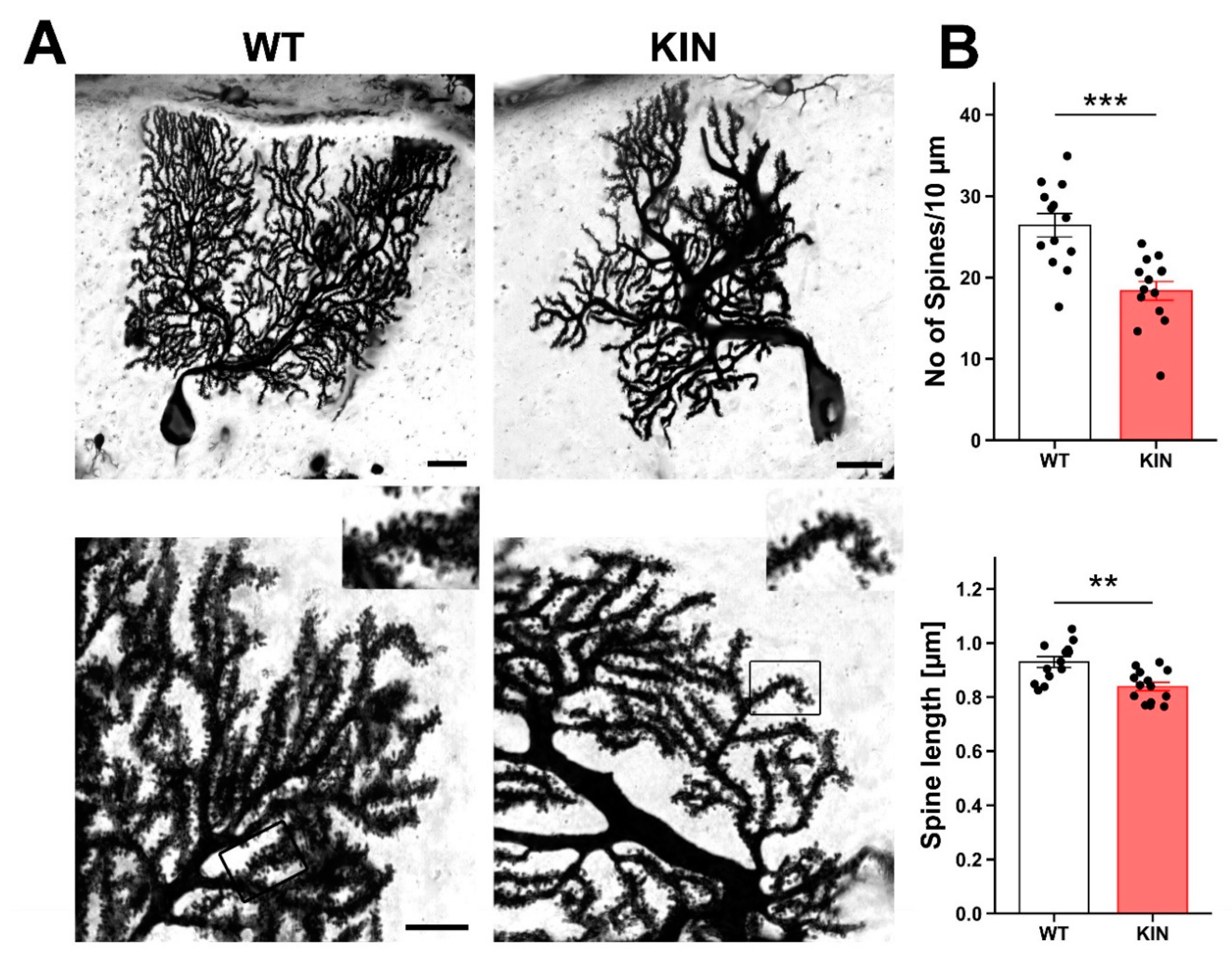
2.7. Morphological Assessment of Purkinje Cell Spines
2.8. Molecular Assessment of Glutamatergic Synapse Strength and Adhesion
2.9. Dissecting the Molecular Signature of SCA2 Pathology to Define Purkinje Neuron Contribution
3. Discussion
4. Materials and Methods
4.1. Animals and Genotyping
4.2. Transcriptome Screening
4.3. Cell Culture and Treatments
4.4. RNA Isolation and Expression Analyses
4.5. Protein Extraction and Quantitative Immunoblots
4.6. Colorimetric Ca2+ Measurement
4.7. Immunostainings
4.8. Silver Impregnation
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| °C | Degrees Celcius |
| µg | Microgram |
| µm | Micrometer |
| μL | Microliter |
| μM | Micromolar |
| ACTB | Beta Actin |
| Adam22 | A Disintegrin And Metalloproteinase Domain 22 |
| Adam23 | A Disintegrin And Metalloproteinase Domain 23 |
| ALS | Amyotrophic lateral sclerosis |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| ANOVA | Analysis of variance |
| AS | Alternative splicing |
| ASO | Antisense oligonucleotide |
| Atf4 | Activating Transcription Factor 4 |
| Atf6 | Activating Transcription Factor 6 |
| Atp2a1 | ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2+ Transporting 1 |
| Atp2a2 | ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2+ Transporting 2 |
| Atp2a3 | ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2+ Transporting 3 |
| Atp2b2 | ATPase Plasma Membrane Ca2+ Transporting 2 |
| ATXN2 | Ataxin-2 |
| ATXN2L | Ataxin-2-Like |
| BiP | Endoplasmic Reticulum Lumenal Ca2+-Binding Protein Grp78 (Hspa5) |
| BSA | Bovine serum albumin |
| Ca2+ | Calcium ion |
| CAA | Cytosine-Adenine-Adenine trinucleotide |
| Cabp7 | Calcium Binding Protein 7 |
| Cacna1a | Calcium Voltage-Gated Channel Subunit alpha-1-A |
| CAG | Cytosine-Adenine-Guanine trinucleotide |
| CaM | Calmodulin |
| CaMK | Ca2+/Calmodulin-Dependent Protein Kinase |
| Camk2a | Ca2+/Calmodulin-Dependent Protein Kinase II alpha |
| Camk2b | Ca2+/Calmodulin-Dependent Protein Kinase II beta |
| Camk2d | Ca2+/Calmodulin-Dependent Protein Kinase II delta |
| Camk2g | Ca2+/Calmodulin-Dependent Protein Kinase II gamma |
| Camk4 | Ca2+/Calmodulin-Dependent Protein Kinase IV |
| CaMKIIα | Ca2+/Calmodulin-Dependent Protein Kinase II alpha |
| CaMKIV | Ca2+/Calmodulin-Dependent Protein Kinase IV |
| Camkk1 | Ca2+/Calmodulin-Dependent Protein Kinase Kinase 1 |
| Camkk2 | Ca2+/Calmodulin-Dependent Protein Kinase Kinase 2 |
| Car8 | Carbonic Anhydrase 8 |
| CaV2.1 | Calcium Voltage-Gated Channel Subunit alpha-1-A |
| Cbln1 | Cerebellin 1 |
| Cbln2 | Cerebellin 2 |
| Cbln3 | Cerebellin 3 |
| Cbln4 | Cerebellin 4 |
| cDNA | Complementary DNA |
| Chop | C/EBP-Homologous Protein (Ddit3) |
| CNS | Central nervous system |
| Ct | Cycle threshold |
| C-terminal | Carboxy-terminal end |
| DAG | Diacylglycerol |
| DAPI | 4′,6-diamidino-2-phenylindole |
| ddH2O | Double-distilled water |
| Dlg4 | Discs Large MAGUK Scaffold Protein 4 |
| DMEM | Dulbecco’s modified essential medium |
| DNA | Deoxyribonucleic acid |
| DNase | Deoxyribonuclease |
| EDTA | Ethylenediaminetetraacetic acid |
| eIF | Eukaryotic Translation Initiation Factor |
| ER | Endoplasmic reticulum |
| ERAD | Endoplasmic reticulum—associated degradation |
| FCS | Fetal calf serum |
| FMR1 | Fragile X Mental Retardation 1 |
| FTLD | Fronto-temporal lobar degeneration |
| FUS | Fused in Sarcoma |
| Fwd | Forward primer |
| FXTAS | Fragile-X-tremor-ataxia syndrome |
| g | Gram |
| GC | Granule cell |
| GCL | Granule cell layer |
| GluA3 | Glutamate Ionotropic Receptor AMPA Type Subunit 3 |
| GluD2 | Glutamate Ionotropic Receptor Delta Type Subunit 2 |
| GO | Gene Ontology |
| Gria3 | Glutamate Ionotropic Receptor AMPA Type Subunit 3 |
| Grid2 | Glutamate Ionotropic Receptor Delta Type Subunit 2 |
| Grin1 | Glutamate Ionotropic Receptor NMDA Type Subunit 1 |
| Grm1 | Glutamate Metabotropic Receptor 1 |
| Grm4 | Glutamate Metabotropic Receptor 4 |
| Grp75 | Glucose-Regulated Protein 75 Kda (Hspa9) |
| h | Hour |
| HCl | Hydrochloric acid |
| i.e., | id est |
| Icmt | Isoprenylcysteine Carboxyl Methyltransferase |
| Igfbp5 | Insulin Like Growth Factor Binding Protein 5 |
| Inpp5a | Inositol Polyphosphate-5-Phosphatase A |
| IP3 | Inositol-1,4,5-trisphosphate |
| IP3R | Inositol-1,4,5-Trisphosphate Receptor Type 1 |
| Ire1 | Inositol-Requiring Protein 1 (Ern1) |
| Itpka | Inositol-Trisphosphate 3-Kinase A |
| Itpr1 | Inositol-1,4,5-Trisphosphate Receptor Type 1 |
| K+ | Potassium ion |
| kD | Kilo Dalton |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| kg | Kilogram |
| Khdrbs1 | KH RNA Binding Domain Containing, Signal Transduction Associated 1 |
| Khdrbs2 | KH RNA Binding Domain Containing, Signal Transduction Associated 2 |
| Khdrbs3 | KH RNA Binding Domain Containing, Signal Transduction Associated 3 |
| KIN | Knock in |
| KO | Knock out |
| l | Liter |
| Lgi1 | Leucine Rich Glioma Inactivated 1 |
| Lgi3 | Leucine Rich Glioma Inactivated 3 |
| LRRTM | Leucine Rich Repeat Transmembrane Protein |
| Lsm | Like-Smith antigen protein domain |
| Lsm-AD | Lsm-associated domain |
| M | Molar |
| MAM | Mitochondria-associated membrane |
| Mcu | Mitochondrial Calcium Uniporter |
| MEF | Mouse embryonal fibroblast |
| Mfn1 | Mitofusin 1 |
| Mfn2 | Mitofusin 2 |
| mg | Milligram |
| mGluR | Glutamate Metabotropic Receptor |
| Micu1 | Mitochondrial Calcium Uptake 1 |
| Micu2 | Mitochondrial Calcium Uptake 2 |
| Micu3 | Mitochondrial Calcium Uptake 3 |
| min | Minute |
| miRNA | Micro RNA |
| ml | Milliliter |
| ML | Molecular layer |
| mm | Millimeter |
| mM | Millimolar |
| mo | Month |
| mRNA | Messenger RNA |
| mTORC1 | Mechanistic Target of Rapamycin Complex 1 |
| n | Number of samples |
| NA | Numerical Aperture |
| NaCl | Sodium chloride |
| NCX | Na+/Ca2+-Exchange Protein (Slc8a2) |
| ng | Nanogram |
| Nlgn1 | Neuroligin 1 |
| Nlgn2 | Neuroligin 2 |
| Nlgn3 | Neuroligin 3 |
| nm | Nanometer |
| NMDA | N-methyl-d-aspartic acid |
| No | Number |
| NP-40 | Nonidet P-40 (nonyl phenoxypolyethoxylethanol) |
| Nrxn1 | Neurexin 1 |
| Nrxn2 | Neurexin 2 |
| Nrxn3 | Neurexin 3 |
| N-terminal | Amino (NH2)-terminal end |
| OPA1 | Optic Atrophy Protein 1 |
| Orai1 | ORAI Calcium Release-Activated Calcium Modulator 1 |
| p | Probability value |
| PABP | Poly(A)-Binding Protein |
| PAM2 | PABP-interacting motif 2 |
| PBS | Phosphate buffered saline |
| PC | Purkinje cell |
| PCL | Purkinje cell layer |
| Pcp2 | Purkinje Cell Protein 2 |
| Pcp4 | Purkinje Cell Protein 4 (PEP-19) |
| PCR | Polymerase chain reaction |
| PD | Parkison’s disease |
| Perk | PRKR-Like Endoplasmic Reticulum Kinase (Eif2ak3) |
| PFA | Paraformaldehyde |
| PINK1 | PTEN Induced Kinase 1 |
| PIP2 | Phosphatidylinositol-4,5-bisphosphate |
| PKC | Protein Kinase C |
| PLC | Phospholipase C |
| Plcb3 | Phospholipase C Beta 3 |
| Plcb4 | Phospholipase C Beta 4 |
| Plcg1 | Phospholipase C Gamma 1 |
| PMCA | Plasma Membrane Ca2+ Pump (Atp2b2) |
| pmol | Picomole |
| PolyQ | Poly-Glutamine |
| PRD | Proline-rich domain |
| Prkcd | Protein Kinase C Delta |
| Prmt8 | Protein Arginine Methyltransferase 8 |
| PSD95 | Postsynaptic Density Protein 95 (Dlg4) |
| Q | Glutamine |
| RBFOX1 | RNA Binding Fox-1 Homolog 1 |
| RBP | RNA-binding protein |
| Rev | Reverse primer |
| Rgs8 | Regulator of G Protein Signaling 8 |
| RIPA | Radioimmunoprecipitation assay |
| RNA | Ribonucleic acid |
| RNase | Ribonuclease |
| Rora | Retinoic Acid Receptor-Related Orphan Receptor Alpha |
| RT | Room temperature |
| RTK | Receptor Tyrosine Kinase |
| RT-qPCR | Reverse-Transcriptase quantitative PCR |
| Ryr1 | Ryanodine Receptor 1 |
| Ryr3 | Ryanodine Receptor 3 |
| sec | Second |
| s.e.m. | Standard error of the mean |
| Sam68 | Src-Associated In Mitosis 68 KDa Protein (Khdrbs1) |
| SCA1 | Spinocerebellar ataxia type 1 |
| SCA2 | Spinocerebellar ataxia type 2 |
| SDS | Sodium dodecyl sulfate |
| Sema7a | Semaphorin 7A |
| SERCA1 | Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase 1 (Atp2a1) |
| SERCA2 | Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase 2 (Atp2a2) |
| SERCA3 | Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase 3 (Atp2a3) |
| SG | Stress granule |
| Shank1 | SH3 And Multiple Ankyrin Repeat Domains 1 |
| Shank2 | SH3 And Multiple Ankyrin Repeat Domains 2 |
| Sigmar1 | Sigma Non-Opioid Intracellular Receptor 1 |
| Slc8a2 | Solute Carrier Family 8 Member A2 |
| Slm1 | Sam68-Like Mammalian Protein 1 (Khdrbs2) |
| Slm2 | Sam68-Like Mammalian Protein 2 (Khdrbs3) |
| Smdt1 | Single-Pass Membrane Protein with Aspartate Rich Tail 1 |
| Stim1 | Stromal Interaction Molecule 1 |
| STRING | Search Tool for the Retrieval of Interacting Genes/Proteins |
| T | Trend |
| Tbp | TATA-binding factor of transcription |
| TBS-T | Tris-buffered saline with Tween20 |
| TCA | Tricarboxylic acid cycle |
| TDP-43 | TAR DNA-Binding Protein 43 (Tardbp) |
| TG | Thapsigargin |
| Tg | Transgenic (mouse) |
| TIA-1 | T-Cell-Restricted Intracellular Antigen-1 |
| TM | Tunicamycin |
| tRNA | Transfer RNA |
| Trpc3 | Transient Receptor Potential Cation Channel Subfamily C Member 3 |
| UniProt | Universal Protein resource |
| UPR | Unfolded protein response |
| Vdac1 | Voltage Dependent Anion Channel 1 |
| vs. | versus |
| wk | Week |
| WT | Wild type |
| Xbp1s | X-Box Binding Protein 1 spliced |
| Xbp1u | X-Box Binding Protein 1 unspliced |
References
- Pulst, S.M.; Nechiporuk, A.; Nechiporuk, T.; Gispert, S.; Chen, X.N.; Lopes-Cendes, I.; Pearlman, S.; Starkman, S.; Orozco-Diaz, G.; Lunkes, A.; et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 1996, 14, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Sanpei, K.; Takano, H.; Igarashi, S.; Sato, T.; Oyake, M.; Sasaki, H.; Wakisaka, A.; Tashiro, K.; Ishida, Y.; Ikeuchi, T.; et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat. Genet. 1996, 14, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Imbert, G.; Saudou, F.; Yvert, G.; Devys, D.; Trottier, Y.; Garnier, J.M.; Weber, C.; Mandel, J.L.; Cancel, G.; Abbas, N.; et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 1996, 14, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Pérez, L.; Seifried, C.; Santos-Falcón, N.; Abele, M.; Ziemann, U.; Almaguer, L.E.; Martínez-Góngora, E.; Sánchez-Cruz, G.; Canales, N.; Pérez-González, R.; et al. Saccade velocity is controlled by polyglutamine size in spinocerebellar ataxia 2. Ann. Neurol. 2004, 56, 444–447. [Google Scholar] [CrossRef]
- Velázquez-Pérez, L.; Rodríguez-Labrada, R.; Canales-Ochoa, N.; Montero, J.M.; Sánchez-Cruz, G.; Aguilera-Rodríguez, R.; Almaguer-Mederos, L.E.; Laffita-Mesa, J.M. Progression of early features of spinocerebellar ataxia type 2 in individuals at risk: A longitudinal study. Lancet Neurol. 2014, 13, 482–489. [Google Scholar] [CrossRef]
- Auburger, G.W.J. Spinocerebellar ataxia type 2. Handb. Clin. Neurol. 2012, 103, 423–436. [Google Scholar] [CrossRef]
- Velázquez-Pérez, L.; Seifried, C.; Abele, M.; Wirjatijasa, F.; Rodríguez-Labrada, R.; Santos-Falcón, N.; Sánchez-Cruz, G.; Almaguer-Mederos, L.; Tejeda, R.; Canales-Ochoa, N.; et al. Saccade velocity is reduced in presymptomatic spinocerebellar ataxia type 2. Clin. Neurophysiol. 2009, 120, 632–635. [Google Scholar] [CrossRef]
- Almaguer-Mederos, L.E.; Aguilera Rodríguez, R.; González Zaldivar, Y.; Almaguer Gotay, D.; Cuello Almarales, D.; Laffita Mesa, J.; Vázquez Mojena, Y.; Zayas Feria, P.; Auburger, G.; Gispert, S.; et al. Estimation of survival in spinocerebellar ataxia type 2 Cuban patients. Clin. Genet. 2013, 83, 293–294. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Gwinn-Hardy, K.; Chen, J.Y.; Liu, H.C.; Liu, T.Y.; Boss, M.; Seltzer, W.; Adam, A.; Singleton, A.; Koroshetz, W.; Waters, C.; et al. Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology 2000, 55, 800–805. [Google Scholar] [CrossRef]
- Ross, O.A.; Rutherford, N.J.; Baker, M.; Soto-Ortolaza, A.I.; Carrasquillo, M.M.; DeJesus-Hernandez, M.; Adamson, J.; Li, M.; Volkening, K.; Finger, E.; et al. Ataxin-2 repeat-length variation and neurodegeneration. Hum. Mol. Genet. 2011, 20, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Kurz, A.; Waibel, S.; Bauer, P.; Liepelt, I.; Geisen, C.; Gitler, A.D.; Becker, T.; Weber, M.; Berg, D.; et al. The modulation of Amyotrophic Lateral Sclerosis risk by ataxin-2 intermediate polyglutamine expansions is a specific effect. Neurobiol. Dis. 2012, 45, 356–361. [Google Scholar] [CrossRef]
- Seidel, K.; Siswanto, S.; Brunt, E.R.P.; den Dunnen, W.; Korf, H.-W.; Rüb, U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012, 124, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Rüb, U.; Del Turco, D.; Del Tredici, K.; de Vos, R.A.I.; Brunt, E.R.; Reifenberger, G.; Seifried, C.; Schultz, C.; Auburger, G.; Braak, H. Thalamic involvement in a spinocerebellar ataxia type 2 (SCA2) and a spinocerebellar ataxia type 3 (SCA3) patient, and its clinical relevance. Brain 2003, 126, 2257–2272. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rüb, U.; Brunt, E.R.; Petrasch-Parwez, E.; Schöls, L.; Theegarten, D.; Auburger, G.; Seidel, K.; Schultz, C.; Gierga, K.; Paulson, H.; et al. Degeneration of ingestion-related brainstem nuclei in spinocerebellar ataxia type 2, 3, 6 and 7. Neuropathol. Appl. Neurobiol. 2006, 32, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Gierga, K.; Bürk, K.; Bauer, M.; Orozco Diaz, G.; Auburger, G.; Schultz, C.; Vuksic, M.; Schöls, L.; de Vos, R.A.I.; Braak, H.; et al. Involvement of the cranial nerves and their nuclei in spinocerebellar ataxia type 2 (SCA2). Acta Neuropathol. 2005, 109, 617–631. [Google Scholar] [CrossRef]
- Hoche, F.; Balikó, L.; den Dunnen, W.; Steinecker, K.; Bartos, L.; Sáfrány, E.; Auburger, G.; Deller, T.; Korf, H.-W.; Klockgether, T.; et al. Spinocerebellar ataxia type 2 (SCA2): Identification of early brain degeneration in one monozygous twin in the initial disease stage. Cerebellum 2011, 10, 245–253. [Google Scholar] [CrossRef]
- Schöls, L.; Reimold, M.; Seidel, K.; Globas, C.; Brockmann, K.; Hauser, T.K.; Auburger, G.; Bürk, K.; den Dunnen, W.; Reischl, G.; et al. No parkinsonism in SCA2 and SCA3 despite severe neurodegeneration of the dopaminergic substantia nigra. Brain 2015, 138, 3316–3326. [Google Scholar] [CrossRef]
- Rüb, U.; Farrag, K.; Seidel, K.; Brunt, E.R.; Heinsen, H.; Bürk, K.; Melegh, B.; von Gall, C.; Auburger, G.; Bohl, J.; et al. Involvement of the cholinergic basal forebrain nuclei in spinocerebellar ataxia type 2 (SCA2). Neuropathol. Appl. Neurobiol. 2013, 39, 634–643. [Google Scholar] [CrossRef]
- Tuin, I.; Voss, U.; Kang, J.-S.; Kessler, K.; Rüb, U.; Nolte, D.; Lochmüller, H.; Tinschert, S.; Claus, D.; Krakow, K.; et al. Stages of sleep pathology in spinocerebellar ataxia type 2 (SCA2). Neurology 2006, 67, 1966–1972. [Google Scholar] [CrossRef]
- Seidel, K.; Siswanto, S.; Fredrich, M.; Bouzrou, M.; den Dunnen, W.F.A.; Özerden, I.; Korf, H.-W.; Melegh, B.; de Vries, J.J.; Brunt, E.R.; et al. On the distribution of intranuclear and cytoplasmic aggregates in the brainstem of patients with spinocerebellar ataxia type 2 and 3. Brain Pathol. 2017, 27, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Damrath, E.; Heck, M.V.; Gispert, S.; Azizov, M.; Nowock, J.; Seifried, C.; Rüb, U.; Walter, M.; Auburger, G. ATXN2-CAG42 sequesters PABPC1 into insolubility and induces FBXW8 in cerebellum of old ataxic knock-in mice. PLoS Genet. 2012, 8, e1002920. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.-E.; Canet-Pons, J.; Halbach, M.V.; Arsovic, A.; Pilatus, U.; Chae, W.-H.; Kaya, Z.-E.; Seidel, K.; Rollmann, E.; Mittelbronn, M.; et al. Generation of an Atxn2-CAG100 knock-in mouse reveals N-acetylaspartate production deficit due to early Nat8l dysregulation. Neurobiol. Dis. 2019, 132, 104559. [Google Scholar] [CrossRef]
- Velázquez-Pérez, L.; Tünnerhoff, J.; Rodríguez-Labrada, R.; Torres-Vega, R.; Ruiz-Gonzalez, Y.; Belardinelli, P.; Medrano-Montero, J.; Canales-Ochoa, N.; González-Zaldivar, Y.; Vazquez-Mojena, Y.; et al. Early corticospinal tract damage in prodromal SCA2 revealed by EEG-EMG and EMG-EMG coherence. Clin. Neurophysiol. 2017, 128, 2493–2502. [Google Scholar] [CrossRef]
- Empson, R.M.; Knöpfel, T. Functional integration of calcium regulatory mechanisms at Purkinje neuron synapses. Cerebellum 2012, 11, 640–650. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hoxha, E.; Tempia, F.; Lippiello, P.; Miniaci, M.C. Modulation, Plasticity and Pathophysiology of the Parallel Fiber-Purkinje Cell Synapse. Front. Synaptic Neurosci. 2016, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, E.; Balbo, I.; Miniaci, M.C.; Tempia, F. Purkinje Cell Signaling Deficits in Animal Models of Ataxia. Front. Synaptic Neurosci. 2018, 10, 6. [Google Scholar] [CrossRef]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H.T. Polyglutamine spinocerebellar ataxias—From genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef]
- Estrada, R.; Galarraga, J.; Orozco, G.; Nodarse, A.; Auburger, G. Spinocerebellar ataxia 2 (SCA2): Morphometric analyses in 11 autopsies. Acta Neuropathol. 1999, 97, 306–310. [Google Scholar] [CrossRef]
- Pang, J.T.; Giunti, P.; Chamberlain, S.; An, S.F.; Vitaliani, R.; Scaravilli, T.; Martinian, L.; Wood, N.W.; Scaravilli, F.; Ansorge, O. Neuronal intranuclear inclusions in SCA2: A genetic, morphological and immunohistochemical study of two cases. Brain 2002, 125, 656–663. [Google Scholar] [CrossRef]
- Nonis, D.; Schmidt, M.H.H.; van de Loo, S.; Eich, F.; Dikic, I.; Nowock, J.; Auburger, G. Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell. Signal. 2008, 20, 1725–1739. [Google Scholar] [CrossRef] [PubMed]
- Auburger, G.; Sen, N.-E.; Meierhofer, D.; Başak, A.-N.; Gitler, A.D. Efficient Prevention of Neurodegenerative Diseases by Depletion of Starvation Response Factor Ataxin-2. Trends Neurosci. 2017, 40, 507–516. [Google Scholar] [CrossRef] [PubMed]
- van de Loo, S.; Eich, F.; Nonis, D.; Auburger, G.; Nowock, J. Ataxin-2 associates with rough endoplasmic reticulum. Exp. Neurol. 2009, 215, 110–118. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Nonis, D.; Eich, F.; Klinkenberg, M.; Gorospe, M.; Kötter, P.; Klein, F.A.C.; Kedersha, N.; Auburger, G. Mammalian ataxin-2 modulates translation control at the pre-initiation complex via PI3K/mTOR and is induced by starvation. Biochim. Biophys. Acta 2016, 1862, 1558–1569. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Nonis, D.; Eich, F.; Leske, O.; Damrath, E.; Brunt, E.R.; Lastres-Becker, I.; Heumann, R.; Nowock, J.; Auburger, G. Ataxin-2 modulates the levels of Grb2 and SRC but not ras signaling. J. Mol. Neurosci. 2013, 51, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Seidel, G.; Meierhofer, D.; Şen, N.-E.; Guenther, A.; Krobitsch, S.; Auburger, G. Quantitative Global Proteomics of Yeast PBP1 Deletion Mutants and Their Stress Responses Identifies Glucose Metabolism, Mitochondrial, and Stress Granule Changes. J. Proteome Res. 2017, 16, 504–515. [Google Scholar] [CrossRef]
- Fittschen, M.; Lastres-Becker, I.; Halbach, M.V.; Damrath, E.; Gispert, S.; Azizov, M.; Walter, M.; Müller, S.; Auburger, G. Genetic ablation of ataxin-2 increases several global translation factors in their transcript abundance but decreases translation rate. Neurogenetics 2015, 16, 181–192. [Google Scholar] [CrossRef]
- Ralser, M.; Albrecht, M.; Nonhoff, U.; Lengauer, T.; Lehrach, H.; Krobitsch, S. An integrative approach to gain insights into the cellular function of human ataxin-2. J. Mol. Biol. 2005, 346, 203–214. [Google Scholar] [CrossRef]
- Nonhoff, U.; Ralser, M.; Welzel, F.; Piccini, I.; Balzereit, D.; Yaspo, M.-L.; Lehrach, H.; Krobitsch, S. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol. Biol. Cell 2007, 18, 1385–1396. [Google Scholar] [CrossRef]
- Hart, M.P.; Brettschneider, J.; Lee, V.M.Y.; Trojanowski, J.Q.; Gitler, A.D. Distinct TDP-43 pathology in ALS patients with ataxin 2 intermediate-length polyQ expansions. Acta Neuropathol. 2012, 124, 221–230. [Google Scholar] [CrossRef]
- Hart, M.P.; Gitler, A.D. ALS-associated ataxin 2 polyQ expansions enhance stress-induced caspase 3 activation and increase TDP-43 pathological modifications. J. Neurosci. 2012, 32, 9133–9142. [Google Scholar] [CrossRef]
- McGurk, L.; Lee, V.M.; Trojanowksi, J.Q.; van Deerlin, V.M.; Lee, E.B.; Bonini, N.M. Poly-A binding protein-1 localization to a subset of TDP-43 inclusions in amyotrophic lateral sclerosis occurs more frequently in patients harboring an expansion in C9orf72. J. Neuropathol. Exp. Neurol. 2014, 73, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Hirsch-Reinshagen, V.; Pottier, C.; Nicholson, A.M.; Baker, M.; Hsiung, G.-Y.R.; Krieger, C.; Sengdy, P.; Boylan, K.B.; Dickson, D.W.; Mesulam, M.; et al. Clinical and neuropathological features of ALS/FTD with TIA1 mutations. Acta Neuropathol. Commun. 2017, 5, 96. [Google Scholar] [CrossRef]
- Hansen, M.; Zeddies, S.; Meinders, M.; Di Summa, F.; van Alphen, F.P.J.; Hoogendijk, A.J.; Moore, K.S.; Halbach, M.; Gutiérrez, L.; van den Biggelaar, M.; et al. The RNA-Binding Protein ATXN2 is Expressed during Megakaryopoiesis and May Control Timing of Gene Expression. Int. J. Mol. Sci. 2020, 21, 967. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.E.; Drost, J.; Gispert, S.; Torres-Odio, S.; Damrath, E.; Klinkenberg, M.; Hamzeiy, H.; Akdal, G.; Güllüoğlu, H.; Başak, A.N.; et al. Search for SCA2 blood RNA biomarkers highlights Ataxin-2 as strong modifier of the mitochondrial factor PINK1 levels. Neurobiol. Dis. 2016, 96, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.-E.; Arsovic, A.; Meierhofer, D.; Brodesser, S.; Oberschmidt, C.; Canet-Pons, J.; Kaya, Z.-E.; Halbach, M.-V.; Gispert, S.; Sandhoff, K.; et al. In Human and Mouse Spino-Cerebellar Tissue, Ataxin-2 Expansion Affects Ceramide-Sphingomyelin Metabolism. Int. J. Mol. Sci. 2019, 20, 5854. [Google Scholar] [CrossRef] [PubMed]
- Meierhofer, D.; Halbach, M.; Şen, N.E.; Gispert, S.; Auburger, G. Ataxin-2 (Atxn2)-Knock-Out Mice Show Branched Chain Amino Acids and Fatty Acids Pathway Alterations. Mol. Cell. Proteom. 2016, 15, 1728–1739. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Brodesser, S.; Lütjohann, D.; Azizov, M.; Buchmann, J.; Hintermann, E.; Sandhoff, K.; Schürmann, A.; Nowock, J.; Auburger, G. Insulin receptor and lipid metabolism pathology in ataxin-2 knock-out mice. Hum. Mol. Genet. 2008, 17, 1465–1481. [Google Scholar] [CrossRef]
- Takahara, T.; Maeda, T. Transient sequestration of TORC1 into stress granules during heat stress. Mol. Cell 2012, 47, 242–252. [Google Scholar] [CrossRef]
- DeMille, D.; Badal, B.D.; Evans, J.B.; Mathis, A.D.; Anderson, J.F.; Grose, J.H. PAS kinase is activated by direct SNF1-dependent phosphorylation and mediates inhibition of TORC1 through the phosphorylation and activation of Pbp1. Mol. Biol. Cell 2015, 26, 569–582. [Google Scholar] [CrossRef]
- Scoles, D.R.; Dansithong, W.; Pflieger, L.T.; Paul, S.; Gandelman, M.; Figueroa, K.P.; Rigo, F.; Bennett, C.F.; Pulst, S.M. ALS-associated genes in SCA2 mouse spinal cord transcriptomes. Hum. Mol. Genet. 2020, 29, 1658–1672. [Google Scholar] [CrossRef] [PubMed]
- Canet-Pons, J.; Sen, N.-E.; Arsovic, A.; Almaguer-Mederos, L.-E.; Halbach, M.V.; Key, J.; Döring, C.; Kerksiek, A.; Picchiarelli, G.; Cassel, R.; et al. Atxn2-CAG100-KnockIn Mouse Spinal Cord Shows Progressive TDP43 Pathology Associated with Cholesterol Biosynthesis Suppression. bioRxiv 2019. [Google Scholar] [CrossRef]
- Key, J.; Harter, P.N.; Sen, N.-E.; Gradhand, E.; Auburger, G.; Gispert, S. Mid-Gestation lethality of Atxn2l-Ablated Mice. Int. J. Mol. Sci. 2020, 21, 5124. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.J. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 2015, 524, 481–484. [Google Scholar] [CrossRef]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Torres-Odio, S.; Key, J.; Hoepken, H.-H.; Canet-Pons, J.; Valek, L.; Roller, B.; Walter, M.; Morales-Gordo, B.; Meierhofer, D.; Harter, P.N.; et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J. Neuroinflamm. 2017, 14, 154. [Google Scholar] [CrossRef]
- Prestori, F.; Moccia, F.; D’Angelo, E. Disrupted Calcium Signaling in Animal Models of Human Spinocerebellar Ataxia (SCA). Int. J. Mol. Sci. 2019, 21, 216. [Google Scholar] [CrossRef]
- Halbach, M.V.; Gispert, S.; Stehning, T.; Damrath, E.; Walter, M.; Auburger, G. Atxn2 Knockout and CAG42-Knock-in Cerebellum Shows Similarly Dysregulated Expression in Calcium Homeostasis Pathway. Cerebellum 2017, 16, 68–81. [Google Scholar] [CrossRef]
- Pflieger, L.T.; Dansithong, W.; Paul, S.; Scoles, D.R.; Figueroa, K.P.; Meera, P.; Otis, T.S.; Facelli, J.C.; Pulst, S.M. Gene co-expression network analysis for identifying modules and functionally enriched pathways in SCA2. Hum. Mol. Genet. 2017, 26, 3069–3080. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Swulius, M.T.; Waxham, M.N. Ca(2+)/calmodulin-dependent protein kinases. Cell. Mol. Life Sci. 2008, 65, 2637–2657. [Google Scholar] [CrossRef]
- Becker, E.B.E. From Mice to Men: TRPC3 in Cerebellar Ataxia. Cerebellum 2017, 16, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.W.; Sachs, A.J.; Nystuen, A.M. Deletion of Inpp5a causes ataxia and cerebellar degeneration in mice. Neurogenetics 2015, 16, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Windhorst, S.; Song, K.; Gazdar, A.F. Inositol-1,4,5-trisphosphate 3-kinase-A (ITPKA) is frequently over-expressed and functions as an oncogene in several tumor types. Biochem. Pharmacol. 2017, 137, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Blundon, J.A.; Rong, Y.; Zakharenko, S.S.; Morgan, J.I. Impaired locomotor learning and altered cerebellar synaptic plasticity in pep-19/PCP4-null mice. Mol. Cell. Biol. 2011, 31, 2838–2844. [Google Scholar] [CrossRef] [PubMed]
- Iijima, T.; Wu, K.; Witte, H.; Hanno-Iijima, Y.; Glatter, T.; Richard, S.; Scheiffele, P. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 2011, 147, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Soo, K.Y.; Warraich, S.T.; Sundaramoorthy, V.; Blair, I.P.; Atkin, J.D. Ataxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Treutlein, B.; Gokce, O.; Quake, S.R.; Südhof, T.C. Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, E1291–E1299. [Google Scholar] [CrossRef]
- Iijima, T.; Hidaka, C.; Iijima, Y. Spatio-temporal regulations and functions of neuronal alternative RNA splicing in developing and adult brains. Neurosci. Res. 2016, 109, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Iijima, T.; Iijima, Y.; Witte, H.; Scheiffele, P. Neuronal cell type-specific alternative splicing is regulated by the KH domain protein SLM1. J. Cell Biol. 2014, 204, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Suzuki, S.; Iijima, Y.; Iijima, T. Neuroligin-induced presynaptic differentiation through SLM2-mediated splicing modifications of neurexin in cerebellar cultures. Biochem. Biophys. Res. Commun. 2017, 493, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Saito, F.; Tang, J.; Satz, J.; Campbell, K.; Südhof, T.C. A stoichiometric complex of neurexins and dystroglycan in brain. J. Cell Biol. 2001, 154, 435–445. [Google Scholar] [CrossRef]
- Reissner, C.; Stahn, J.; Breuer, D.; Klose, M.; Pohlentz, G.; Mormann, M.; Missler, M. Dystroglycan binding to α-neurexin competes with neurexophilin-1 and neuroligin in the brain. J. Biol. Chem. 2014, 289, 27585–27603. [Google Scholar] [CrossRef]
- Fukata, Y.; Lovero, K.L.; Iwanaga, T.; Watanabe, A.; Yokoi, N.; Tabuchi, K.; Shigemoto, R.; Nicoll, R.A.; Fukata, M. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc. Natl. Acad. Sci. USA 2010, 107, 3799–3804. [Google Scholar] [CrossRef]
- Zhang, P.; Lisman, J.E. Activity-dependent regulation of synaptic strength by PSD-95 in CA1 neurons. J. Neurophysiol. 2012, 107, 1058–1066. [Google Scholar] [CrossRef]
- Huynh, D.P.; Figueroa, K.; Hoang, N.; Pulst, S.M. Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet. 2000, 26, 44–50. [Google Scholar] [CrossRef]
- Liu, J.; Tang, T.-S.; Tu, H.; Nelson, O.; Herndon, E.; Huynh, D.P.; Pulst, S.M.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J. Neurosci. 2009, 29, 9148–9162. [Google Scholar] [CrossRef]
- Wan, C.; Borgeson, B.; Phanse, S.; Tu, F.; Drew, K.; Clark, G.; Xiong, X.; Kagan, O.; Kwan, J.; Bezginov, A.; et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525, 339–344. [Google Scholar] [CrossRef]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010, 29, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Younts, T.J.; Castillo, P.E.; Jordan, B.A. RNA-binding protein Sam68 controls synapse number and local β-actin mRNA metabolism in dendrites. Proc. Natl. Acad. Sci. USA 2013, 110, 3125–3130. [Google Scholar] [CrossRef] [PubMed]
- Sudhakaran, I.P.; Hillebrand, J.; Dervan, A.; Das, S.; Holohan, E.E.; Hülsmeier, J.; Sarov, M.; Parker, R.; VijayRaghavan, K.; Ramaswami, M. FMRP and Ataxin-2 function together in long-term olfactory habituation and neuronal translational control. Proc. Natl. Acad. Sci. USA 2014, 111, E99–E108. [Google Scholar] [CrossRef] [PubMed]
- Herring, B.E.; Nicoll, R.A. Long-Term Potentiation: From CaMKII to AMPA Receptor Trafficking. Annu. Rev. Physiol. 2016, 78, 351–365. [Google Scholar] [CrossRef]
- Bayer, K.U.; Schulman, H. CaM Kinase: Still Inspiring at 40. Neuron 2019, 103, 380–394. [Google Scholar] [CrossRef]
- Borovac, J.; Bosch, M.; Okamoto, K. Regulation of actin dynamics during structural plasticity of dendritic spines: Signaling messengers and actin-binding proteins. Mol. Cell. Neurosci. 2018, 91, 122–130. [Google Scholar] [CrossRef]
- Zalcman, G.; Federman, N.; Romano, A. CaMKII Isoforms in Learning and Memory: Localization and Function. Front. Mol. Neurosci. 2018, 11, 445. [Google Scholar] [CrossRef]
- Ibata, K.; Kono, M.; Narumi, S.; Motohashi, J.; Kakegawa, W.; Kohda, K.; Yuzaki, M. Activity-Dependent Secretion of Synaptic Organizer Cbln1 from Lysosomes in Granule Cell Axons. Neuron 2019, 102, 1184–1198.e10. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, L.Y.; Liu, X.; Maxeiner, S.; Lee, S.-J.; Gokce, O.; Südhof, T.C. Neuroligins Sculpt Cerebellar Purkinje-Cell Circuits by Differential Control of Distinct Classes of Synapses. Neuron 2015, 87, 781–796. [Google Scholar] [CrossRef]
- Hansen, S.T.; Meera, P.; Otis, T.S.; Pulst, S.M. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet. 2013, 22, 271–283. [Google Scholar] [CrossRef]
- Kasumu, A.W.; Liang, X.; Egorova, P.; Vorontsova, D.; Bezprozvanny, I. Chronic suppression of inositol 1,4,5-triphosphate receptor-mediated calcium signaling in cerebellar Purkinje cells alleviates pathological phenotype in spinocerebellar ataxia 2 mice. J. Neurosci. 2012, 32, 12786–12796. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Breuer, P.; Peitz, M.; Jungverdorben, J.; Kesavan, J.; Poppe, D.; Doerr, J.; Ladewig, J.; Mertens, J.; Tüting, T.; et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 2011, 480, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Al-Ramahi, I.; Am, P.; Lim, J.; Zhang, M.; Sorensen, R.; de Haro, M.; Branco, J.; Pulst, S.M.; Zoghbi, H.Y.; Botas, J. dAtaxin-2 mediates expanded Ataxin-1-induced neurodegeneration in a Drosophila model of SCA1. PLoS Genet. 2007, 3, e234. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; Feany, M.B. Genetic modifiers of tauopathy in Drosophila. Genetics 2003, 165, 1233–1242. [Google Scholar] [PubMed]
- McCann, C.; Holohan, E.E.; Das, S.; Dervan, A.; Larkin, A.; Lee, J.A.; Rodrigues, V.; Parker, R.; Ramaswami, M. The Ataxin-2 protein is required for microRNA function and synapse-specific long-term olfactory habituation. Proc. Natl. Acad. Sci. USA 2011, 108, E655–E662. [Google Scholar] [CrossRef]
- Rais, M.; Binder, D.K.; Razak, K.A.; Ethell, I.M. Sensory Processing Phenotypes in Fragile X Syndrome. ASN Neuro 2018, 10, 1759091418801092. [Google Scholar] [CrossRef]
- Twick, I.; Lee, J.A.; Ramaswami, M. Olfactory habituation in Drosophila-odor encoding and its plasticity in the antennal lobe. Prog. Brain Res. 2014, 208, 3–38. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Nguyen, T.-M.; Schreiner, D.; Le, X.; Traunmüller, L.; Bornmann, C.; Scheiffele, P. An alternative splicing switch shapes neurexin repertoires in principal neurons versus interneurons in the mouse hippocampus. Elife 2016, 5, e22757. [Google Scholar] [CrossRef]
- Czechowska, N.; van Rienen, A.; Lang, F.; Eiberger, B.; Baader, S.L. An update on the Golgi staining technique improving cerebellar cell type specificity. Histochem. Cell Biol. 2019, 151, 327–341. [Google Scholar] [CrossRef]
- Forster, B.; van de Ville, D.; Berent, J.; Sage, D.; Unser, M. Complex wavelets for extended depth-of-field: A new method for the fusion of multichannel microscopy images. Microsc. Res. Tech. 2004, 65, 33–42. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Assay ID | Transcript | Assay ID | Transcript | Assay ID |
|---|---|---|---|---|---|
| Adam22 | Mm01316488_m1 | Ern1(Ire1) | Mm00470233_m1 | Nrxn1 | Mm03011136_m1 |
| Adam23 | Mm00478606_m1 | Gria3 | Mm00497506_m1 | Nrxn2 | Mm01236851_m1 |
| Atf4 | Mm00515324_m1 | Grid2 | Mm00515053_m1 | Nrxn3 | Mm00553213_m1 |
| Atf6 | Mm01295319_m1 | Grm1 | Mm00810219_m1 | Orai1 | Mm00774349_m1 |
| Atp2a1 | Mm01275320_m1 | Grm4 | Mm01306128_m1 | Pcp4 | Mm00500973_m1 |
| Atp2a2 | Mm01201431_m1 | Hspa5(Bip) | Mm00517691_m1 | Plcb3 | Mm00476953_m1 |
| Atp2a3 | Mm00443898_m1 | Hspa9(Grp75) | Mm00477716_g1 | Plcb4 | Mm00649825_m1 |
| Atp2b2 | Mm00437640_m1 | Inpp5a | Mm00805812_m1 | Plcg1 | Mm01247293_m1 |
| Atxn2 | Mm01199894_m1 | Itpka | Mm00525139_m1 | Prkcd | Mm00440891_m1 |
| Cacna1a | Mm00432190_m1 | Itpr1 | Mm00439907_m1 | Ryr1 | Mm01175211_m1 |
| Camk2a | Mm00437967_m1 | Khdrbs1 | Mm00516130_m1 | Ryr3 | Mm01328421_m1 |
| Camk2b | Mm00432284_m1 | Khdrbs2 | Mm00504671_m1 | Shank1 | Mm01206737_m1 |
| Camk2d | Mm00499266_m1 | Khdrbs3 | Mm00501666_m1 | Shank2 | Mm00683065_m1 |
| Camk2g | Mm00618047_m1 | Lgi1 | Mm01198941_m1 | Sigmar1 | Mm00448086_m1 |
| Camk4 | Mm01135329_m1 | Lgi3 | Mm00507490_m1 | Slc8a2 | Mm00455836_m1 |
| Camkk1 | Mm00517053_m1 | Mcu | Mm01168773_m1 | Smdt1 | Mm01306306_m1 |
| Camkk2 | Mm00520236_m1 | Mfn1 | Mm00612599_m1 | Stim1 | Mm01158413_m1 |
| Cbln1 | Mm01247194_g1 | Mfn2 | Mm00500120_m1 | Tbp | Mm00446973_m1 |
| Cbln2 | Mm01261557_g1 | Micu1 | Mm00522783_m1 | Trpc3 | Mm00444690-m1 |
| Cbln3 | Mm00490772_g1 | Micu2 | Mm00551312_m1 | Vdac1 | Mm00834272_m1 |
| Cbln4 | Mm00558663_m1 | Micu3 | Mm01194824_m1 | Xbp1s | Mm03464496_m1 |
| Ddit3(Chop) | Mm00499207_m1 | Nlgn1 | Mm02344307_m1 | Xbp1u | Mm03464497_s1 |
| Dlg4(Psd-95) | Mm00492193_m1 | Nlgn2 | Mm01703404_m1 | ||
| Eif2ak3(Perk) | Mm00438708_m1 | Nlgn3 | Mm01225951_m1 |
| Primer Name | Sequence (5′ → 3′) | Reference |
|---|---|---|
| AS1 − Fwd | ACTGCAGCCAAGGAAAAGAAGAGTA | |
| AS1 − Rev | GTTTTAAAGGACAGAGTTATTTCAT | |
| AS2 − Fwd | TCTGCGTCAGGTGACAATATCAG | |
| AS2 + Fwd | CTCAGGCATTGGACACGCTA | |
| AS2 −/+ Rev | GAAGGTCGGCTGTGCTGGGG | Nguyen et al. 2016 [99] |
| AS3 − Fwd | TCAATCTAGGCAAAGGTCCTG | |
| AS3 + Fwd | TTGTATCAGGATTAACTGTAATTCC | |
| AS3 −/+ Rev | TTTCCTCGCCGAACCACACG | Iijima et al. 2011 [67] |
| AS4 − Fwd | CGCTACCCTGCAGGGCGTCAGCTCAC | |
| AS4 + Fwd | TAGTTGATGAATGGCTACTCGACAAA | Iijima et al. 2011 [67] |
| AS4 −/+ Rev | GACTCAGTTGTCATAGAGGAAGGCAC | Iijima et al. 2011 [67] |
| AS5 − Fwd | AGCCAGCCAACCCCACCAGAGTA | |
| AS5 + Fwd | AGATGACATCCTTGTGGCCT | |
| AS5 −/+ Rev | ACCATGCCAGTGGTACTGCT | |
| AS6 − Fwd | ATGCGAAGGGCCCAGCA | |
| AS6 + Fwd | GCATTGATGAAAGCTGACTTGC | |
| AS6 −/+ Rev | GGAAGTCATGCTACAGTCACAGC | |
| Actb Fwd | GGAAATCGTGCGTGACATCAAAG | |
| Actb Rev | CATACCCAAGAAGGAAGGCTGG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arsović, A.; Halbach, M.V.; Canet-Pons, J.; Esen-Sehir, D.; Döring, C.; Freudenberg, F.; Czechowska, N.; Seidel, K.; Baader, S.L.; Gispert, S.; et al. Mouse Ataxin-2 Expansion Downregulates CamKII and Other Calcium Signaling Factors, Impairing Granule—Purkinje Neuron Synaptic Strength. Int. J. Mol. Sci. 2020, 21, 6673. https://doi.org/10.3390/ijms21186673
Arsović A, Halbach MV, Canet-Pons J, Esen-Sehir D, Döring C, Freudenberg F, Czechowska N, Seidel K, Baader SL, Gispert S, et al. Mouse Ataxin-2 Expansion Downregulates CamKII and Other Calcium Signaling Factors, Impairing Granule—Purkinje Neuron Synaptic Strength. International Journal of Molecular Sciences. 2020; 21(18):6673. https://doi.org/10.3390/ijms21186673
Chicago/Turabian StyleArsović, Aleksandar, Melanie Vanessa Halbach, Júlia Canet-Pons, Dilhan Esen-Sehir, Claudia Döring, Florian Freudenberg, Nicoletta Czechowska, Kay Seidel, Stephan L. Baader, Suzana Gispert, and et al. 2020. "Mouse Ataxin-2 Expansion Downregulates CamKII and Other Calcium Signaling Factors, Impairing Granule—Purkinje Neuron Synaptic Strength" International Journal of Molecular Sciences 21, no. 18: 6673. https://doi.org/10.3390/ijms21186673
APA StyleArsović, A., Halbach, M. V., Canet-Pons, J., Esen-Sehir, D., Döring, C., Freudenberg, F., Czechowska, N., Seidel, K., Baader, S. L., Gispert, S., Sen, N.-E., & Auburger, G. (2020). Mouse Ataxin-2 Expansion Downregulates CamKII and Other Calcium Signaling Factors, Impairing Granule—Purkinje Neuron Synaptic Strength. International Journal of Molecular Sciences, 21(18), 6673. https://doi.org/10.3390/ijms21186673