MuRF1/TRIM63, Master Regulator of Muscle Mass




Abstract

1. Introduction
2. Structure
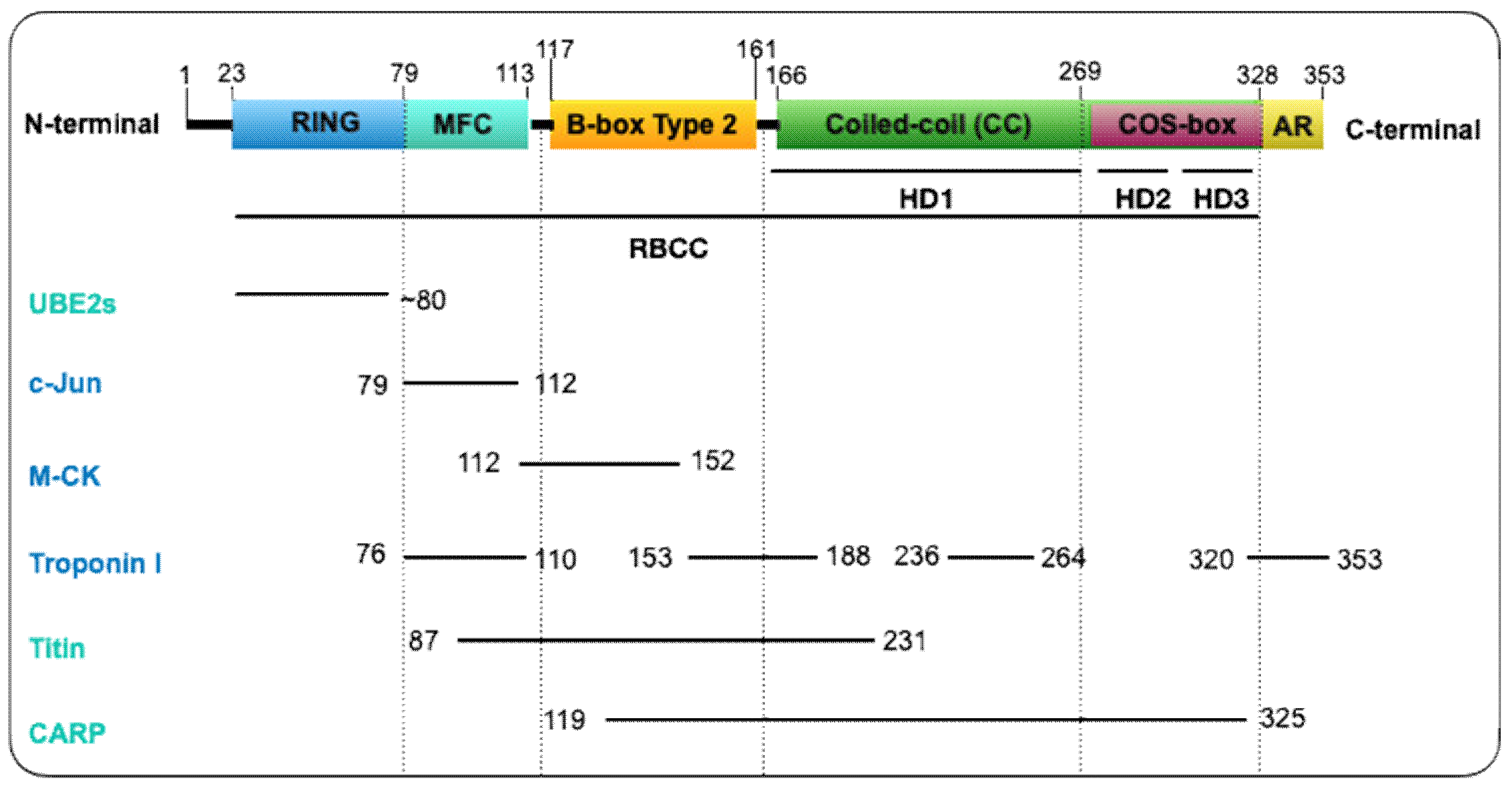
2.1. MuRF1 Belongs to the Tripartite Motif (TRIM) Family of Proteins
2.2. What Is Known about the Tridimensional Structure of MuRF1?
2.3. Two Other MuRF Proteins
2.3.1. MuRF2
2.3.2. MuRF3
3. Regulation of MuRF1
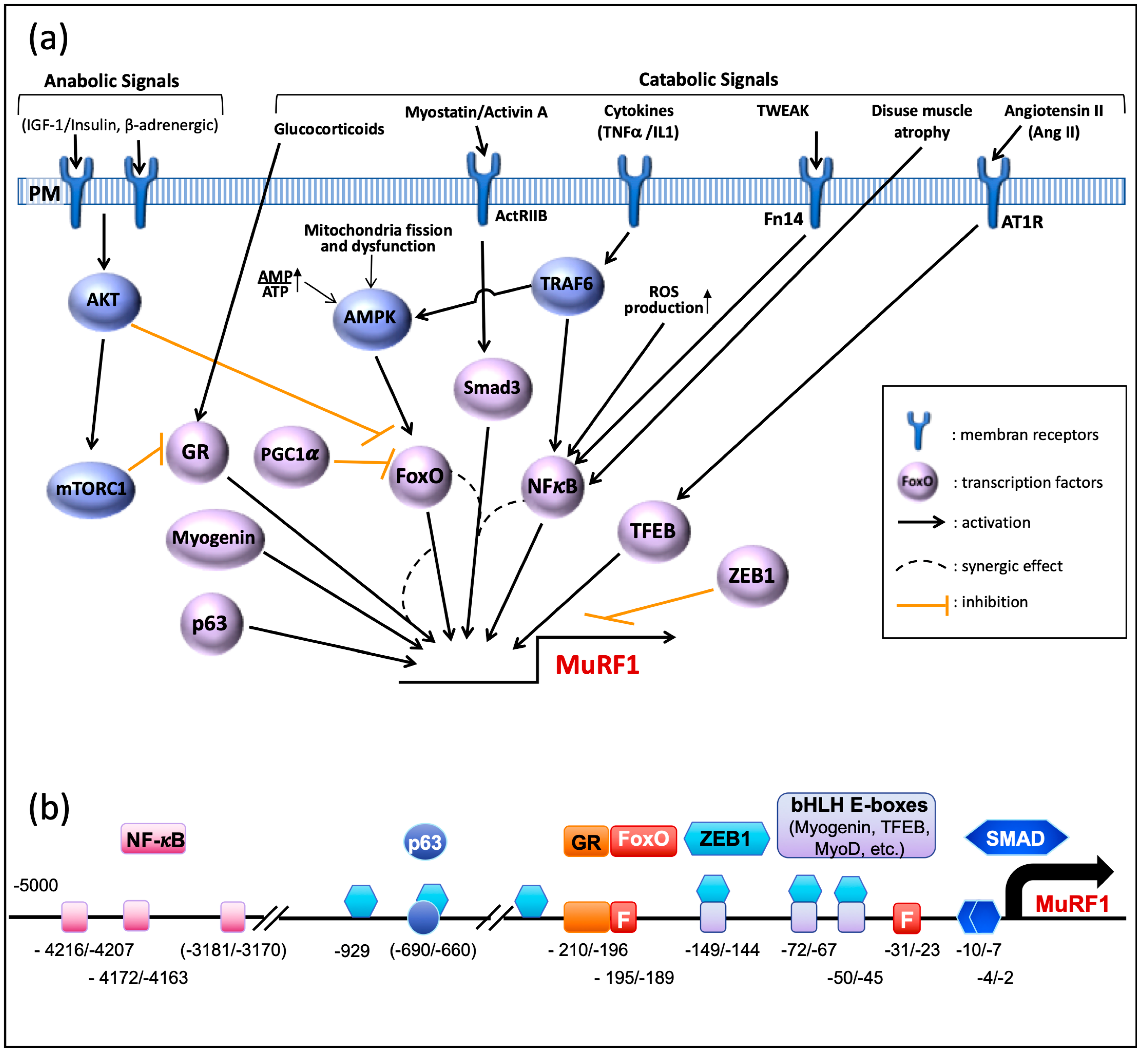
3.1. Transcriptional Regulation
3.2. Post-Translational Modifications
4. MuRF1 Location
4.1. Tissue/Fibrillar Location
4.2. Subcellular Location
5. MuRF1, Mutants, and Phenotypes
5.1. Phenotype of MuRF1-Knock-Out (KO) Mice
5.1.1. Skeletal Muscle
5.1.2. Cardiac Muscle Phenotype of MuRF1-KO Mice
5.2. MuRF1 Mutation in Humans
5.3. MuRF1 Dominant Negative Mutant
5.4. Mutant Over-Expressing MuRF1
5.4.1. Skeletal Muscle
5.4.2. Heart
5.5. MuRF1/MuRF2 Double Knock-Out
5.6. MuRF1/MuRF3 Double Knock-Out
6. Interacting Partners
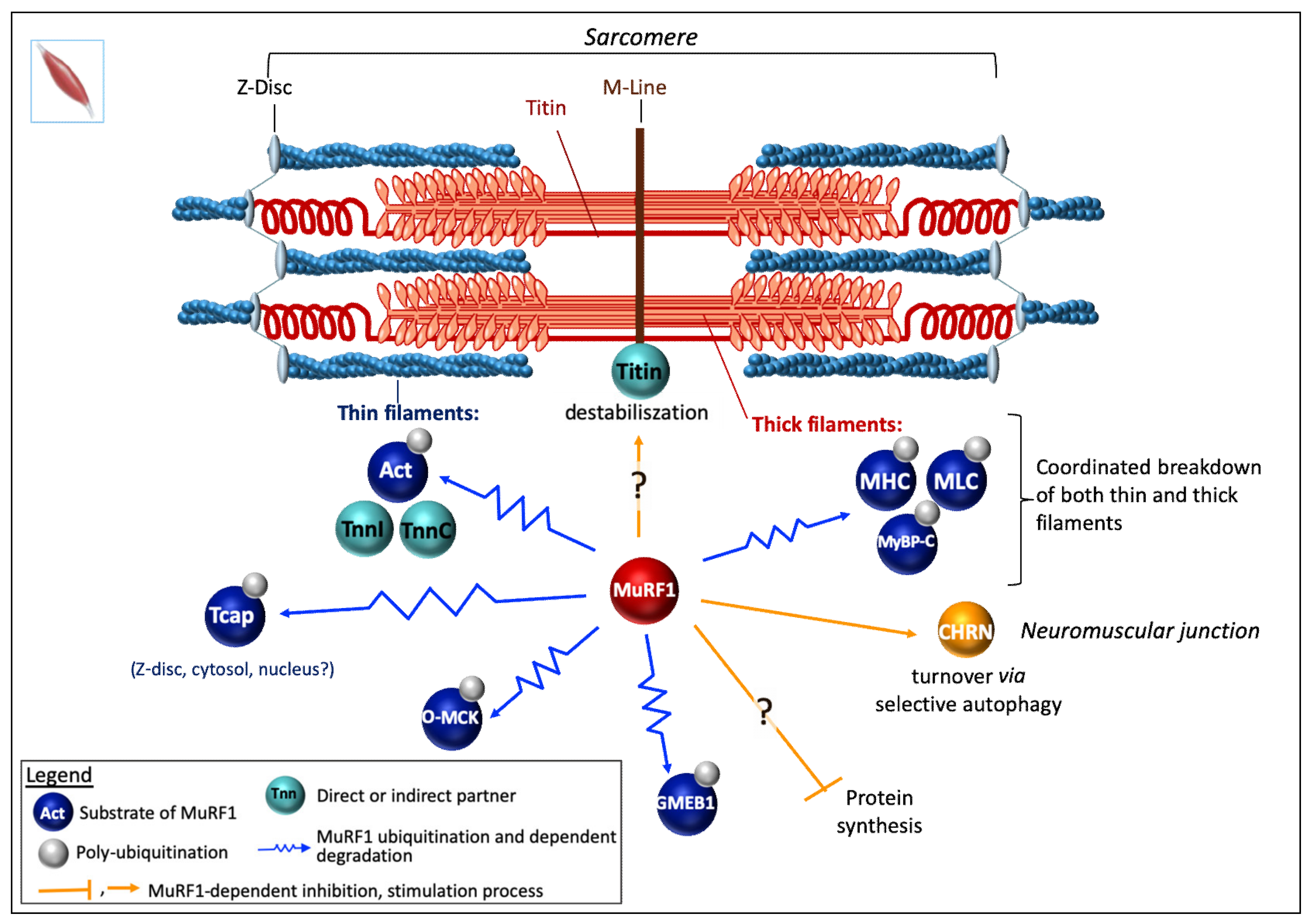
6.1. MuRF1 Substrates
6.1.1. In Skeletal Muscle
Myofibrillar/Contractile Proteins
Miscellaneous
6.1.2. In the Heart
Myofibrillar Proteins
Signaling and Protein Synthesis
6.2. Other MuRF1 Interactors (Not or Not Yet Substrates)
6.3. Ubiquitin Conjugating Enzymes, E2s
7. MuRF1 Functions
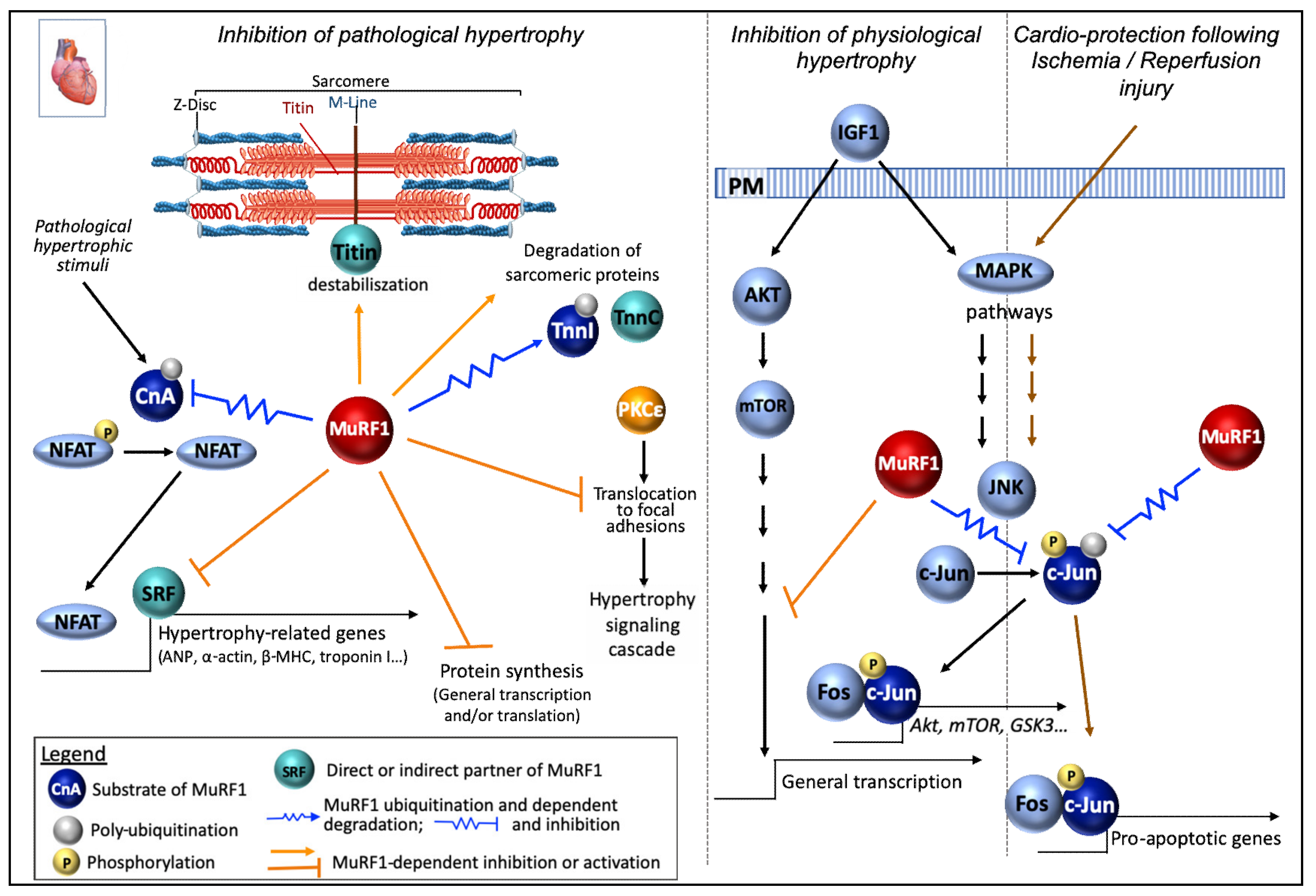
7.1. Cardiac and Skeletal Muscle Functions
7.2. Functional Redundancy of the MuRF Proteins
7.3. MuRF1 Dual Functions
8. MuRF1 Inhibitors/Modulators
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| a-actin | alpha-actin |
| ActRIIB | activin receptor type 2B |
| ALS | amyotrophic lateral sclerosis |
| Ang II | angiotensin II |
| ANP | atrial natriuretic peptide |
| AT | acidic tail |
| AT1R | angiotensin II receptor type 1 |
| Bb2 | B-box type 2 domain |
| bHLH | basic helix-loop-helix |
| Bif-1 | BAX interacting factor 1 |
| C/EBP | CCAAT/enhancer binding protein |
| CARP | ubiquitin carboxyl-terminal hydrolase MINDY-3 |
| CC | coiled-coil domain |
| CHOP | C/EBP homologous protein |
| CHRN | cholinergic receptor nicotinic/nicotinic acetylcholine receptor |
| CK | creatine kinase |
| cMyBP-C | cardiac MyBP-C isoform |
| can | calcineurin |
| COS box | C-terminal subgroup one signature-box |
| CRL4 | Cullin-RING-based E3 ubiquitin-protein ligase 4 |
| CSA | cross-sectional area; Dex, dexamethasone |
| dKO | double knock-out |
| EDL | extensor digitorium longus muscle |
| EndoB1 | endophilin B1 |
| EPR | electron paramagnetic resonance |
| FoxO | forkhead box family of transcription factors |
| GME | glucocorticoid modulatory elements |
| GMEB1 | glucocorticoid modulatory element binding protein-1 |
| GR | glucocorticoid receptor |
| HCM | hypertrophic cardiomyopathy |
| HD | helical domain |
| HIBADH | 3-hydroxyisobutyrate dehydrogenase |
| ICU | intensive care unit |
| IGF-1 | insulin-like growth factor 1 |
| IL1 | interleukine 1 |
| I/R | ischemia/reperfusion |
| JNK | c-Jun N-terminal kinases |
| KO | knock-out |
| LAMP1 | lyososomal-associated membrane protein 1 |
| LC3 | microtubule-associated proteins 1A/1B light chain 3b |
| M1/M2 dKO | homozygous constitutive null mutants for MuRF1 or MuRF2 |
| MAFbx | muscle atrophy F-box protein |
| MCT | monocrotaline |
| MEF2 | myocyte enhancer factor-2 |
| MFC | MuRF family specific motif |
| MHC | myosin heavy chain |
| MHCIIa | myosin heavy chain type IIa |
| MI | myocardial infarction |
| MLC | myosin light chain |
| MLP/Csrp3 | muscle limp protein/cystine and glycine-rich protein 3 |
| MCK | muscle-type creatine kinase |
| mTORC1 | mammalian target of rapamycin complex 1 |
| MuRF | muscle-specific RING finger protein |
| MuRF1-KO | transgenic mice specifically silenced for MuRF1 |
| MuRF1-TG | transgenic mice specifically over-expressing MuRF1 in skeletal muscle |
| MuRF1-Tg+ | transgenic mice expressing increased levels of MuRF1 specifically in the heart |
| MuRF1-ΔRING | transgenic mice with a mutant dominant negative form of MuRF1 corresponding to a RING-deletion of MuRF1 |
| MyBP-C | myosin binding prot-C |
| MHC | myosin heavy chain |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFAT | nuclear factor of activated T cells |
| NRVM | neonatal rat ventricular myocytes |
| O-MCK | oxidized form of MCK |
| PE | phenylephrine |
| PDH | pyruvate dehydrogenase |
| PDK2 | pyruvate dehydrogenase kinase isoform 2 |
| PI3K | phosphoinositide 3-kinase |
| polyUb | polyubiquitination |
| PMA | plasma membrane ATPase |
| PPARα | peroxisome proliferator activated receptors |
| PPRE | PPAR response element |
| RACK1 | receptor of activated protein C kinase 1 |
| RBCC | RING-B box-coiled-coil |
| RING | really interesting new gene |
| ROS | reactive oxygen species |
| SPR | surface plasmon resonance |
| SQTM1/p62 | Sequestosome-1 |
| SRF | serum response factor |
| SUMO | small ubiquitin-related modifier |
| TAC | transaortic constriction model |
| TFEB | transcription factor EB |
| TNF | transforming growth factor |
| TnnI, TnnC | troponin I and C |
| TRAF6 | TNF receptor associated factors 6 |
| TRIP12 | thyroid receptor-interacting protein 12 |
| TRIM | tripartite motif family |
| UPS | ubiquitin-proteasome system |
| WT | wild-type |
| Y2H | yeast two-hybrid. |
References
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Centner, T.; Yano, J.; Kimura, E.; McElhinny, A.S.; Pelin, K.; Witt, C.C.; Bang, M.L.; Trombitas, K.; Granzier, H.; Gregorio, C.C.; et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J. Mol. Biol. 2001, 306, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Taillandier, D.; Polge, C. Skeletal muscle atrogenes: From rodent models to human pathologies. Biochimie 2019, 166, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.A.; Etkin, L.D.; Freemont, P.S. A novel zinc finger coiled-coil domain in a family of nuclear proteins. Trends Biochem. Sci. 1992, 17, 344–345. [Google Scholar] [CrossRef]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. BioEssays 2005, 27, 1147–1157. [Google Scholar] [CrossRef]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef]
- Short, K.M.; Cox, T.C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 2006, 281, 8970–8980. [Google Scholar] [CrossRef]
- Caratozzolo, M.F.; Marzano, F.; Mastropasqua, F.; Sbisà, E.; Tullo, A. TRIM8: Making the Right Decision between the Oncogene and Tumour Suppressor Role. Genes 2017, 8, 354. [Google Scholar] [CrossRef]
- Borlepawar, A.; Frey, N.; Rangrez, A.Y. A systematic view on E3 ligase Ring TRIMmers with a focus on cardiac function and disease. Trends Cardiovasc. Med. 2019, 29, 1–8. [Google Scholar] [CrossRef]
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 2008, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM Gene Expression in Response to Interferons. PLoS ONE 2009, 4, e4894. [Google Scholar] [CrossRef] [PubMed]
- Marín, I. Origin and Diversification of TRIM Ubiquitin Ligases. PLoS ONE 2012, 7, e50030. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.-M.; Chang, T.-H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A.P. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Esposito, D.; Koliopoulos, M.G.; Rittinger, K. Structural determinants of TRIM protein function. Biochem. Soc. Trans. 2017, 45, 183–191. [Google Scholar] [CrossRef]
- Mrosek, M.; Meier, S.; Ucurum-Fotiadis, Z.; von Castelmur, E.; Hedbom, E.; Lustig, A.; Grzesiek, S.; Labeit, D.; Labeit, S.; Mayans, O. Structural analysis of B-Box 2 from MuRF1: Identification of a novel self-association pattern in a RING-like fold. Biochemistry 2008, 47, 10722–10730. [Google Scholar] [CrossRef]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschellà, G. Zinc-finger proteins in health and disease. Cell Death Discov. 2017, 3, 1–12. [Google Scholar] [CrossRef]
- Budhidarmo, R.; Nakatani, Y.; Day, C.L. RINGs hold the key to ubiquitin transfer. Trends Biochem. Sci. 2012, 37, 58–65. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H.; Wu, W.; Zhuo, W.; Liu, W.; Zhang, Y.; Cheng, M.; Chen, Y.-G.; Gao, N.; Yu, H.; et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014, 24, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-H.; Du, J.; Fan, Y.-N.; Zhang, M.-L.; Liu, D.-P.; Li, L.; Lockyer, P.; Kang, E.Y.; Patterson, C.; Willis, M.S. The ubiquitin ligase MuRF1 protects against cardiac ischemia/reperfusion injury by its proteasome-dependent degradation of phospho-c-Jun. Am. J. Pathol. 2011, 178, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Franke, B.; Skorupka, K.A.; Cafiso, D.S.; Pornillos, O.; Mayans, O.; Norman, D.G. Exploration of the TRIM Fold of MuRF1 Using EPR Reveals a Canonical Antiparallel Structure and Extended COS-Box. J. Mol. Biol. 2019, 431, 2900–2909. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.A.; Eliazer, S.; Ilaria, R.L.; Richardson, J.A.; Olson, E.N. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. J. Cell Biol. 2000, 150, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Roganowicz, M.D.; Skorupka, K.; Alam, S.L.; Christensen, D.; Doss, G.; Wan, Y.; Frank, G.A.; Ganser-Pornillos, B.K.; Sundquist, W.I.; et al. Mechanism of B-box 2 domain-mediated higher-order assembly of the retroviral restriction factor TRIM5α. Elife 2016, 5, e16309. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Borden, K.L.; Freemont, P.S.; Etkin, L.D. Involvement of the rfp tripartite motif in protein-protein interactions and subcellular distribution. J. Cell Sci. 1997, 110 Pt 14, 1563–1571. [Google Scholar]
- Franke, B.; Gasch, A.; Rodriguez, D.; Chami, M.; Khan, M.M.; Rudolf, R.; Bibby, J.; Hanashima, A.; Bogomolovas, J.; von Castelmur, E.; et al. Molecular basis for the fold organization and sarcomeric targeting of the muscle atrogin MuRF1. Open Biol. 2014, 4. [Google Scholar] [CrossRef]
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.-H.; Rockman, H.A.; Patterson, C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140. [Google Scholar] [CrossRef]
- Koyama, S.; Hata, S.; Witt, C.C.; Ono, Y.; Lerche, S.; Ojima, K.; Chiba, T.; Doi, N.; Kitamura, F.; Tanaka, K.; et al. Muscle RING-finger protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J. Mol. Biol. 2008, 376, 1224–1236. [Google Scholar] [CrossRef]
- Gundogdu, M.; Walden, H. Structural basis of generic versus specific E2–RING E3 interactions in protein ubiquitination. Protein Sci. 2019, 28, 1758–1770. [Google Scholar] [CrossRef]
- Witt, C.C.; Witt, S.H.; Lerche, S.; Labeit, D.; Back, W.; Labeit, S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2008, 27, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.; Mankoo, B.; Gautel, M. Developmental regulation of MURF E3 ubiquitin ligases in skeletal muscle. J. Muscle Res. Cell Motil. 2012, 33, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Pizon, V.; Iakovenko, A.; van der Ven, P.F.M.; Kelly, R.; Fatu, C.; Fürst, D.O.; Karsenti, E.; Gautel, M. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J. Cell Sci. 2002, 115, 4469–4482. [Google Scholar] [CrossRef]
- Ochala, J.; Gustafson, A.-M.; Diez, M.L.; Renaud, G.; Li, M.; Aare, S.; Qaisar, R.; Banduseela, V.C.; Hedström, Y.; Tang, X.; et al. Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: Underlying mechanisms. J. Physiol. 2011, 589, 2007–2026. [Google Scholar] [CrossRef]
- Nguyen, T.; Bowen, T.S.; Augstein, A.; Schauer, A.; Gasch, A.; Linke, A.; Labeit, S.; Adams, V. Expression of MuRF1 or MuRF2 is essential for the induction of skeletal muscle atrophy and dysfunction in a murine pulmonary hypertension model. Skelet. Muscle 2020, 10, 12. [Google Scholar] [CrossRef]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef]
- Jagoe, R.T.; Lecker, S.H.; Gomes, M.; Goldberg, A.L. Patterns of gene expression in atrophying skeletal muscles: Response to food deprivation. FASEB J. 2002, 16, 1697–1712. [Google Scholar] [CrossRef]
- Polge, C.; Koulmann, N.; Claustre, A.; Jarzaguet, M.; Serrurier, B.; Combaret, L.; Béchet, D.; Bigard, X.; Attaix, D.; Taillandier, D. UBE2D2 is not involved in MuRF1-dependent muscle wasting during hindlimb suspension. Int. J. Biochem. Cell Biol. 2016, 79, 488–493. [Google Scholar] [CrossRef]
- Seaborne, R.A.; Hughes, D.C.; Turner, D.C.; Owens, D.J.; Baehr, L.M.; Gorski, P.; Semenova, E.A.; Borisov, O.V.; Larin, A.K.; Popov, D.V.; et al. UBR5 is a novel E3 ubiquitin ligase involved in skeletal muscle hypertrophy and recovery from atrophy. J. Physiol. 2019, 597, 3727–3749. [Google Scholar] [CrossRef]
- Pinheiro-Dardis, C.M.; Erbereli, B.T.; Gigo-Benato, D.; Castro, P.A.T.S.; Russo, T.L. Electrical stimulation delays reinnervation in denervated rat muscle. Muscle Nerve 2017, 56, 108–118. [Google Scholar] [CrossRef]
- Fisher, A.G.; Seaborne, R.A.; Hughes, T.M.; Gutteridge, A.; Stewart, C.; Coulson, J.M.; Sharples, A.P.; Jarvis, J.C. Transcriptomic and epigenetic regulation of disuse atrophy and the return to activity in skeletal muscle. FASEB J. 2017, 31, 5268–5282. [Google Scholar] [CrossRef] [PubMed]
- Baptista, I.L.; Leal, M.L.; Artioli, G.G.; Aoki, M.S.; Fiamoncini, J.; Turri, A.O.; Curi, R.; Miyabara, E.H.; Moriscot, A.S. Leucine attenuates skeletal muscle wasting via inhibition of ubiquitin ligases. Muscle Nerve 2010, 41, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Baptista, I.L.; Silva, W.J.; Artioli, G.G.; Guilherme, J.P.L.F.; Leal, M.L.; Aoki, M.S.; Miyabara, E.H.; Moriscot, A.S. Leucine and HMB differentially modulate proteasome system in skeletal muscle under different sarcopenic conditions. PLoS ONE 2013, 8, e76752. [Google Scholar] [CrossRef]
- Baptista, I.L.; Silvestre, J.G.; Silva, W.J.; Labeit, S.; Moriscot, A.S. FoxO3a suppression and VPS34 activity are essential to anti-atrophic effects of leucine in skeletal muscle. Cell Tissue Res. 2017, 369, 381–394. [Google Scholar] [CrossRef]
- Sacheck, J.M.; Ohtsuka, A.; McLary, S.C.; Goldberg, A.L. IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am. J. Physiol. Endocrinol. Metab. 2004, 287, 591–601. [Google Scholar] [CrossRef]
- Schakman, O.; Kalista, S.; Bertrand, L.; Lause, P.; Verniers, J.; Ketelslegers, J.M.; Thissen, J.P. Role of Akt/GSK-3beta/beta-catenin transduction pathway in the muscle anti-atrophy action of insulin-like growth factor-I in glucocorticoid-treated rats. Endocrinology 2008, 149, 3900–3908. [Google Scholar] [CrossRef]
- Cleveland, B.M.; Weber, G.M.; Blemings, K.P.; Silverstein, J.T. Insulin-like growth factor-I and genetic effects on indexes of protein degradation in response to feed deprivation in rainbow trout (Oncorhynchus mykiss). Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1332–R1342. [Google Scholar] [CrossRef]
- Song, J.; Baer, L.A.; Threlkeld, M.R.S.; Geng, C.; Wade, C.E.; Wolf, S.E. Insulin and exercise improved muscle function in rats with severe burns and hindlimb unloading. Physiol. Rep. 2019, 7, e14158. [Google Scholar] [CrossRef]
- Yoshida, T.; Semprun-Prieto, L.; Sukhanov, S.; Delafontaine, P. IGF-1 prevents ANG II-induced skeletal muscle atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin ligase atrogin-1 expression. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1565–H1570. [Google Scholar] [CrossRef]
- Schakman, O.; Dehoux, M.; Bouchuari, S.; Delaere, S.; Lause, P.; Decroly, N.; Shoelson, S.E.; Thissen, J.-P. Role of IGF-I and the TNFα/NF-κB pathway in the induction of muscle atrogenes by acute inflammation. Am. J. Physiol. Endocrinol. Metab. 2012, 303, 729–739. [Google Scholar] [CrossRef]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, T.; Kitayama, K.; Yamashita, H.; Mori, N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem. J. 2003, 375, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.-S.; Huang, B.; Unterman, T.G.; Harris, R.A. Protein Kinase B-α Inhibits Human Pyruvate Dehydrogenase Kinase-4 Gene Induction by Dexamethasone Through Inactivation of FOXO Transcription Factors. Diabetes 2004, 53, 899–910. [Google Scholar] [CrossRef]
- Waddell, D.S.; Baehr, L.M.; van den Brandt, J.; Johnsen, S.A.; Reichardt, H.M.; Furlow, J.D.; Bodine, S.C. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am. J. Physiol. Endocrinol. Metab. 2008, 295, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.J.; Alamdari, N.; O’Neal, P.; Gonnella, P.; Aversa, Z.; Hasselgren, P.-O. Sepsis increases the expression and activity of the transcription factor Forkhead Box O 1 (FOXO1) in skeletal muscle by a glucocorticoid-dependent mechanism. Int. J. Biochem. Cell Biol. 2010, 42, 701–711. [Google Scholar] [CrossRef]
- McLoughlin, T.J.; Smith, S.M.; DeLong, A.D.; Wang, H.; Unterman, T.G.; Esser, K.A. FoxO1 induces apoptosis in skeletal myotubes in a DNA-binding-dependent manner. Am. J. Physiol. Cell Physiol. 2009, 297, C548–C555. [Google Scholar] [CrossRef]
- Xu, J.; Li, R.; Workeneh, B.; Dong, Y.; Wang, X.; Hu, Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012, 82, 401–411. [Google Scholar] [CrossRef]
- Goodman, C.A.; McNally, R.M.; Hoffmann, F.M.; Hornberger, T.A. Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol. Endocrinol. 2013, 27, 1946–1957. [Google Scholar] [CrossRef]
- Bollinger, L.M.; Witczak, C.A.; Houmard, J.A.; Brault, J.J. SMAD3 augments FoxO3-induced MuRF-1 promoter activity in a DNA-binding-dependent manner. Am. J. Physiol. Cell Physiol. 2014, 307, C278–C287. [Google Scholar] [CrossRef]
- Kang, S.-H.; Lee, H.-A.; Kim, M.; Lee, E.; Sohn, U.D.; Kim, I. Forkhead box O3 plays a role in skeletal muscle atrophy through expression of E3 ubiquitin ligases MuRF-1 and atrogin-1 in Cushing’s syndrome. Am. J. Physiol. Endocrinol. Metab. 2017, 312, 495–507. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin–proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Moylan, J.S.; Smith, J.D.; Chambers, M.A.; McLoughlin, T.J.; Reid, M.B. TNF induction of atrogin-1/MAFbx mRNA depends on Foxo4 expression but not AKT-Foxo1/3 signaling. Am. J. Physiol. Cell Physiol. 2008, 295, C986–C993. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.T.; Bhardwaj, G.; Penniman, C.M.; Krumpoch, M.T.; Beltran, P.A.S.; Klaus, K.; Poro, K.; Li, M.; Pan, H.; Dreyfuss, J.M.; et al. FoxO Transcription Factors Are Critical Regulators of Diabetes-Related Muscle Atrophy. Diabetes 2019, 68, 556–570. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1–PGC-1α interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef]
- Watson, M.L.; Baehr, L.M.; Reichardt, H.M.; Tuckermann, J.P.; Bodine, S.C.; Furlow, J.D. A cell-autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. Am. J. Physiol. Endocrinol. Metab. 2012, 302, 1210–1220. [Google Scholar] [CrossRef]
- Wu, C.-L.; Cornwell, E.W.; Jackman, R.W.; Kandarian, S.C. NF-κB but not FoxO sites in the MuRF1 promoter are required for transcriptional activation in disuse muscle atrophy. Am. J. Physiol. Cell Physiol. 2014, 306, C762–C767. [Google Scholar] [CrossRef]
- Yarar-Fisher, C.; Bickel, C.S.; Kelly, N.A.; Stec, M.J.; Windham, S.T.; McLain, A.B.; Oster, R.A.; Bamman, M.M. Heightened TWEAK-NF-κB signaling and inflammation-associated fibrosis in paralyzed muscles of men with chronic spinal cord injury. Am. J. Physiol. Endocrinol. Metab. 2016, 310, 754–761. [Google Scholar] [CrossRef]
- Vainshtein, A.; Sandri, M. Signaling Pathways that Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef]
- Files, D.C.; D’Alessio, F.R.; Johnston, L.F.; Kesari, P.; Aggarwal, N.R.; Garibaldi, B.T.; Mock, J.R.; Simmers, J.L.; DeGorordo, A.; Murdoch, J.; et al. A critical role for muscle ring finger-1 in acute lung injury-associated skeletal muscle wasting. Am. J. Respir. Crit. Care Med. 2012, 185, 825–834. [Google Scholar] [CrossRef]
- Sriram, S.; Subramanian, S.; Juvvuna, P.K.; Ge, X.; Lokireddy, S.; McFarlane, C.D.; Wahli, W.; Kambadur, R.; Sharma, M. Myostatin augments muscle-specific ring finger protein-1 expression through an NF-kB independent mechanism in SMAD3 null muscle. Mol. Endocrinol. 2014, 28, 317–330. [Google Scholar] [CrossRef]
- Arrowsmith, C.H. Structure and function in the p53 family. Cell Death Differ. 1999, 6, 1169–1173. [Google Scholar] [CrossRef]
- Benosman, S.; Meng, X.; Grabowiecki, Y.V.; Palamiuc, L.; Hritcu, L.; Gross, I.; Mellitzer, G.; Taya, Y.; Loeffler, J.-P.; Gaiddon, C. Complex regulation of p73 isoforms after alteration of the amyloid precursor polypeptide (APP) function and DNA damages in neurons. J. Biol. Chem. 2011. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef]
- Von Grabowiecki, Y.; Abreu, P.; Blanchard, O.; Palamiuc, L.; Benosman, S.; Mériaux, S.; Devignot, V.; Gross, I.; Mellitzer, G.; de Aguilar, J.L.G.; et al. Transcriptional activator TAp63 is upregulated in muscular atrophy during ALS and induces the pro-atrophic ubiquitin ligase Trim63. Elife 2016, 5. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef]
- Bois, P.D.; Tortola, C.P.; Lodka, D.; Kny, M.; Schmidt, F.; Song, K.; Schmidt, S.; Bassel-Duby, R.; Olson, E.N.; Fielitz, J. Angiotensin II Induces Skeletal Muscle Atrophy by Activating TFEB-Mediated MuRF1 Expression. Circ. Res. 2015, 117, 424–436. [Google Scholar] [CrossRef]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993, 364, 501–506. [Google Scholar] [CrossRef]
- Nabeshima, Y.; Hanaoka, K.; Hayasaka, M.; Esuml, E.; Li, S.; Nonaka, I.; Nabeshima, Y. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 1993, 364, 532–535. [Google Scholar] [CrossRef]
- Moresi, V.; Williams, A.H.; Meadows, E.; Flynn, J.M.; Potthoff, M.J.; McAnally, J.; Shelton, J.M.; Backs, J.; Klein, W.H.; Richardson, J.A.; et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 2010, 143, 35–45. [Google Scholar] [CrossRef]
- Ninfali, C.; Siles, L.; Darling, D.S.; Postigo, A. Regulation of muscle atrophy-related genes by the opposing transcriptional activities of ZEB1/CtBP and FOXO3. Nucleic Acids Res. 2018, 46, 10697–10708. [Google Scholar] [CrossRef]
- Nerlov, C. The C/EBP family of transcription factors: A paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007, 17, 318–324. [Google Scholar] [CrossRef]
- Gonnella, P.; Alamdari, N.; Tizio, S.; Aversa, Z.; Petkova, V.; Hasselgren, P.-O. C/EBPβ regulates dexamethasone-induced muscle cell atrophy and expression of atrogin-1 and MuRF1. J. Cell. Biochem. 2011, 112, 1737–1748. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, G.; Sin, K.W.T.; Liu, Z.; Lin, R.-K.; Li, M.; Li, Y.-P. Activin A induces skeletal muscle catabolism via p38β mitogen-activated protein kinase. J. Cachexia Sarcopenia Muscle 2017, 8, 202–212. [Google Scholar] [CrossRef]
- Silva, K.A.S.; Dong, J.; Dong, Y.; Dong, Y.; Schor, N.; Tweardy, D.J.; Zhang, L.; Mitch, W.E. Inhibition of Stat3 Activation Suppresses Caspase-3 and the Ubiquitin-Proteasome System, Leading to Preservation of Muscle Mass in Cancer Cachexia. J. Biol. Chem. 2015, 290, 11177–11187. [Google Scholar] [CrossRef]
- Fang, C.X.; Dong, F.; Thomas, D.P.; Ma, H.; He, L.; Ren, J. Hypertrophic cardiomyopathy in high-fat diet-induced obesity: Role of suppression of forkhead transcription factor and atrophy gene transcription. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1206–H1215. [Google Scholar] [CrossRef]
- Shimizu, H.; Langenbacher, A.D.; Huang, J.; Wang, K.; Otto, G.; Geisler, R.; Wang, Y.; Chen, J.-N. The Calcineurin-FoxO-MuRF1 signaling pathway regulates myofibril integrity in cardiomyocytes. Elife 2017, 6. [Google Scholar] [CrossRef]
- Baskin, K.K.; Taegtmeyer, H. AMP-activated protein kinase regulates E3 ligases in rodent heart. Circ. Res. 2011, 109, 1153–1161. [Google Scholar] [CrossRef]
- Jaitovich, A.; Angulo, M.; Lecuona, E.; Dada, L.A.; Welch, L.C.; Cheng, Y.; Gusarova, G.; Ceco, E.; Liu, C.; Shigemura, M.; et al. High CO2 levels cause skeletal muscle atrophy via AMP-activated kinase (AMPK), FoxO3a protein, and muscle-specific Ring finger protein 1 (MuRF1). J. Biol. Chem. 2015, 290, 9183–9194. [Google Scholar] [CrossRef]
- Namuduri, A.V.; Heras, G.; Mi, J.; Cacciani, N.; Hörnaeus, K.; Konzer, A.; Lind, S.B.; Larsson, L.; Gastaldello, S. A Proteomic Approach to Identify Alterations in the Small Ubiquitin-like Modifier (SUMO) Network during Controlled Mechanical Ventilation in Rat Diaphragm Muscle. Mol. Cell. Proteom. 2017, 16, 1081–1097. [Google Scholar] [CrossRef]
- Heras, G.; Namuduri, A.V.; Traini, L.; Shevchenko, G.; Falk, A.; Lind, S.B.; Jia, M.; Tian, G.; Gastaldello, S. Muscle RING-finger protein-1 (MuRF1) functions and cellular localization are regulated by SUMO1 post-translational modification. J. Mol. Cell Biol. 2019, 11, 356–370. [Google Scholar] [CrossRef]
- Kim, H.T.; Kim, K.P.; Lledias, F.; Kisselev, A.F.; Scaglione, K.M.; Skowyra, D.; Gygi, S.P.; Goldberg, A.L. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem. 2007, 282, 17375–17386. [Google Scholar] [CrossRef]
- Bowen, T.S.; Adams, V.; Werner, S.; Fischer, T.; Vinke, P.; Brogger, M.N.; Mangner, N.; Linke, A.; Sehr, P.; Lewis, J.; et al. Small-molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 939–953. [Google Scholar] [CrossRef]
- Mota, R.; Rodríguez, J.E.; Bonetto, A.; O’Connell, T.M.; Asher, S.A.; Parry, T.L.; Lockyer, P.; McCudden, C.R.; Couch, M.E.; Willis, M.S. Post-translationally modified muscle-specific ubiquitin ligases as circulating biomarkers in experimental cancer cachexia. Am. J. Cancer Res. 2017, 7, 1948–1958. [Google Scholar]
- Bdolah, Y.; Segal, A.; Tanksale, P.; Karumanchi, S.A.; Lecker, S.H. Atrophy-related ubiquitin ligases atrogin-1 and MuRF-1 are associated with uterine smooth muscle involution in the postpartum period. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R971–R976. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Moriscot, A.S.; Baptista, I.L.; Bogomolovas, J.; Witt, C.; Hirner, S.; Granzier, H.; Labeit, S. MuRF1 is a muscle fiber-type II associated factor and together with MuRF2 regulates type-II fiber trophicity and maintenance. J. Struct. Biol. 2010, 170, 344–353. [Google Scholar] [CrossRef]
- Polge, C.; Leulmi, R.; Jarzaguet, M.; Claustre, A.; Combaret, L.; Béchet, D.; Heng, A.-E.; Attaix, D.; Taillandier, D. UBE2B is implicated in myofibrillar protein loss in catabolic C2C12 myotubes: UBE2B and myofibrillar protein degradation. J. Cachexia Sarcopenia Muscle 2016, 7, 377–387. [Google Scholar] [CrossRef]
- Belova, S.P.; Mochalova, E.P.; Kostrominova, T.Y.; Shenkman, B.S.; Nemirovskaya, T.L. P38α-MAPK Signaling Inhibition Attenuates Soleus Atrophy during Early Stages of Muscle Unloading. Int. J. Mol. Sci. 2020, 21, 2756. [Google Scholar] [CrossRef]
- Macpherson, P.C.D.; Wang, X.; Goldman, D. Myogenin regulates denervation-dependent muscle atrophy in mouse soleus muscle. J. Cell. Biochem. 2011, 112, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Atherton, P.J.; Greenhaff, P.L.; Phillips, S.M.; Bodine, S.C.; Adams, C.M.; Lang, C.H. Control of skeletal muscle atrophy in response to disuse: Clinical/preclinical contentions and fallacies of evidence. Am. J. Physiol. Endocrinol. Metab. 2016, 311, 594–604. [Google Scholar] [CrossRef]
- McElhinny, A.S.; Kakinuma, K.; Sorimachi, H.; Labeit, S.; Gregorio, C.C. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J. Cell Biol. 2002, 157, 125–136. [Google Scholar] [CrossRef]
- Chen, S.N.; Czernuszewicz, G.; Tan, Y.; Lombardi, R.; Jin, J.; Willerson, J.T.; Marian, A.J. Human molecular genetic and functional studies identify TRIM63, encoding Muscle RING Finger Protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ. Res. 2012, 111, 907–919. [Google Scholar] [CrossRef]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef]
- Arya, R.; Kedar, V.; Hwang, J.R.; McDonough, H.; Li, H.-H.; Taylor, J.; Patterson, C. Muscle ring finger protein-1 inhibits PKC{epsilon} activation and prevents cardiomyocyte hypertrophy. J. Cell Biol. 2004, 167, 1147–1159. [Google Scholar] [CrossRef]
- Savarese, M.; Jonson, P.H.; Huovinen, S.; Paulin, L.; Auvinen, P.; Udd, B.; Hackman, P. The complexity of titin splicing pattern in human adult skeletal muscles. Skelet. Muscle 2018, 8, 11. [Google Scholar] [CrossRef]
- Lange, S.; Xiang, F.; Yakovenko, A.; Vihola, A.; Hackman, P.; Rostkova, E.; Kristensen, J.; Brandmeier, B.; Franzen, G.; Hedberg, B.; et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science 2005, 308, 1599–1603. [Google Scholar] [CrossRef]
- Olivé, M.; Abdul-Hussein, S.; Oldfors, A.; González-Costello, J.; van der Ven, P.F.M.; Fürst, D.O.; González, L.; Moreno, D.; Torrejón-Escribano, B.; Alió, J.; et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum. Mol. Genet. 2015, 24, 3638–3650. [Google Scholar] [CrossRef]
- Hwee, D.T.; Baehr, L.M.; Philp, A.; Baar, K.; Bodine, S.C. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell 2014, 13, 92–101. [Google Scholar] [CrossRef]
- Labeit, S.; Kohl, C.H.; Witt, C.C.; Labeit, D.; Jung, J.; Granzier, H. Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1-KO mice. J. Biomed. Biotechnol. 2010, 2010, 693741. [Google Scholar] [CrossRef] [PubMed]
- Sartori, R.; Schirwis, E.; Blaauw, B.; Bortolanza, S.; Zhao, J.; Enzo, E.; Stantzou, A.; Mouisel, E.; Toniolo, L.; Ferry, A.; et al. BMP signaling controls muscle mass. Nat. Genet. 2013, 45, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.K.; Bhatnagar, S.; Mishra, V.; Srivastava, S.; Darnay, B.G.; Choi, Y.; Kumar, A. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol. Cell. Biol. 2012, 32, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, P.; Plant, P.J.; Correa, J.; Bain, A.; Takeda, M.; Kawabe, H.; Rotin, D.; Bain, J.R.; Batt, J.A.E. The ubiquitin ligase Nedd4-1 participates in denervation-induced skeletal muscle atrophy in mice. PLoS ONE 2012, 7, e46427. [Google Scholar] [CrossRef]
- An, C.-I.; Ganio, E.; Hagiwara, N. Trip12, a HECT domain E3 ubiquitin ligase, targets Sox6 for proteasomal degradation and affects fiber type-specific gene expression in muscle cells. Skelet. Muscle 2013, 3, 11. [Google Scholar] [CrossRef]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 Degrades Myosin Heavy Chain Protein in Dexamethasone-Treated Skeletal Muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef]
- Baehr, L.M.; Furlow, J.D.; Bodine, S.C. Muscle sparing in muscle RING finger 1 null mice: Response to synthetic glucocorticoids. J. Physiol. 2011, 589, 4759–4776. [Google Scholar] [CrossRef]
- Shoji, S.; Pennington, R.J. The effect of cortisone on protein breakdown and synthesis in rat skeletal muscle. Mol. Cell. Endocrinol. 1977, 6, 159–169. [Google Scholar] [CrossRef]
- Maejima, Y.; Usui, S.; Zhai, P.; Takamura, M.; Kaneko, S.; Zablocki, D.; Yokota, M.; Isobe, M.; Sadoshima, J. Muscle-specific RING finger 1 negatively regulates pathological cardiac hypertrophy through downregulation of calcineurin A. Circ. Heart Fail. 2014, 7, 479–490. [Google Scholar] [CrossRef]
- Willis, M.S.; Ike, C.; Li, L.; Wang, D.-Z.; Glass, D.J.; Patterson, C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ. Res. 2007, 100, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Schisler, J.C.; Li, L.; Rodríguez, J.E.; Hilliard, E.G.; Charles, P.C.; Patterson, C. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ. Res. 2009, 105, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Parry, T.L.; Brown, D.I.; Mota, R.I.; Huang, W.; Beak, J.Y.; Sola, M.; Zhou, C.; Hicks, S.T.; Caughey, M.C.; et al. Doxorubicin Exposure Causes Subacute Cardiac Atrophy Dependent on the Striated Muscle-Specific Ubiquitin Ligase MuRF1. Circ. Heart Fail. 2019, 12, e005234. [Google Scholar] [CrossRef]
- Verdecchia, P.; Angeli, F.; Borgioni, C.; Gattobigio, R.; de Simone, G.; Devereux, R.B.; Porcellati, C. Changes in cardiovascular risk by reduction of left ventricular mass in hypertension: A meta-analysis. Am. J. Hypertens. 2003, 16, 895–899. [Google Scholar] [CrossRef]
- Conraads, V.M.; Vrints, C.J.; Rodrigus, I.E.; Hoymans, V.Y.; van Craenenbroeck, E.M.; Bosmans, J.; Claeys, M.J.; van Herck, P.; Linke, A.; Schuler, G.; et al. Depressed expression of MuRF1 and MAFbx in areas remote of recent myocardial infarction: A mechanism contributing to myocardial remodeling? Basic Res. Cardiol. 2010, 105, 219–226. [Google Scholar] [CrossRef]
- Jokela, M.; Baumann, P.; Huovinen, S.; Penttilä, S.; Udd, B. Homozygous Nonsense Mutation p.Q274X in TRIM63 (MuRF1) in a Patient with Mild Skeletal Myopathy and Cardiac Hypertrophy. J. Neuromuscul. Dis. 2019, 6, 143–146. [Google Scholar] [CrossRef]
- Hirner, S.; Krohne, C.; Schuster, A.; Hoffmann, S.; Witt, S.; Erber, R.; Sticht, C.; Gasch, A.; Labeit, S.; Labeit, D. MuRF1-dependent regulation of systemic carbohydrate metabolism as revealed from transgenic mouse studies. J. Mol. Biol. 2008, 379, 666–677. [Google Scholar] [CrossRef]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef]
- Fielitz, J.; Kim, M.-S.; Shelton, J.M.; Latif, S.; Spencer, J.A.; Glass, D.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Investig. 2007, 117, 2486–2495. [Google Scholar] [CrossRef]
- Eble, D.M.; Spragia, M.L.; Ferguson, A.G.; Samarel, A.M. Sarcomeric myosin heavy chain is degraded by the proteasome. Cell Tissue Res. 1999, 296, 541–548. [Google Scholar] [CrossRef]
- Ikemoto, M.; Nikawa, T.; Takeda, S.; Watanabe, C.; Kitano, T.; Baldwin, K.M.; Izumi, R.; Nonaka, I.; Towatari, T.; Teshima, S.; et al. Space shuttle flight (STS-90) enhances degradation of rat myosin heavy chain in association with activation of ubiquitin-proteasome pathway. FASEB J. 2001, 15, 1279–1281. [Google Scholar] [CrossRef] [PubMed]
- Mearini, G.; Gedicke, C.; Schlossarek, S.; Witt, C.C.; Krämer, E.; Cao, P.; Gomes, M.D.; Lecker, S.H.; Labeit, S.; Willis, M.S.; et al. Atrogin-1 and MuRF1 regulate cardiac MyBP-C levels via different mechanisms. Cardiovasc. Res. 2010, 85, 357–366. [Google Scholar] [CrossRef]
- Haus, J.M.; Carrithers, J.A.; Carroll, C.C.; Tesch, P.A.; Trappe, T.A. Contractile and connective tissue protein content of human skeletal muscle: Effects of 35 and 90 days of simulated microgravity and exercise countermeasures, American Journal of Physiology-Regulatory. Integr. Comp. Physiol. 2007, 293, R1722–R1727. [Google Scholar] [CrossRef] [PubMed]
- Borina, E.; Pellegrino, M.A.; D’Antona, G.; Bottinelli, R. Myosin and actin content of human skeletal muscle fibers following 35 days bed rest. Scand. J. Med. Sci. Sports 2010, 20, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Cosper, P.F.; Leinwand, L.A. Myosin heavy chain is not selectively decreased in murine cancer cachexia. Int. J. Cancer 2012, 130, 2722–2727. [Google Scholar] [CrossRef]
- Polge, C.; Heng, A.-E.; Jarzaguet, M.; Ventadour, S.; Claustre, A.; Combaret, L.; Bechet, D.; Matondo, M.; Uttenweiler-Joseph, S.; Monsarrat, B.; et al. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J. 2011, 25, 3790–3802. [Google Scholar] [CrossRef]
- Ventadour, S.; Jarzaguet, M.; Wing, S.S.; Chambon, C.; Combaret, L.; Béchet, D.; Attaix, D.; Taillandier, D. A new method of purification of proteasome substrates reveals polyubiquitination of 20 S proteasome subunits. J. Biol. Chem. 2007, 282, 5302–5309. [Google Scholar] [CrossRef]
- Polge, C.; Cabantous, S.; Deval, C.; Claustre, A.; Hauvette, A.; Bouchenot, C.; Aniort, J.; Béchet, D.; Combaret, L.; Attaix, D.; et al. A muscle-specific MuRF1-E2 network requires stabilization of MuRF1-E2 complexes by telethonin, a newly identified substrate. J. Cachexia Sarcopenia Muscle 2018, 9, 129–145. [Google Scholar] [CrossRef]
- Vainzof, M.; Moreira, E.S.; Suzuki, O.T.; Faulkner, G.; Valle, G.; Beggs, A.H.; Carpen, O.; Ribeiro, A.F.; Zanoteli, E.; Gurgel-Gianneti, J.; et al. Telethonin protein expression in neuromuscular disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2002, 1588, 33–40. [Google Scholar] [CrossRef]
- Bos, J.M.; Poley, R.N.; Ny, M.; Tester, D.J.; Xu, X.; Vatta, M.; Towbin, J.A.; Gersh, B.J.; Ommen, S.R.; Ackerman, M.J. Genotype—phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol. Genet. Metab. 2006, 88, 78–85. [Google Scholar] [CrossRef] [PubMed]
- De la Hoz, C.P.de.; Hernández-Laín, A.; Olivé, M.; Fernández-Marmiesse, A.; Domínguez-González, C. Novel mutation in TCAP manifesting with asymmetric calves and early-onset joint retractions. Neuromuscul. Disord. 2016, 26, 749–753. [Google Scholar] [CrossRef]
- Knöll, R.; Linke, W.A.; Zou, P.; Miocic, S.; Kostin, S.; Buyandelger, B.; Ku, C.-H.; Neef, S.; Bug, M.; Schäfer, K.; et al. Telethonin deficiency is associated with maladaptation to biomechanical stress in the mammalian heart. Circ. Res. 2011, 109, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Knöll, R.; Hoshijima, M.; Hoffman, H.M.; Person, V.; Lorenzen-Schmidt, I.; Bang, M.-L.; Hayashi, T.; Shiga, N.; Yasukawa, H.; Schaper, W.; et al. The Cardiac Mechanical Stretch Sensor Machinery Involves a Z Disc Complex that Is Defective in a Subset of Human Dilated Cardiomyopathy. Cell 2002, 111, 943–955. [Google Scholar] [CrossRef]
- Nicholas, G.; Thomas, M.; Langley, B.; Somers, W.; Patel, K.; Kemp, C.F.; Sharma, M.; Kambadur, R. Titin-cap associates with, and regulates secretion of, Myostatin. J. Cell. Physiol. 2002, 193, 120–131. [Google Scholar] [CrossRef]
- Kojic, S.; Medeot, E.; Guccione, E.; Krmac, H.; Zara, I.; Martinelli, V.; Valle, G.; Faulkner, G. The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle. J. Mol. Biol. 2004, 339, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Olson, E.N. Calsarcin-3, a novel skeletal muscle-specific member of the calsarcin family, interacts with multiple Z-disc proteins. J. Biol. Chem. 2002, 277, 13998–14004. [Google Scholar] [CrossRef] [PubMed]
- Heng, A.E.; Ventadour, S.; Jarzaguet, M.; Pouch-Pélissier, M.N.; Guezennec, C.Y.; Bigard, X.; Attaix, D.; Taillandier, D. Coordinate expression of the 19S regulatory complex and evidence for ubiquitin-dependent telethonin degradation in the unloaded soleus muscle. Int. J. Biochem. Cell Biol. 2008, 40, 2544–2552. [Google Scholar] [CrossRef]
- Wallimann, T.; Schlösser, T.; Eppenberger, H.M. Function of M-line-bound creatine kinase as intramyofibrillar ATP regenerator at the receiving end of the phosphorylcreatine shuttle in muscle. J. Biol. Chem. 1984, 259, 5238–5246. [Google Scholar]
- Ventura-Clapier, R.; Veksler, V.; Hoerter, J.A. Myofibrillar creatine kinase and cardiac contraction. Mol. Cell. Biochem. 1994, 133, 125–144. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef]
- Lin, G.; Liu, Y.; MacLeod, K.M. Regulation of muscle creatine kinase by phosphorylation in normal and diabetic hearts. Cell. Mol. Life Sci. 2009, 66, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Dieni, C.A.; Storey, K.B. Creatine kinase regulation by reversible phosphorylation in frog muscle. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2009, 152, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.-J.; Yan, Y.-B.; Liu, Y.; Zhou, H.-M. The generation of the oxidized form of creatine kinase is a negative regulation on muscle creatine kinase. J. Biol. Chem. 2007, 282, 12022–12029. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Tsuruma, K.; Uehara, T.; Nomura, Y. GMEB1, a novel endogenous caspase inhibitor, prevents hypoxia- and oxidative stress-induced neuronal apoptosis. Neurosci. Lett. 2008, 438, 34–37. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Duran, M.; Loupatty, F.J. Enzymology of the branched-chain amino acid oxidation disorders: The valine pathway. J. Inherit. Metab. Dis. 2012, 35, 5–12. [Google Scholar] [CrossRef]
- Oka, T.; Dai, Y.-S.; Molkentin, J.D. Regulation of calcineurin through transcriptional induction of the calcineurin a beta promoter in vitro and in vivo. Mol. Cell. Biol. 2005, 25, 6649–6659. [Google Scholar] [CrossRef]
- Rothermel, B.A.; McKinsey, T.A.; Vega, R.B.; Nicol, R.L.; Mammen, P.; Yang, J.; Antos, C.L.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N.; et al. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 3328–3333. [Google Scholar] [CrossRef]
- Heineke, J.; Auger-Messier, M.; Correll, R.N.; Xu, J.; Benard, M.J.; Yuan, W.; Drexler, H.; Parise, L.V.; Molkentin, J.D. CIB1 is a regulator of pathological cardiac hypertrophy. Nat. Med. 2010, 16, 872–879. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Molkentin, J.D. Calcineurin–NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475. [Google Scholar] [CrossRef]
- Ferrandi, C.; Ballerio, R.; Gaillard, P.; Giachetti, C.; Carboni, S.; Vitte, P.-A.; Gotteland, J.-P.; Cirillo, R. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br. J. Pharmacol. 2004, 142, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Milano, G.; Morel, S.; Bonny, C.; Samaja, M.; von Segesser, L.K.; Nicod, P.; Vassalli, G. A peptide inhibitor of c-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivo. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1828–H1835. [Google Scholar] [CrossRef]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Rodríguez, J.E.; Hite, R.L.; Min, J.; Walton, B.L.; Willis, M.S. Muscle RING finger-1 attenuates IGF-I-dependent cardiomyocyte hypertrophy by inhibiting JNK signaling. Am. J. Physiol. Endocrinol. Metab. 2014, 306, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chow, A.; Powers, J.; Fajardo, G.; Bernstein, D. Microarray analysis of gene expression after transverse aortic constriction in mice. Physiol. Genom. 2004, 19, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Bogomolovas, J.; Strack, S.; Choi, K.-R.; Khan, M.M.; Wagner, A.; Brohm, K.; Hanashima, A.; Gasch, A.; Labeit, D.; et al. Regulation of nicotinic acetylcholine receptor turnover by MuRF1 connects muscle activity to endo/lysosomal and atrophy pathways. Age 2013, 35, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Kjaerulff, O.; Brodin, L.; Jung, A. The structure and function of endophilin proteins. Cell Biochem. Biophys. 2011, 60, 137–154. [Google Scholar] [CrossRef]
- Li, J.; Barylko, B.; Eichorst, J.P.; Mueller, J.D.; Albanesi, J.P.; Chen, Y. Association of Endophilin B1 with Cytoplasmic Vesicles. Biophys. J. 2016, 111, 565–576. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Khan, M.M.; Strack, S.; Wild, F.; Hanashima, A.; Gasch, A.; Brohm, K.; Reischl, M.; Carnio, S.; Labeit, D.; Sandri, M.; et al. Role of autophagy, SQSTM1, SH3GLB1, and TRIM63 in the turnover of nicotinic acetylcholine receptors. Autophagy 2014, 10, 123–136. [Google Scholar] [CrossRef]
- Li, G.; Ji, T.; Chen, J.; Fu, Y.; Hou, L.; Feng, Y.; Zhang, T.; Song, T.; Zhao, J.; Endo, Y.; et al. CRL4DCAF8 Ubiquitin Ligase Targets Histone H3K79 and Promotes H3K9 Methylation in the Liver. Cell Rep. 2017, 18, 1499–1511. [Google Scholar] [CrossRef]
- Nowak, M.; Suenkel, B.; Porras, P.; Migotti, R.; Schmidt, F.; Kny, M.; Zhu, X.; Wanker, E.E.; Dittmar, G.; Fielitz, J.; et al. DCAF8, a novel MuRF1 interaction partner, promotes muscle atrophy. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Ibanez, M.R.; Gatica, A.E.; Yang, S.; Wei, S.; Mei, S.; Falck, J.R.; Capdevila, J.H. Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. J. Biol. Chem. 2007, 282, 17685–17695. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.E.; Liao, J.-Y.; He, J.; Schisler, J.C.; Newgard, C.B.; Drujan, D.; Glass, D.J.; Frederick, C.B.; Yoder, B.C.; Lalush, D.S.; et al. The ubiquitin ligase MuRF1 regulates PPARα activity in the heart by enhancing nuclear export via monoubiquitination. Mol. Cell. Endocrinol. 2015, 413, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Duff, D.A. Long, Roles for RACK1 in cancer cell migration and invasion. Cell. Signal 2017, 35, 250–255. [Google Scholar] [CrossRef]
- Mochly-Rosen, D.; Wu, G.; Hahn, H.; Osinska, H.; Liron, T.; Lorenz, J.N.; Yatani, A.; Robbins, J.; Dorn, G.W. Cardiotrophic effects of protein kinase C epsilon: Analysis by in vivo modulation of PKCepsilon translocation. Circ. Res. 2000, 86, 1173–1179. [Google Scholar] [CrossRef]
- Mochly-Rosen, D. Localization of protein kinases by anchoring proteins: A theme in signal transduction. Science 1995, 268, 247–251. [Google Scholar] [CrossRef]
- Gregorio, C.C.; Granzier, H.; Sorimachi, H.; Labeit, S. Muscle assembly: A titanic achievement? Curr. Opin. Cell Biol. 1999, 11, 18–25. [Google Scholar] [CrossRef]
- Machado, C.; Andrew, D.J. D-Titin: A giant protein with dual roles in chromosomes and muscles. J. Cell Biol. 2000, 151, 639–652. [Google Scholar] [CrossRef]
- Obermann, W.M.; Gautel, M.; Weber, K.; Fürst, D.O. Molecular structure of the sarcomeric M band: Mapping of titin and myosin binding domains in myomesin and the identification of a potential regulatory phosphorylation site in myomesin. EMBO J. 1997, 16, 211–220. [Google Scholar] [CrossRef]
- Higashikuse, Y.; Mittal, N.; Arimura, T.; Yoon, S.H.; Oda, M.; Enomoto, H.; Kaneda, R.; Hattori, F.; Suzuki, T.; Kawakami, A.; et al. Perturbation of the titin/MURF1 signaling complex is associated with hypertrophic cardiomyopathy in a fish model and in human patients. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Brückner, A.; Polge, C.; Lentze, N.; Auerbach, D.; Schlattner, U. Yeast Two-Hybrid, a Powerful Tool for Systems Biology. Int. J. Mol. Sci. 2009, 10, 2763–2788. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Cécile, P.; Julien, A.; Andrea, A.; Agnès, C.; Cécile, C.-G.; Clara, T.; Christiane, D.; Lydie, C.; Daniel, B.; Marco, S.; et al. UBE2E1 Is Preferentially Expressed in the Cytoplasm of Slow-Twitch Fibers and Protects Skeletal Muscles from Exacerbated Atrophy upon Dexamethasone Treatment. Cells 2018, 7, 214. [Google Scholar] [CrossRef]
- Marblestone, J.G.; Butt, S.; McKelvey, D.M.; Sterner, D.E.; Mattern, M.R.; Nicholson, B.; Eddins, M.J. Comprehensive ubiquitin E2 profiling of ten ubiquitin E3 ligases. Cell Biochem. Biophys. 2013, 67, 161–167. [Google Scholar] [CrossRef]
- Van Wijk, S.J.L.; de Vries, S.J.; Kemmeren, P.; Huang, A.; Boelens, R.; Bonvin, A.M.J.J.; Timmers, H.T.M. A comprehensive framework of E2-RING E3 interactions of the human ubiquitin-proteasome system. Mol. Syst. Biol. 2009, 5, 295. [Google Scholar] [CrossRef]
- Markson, G.; Kiel, C.; Hyde, R.; Brown, S.; Charalabous, P.; Bremm, A.; Semple, J.; Woodsmith, J.; Duley, S.; Salehi-Ashtiani, K.; et al. Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res. 2009, 19, 1905–1911. [Google Scholar] [CrossRef]
- Napolitano, L.M.; Jaffray, E.G.; Hay, R.T.; Meroni, G. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem. J. 2011, 434, 309–319. [Google Scholar] [CrossRef]
- Banerjee, R.; He, J.; Spaniel, C.; Quintana, M.T.; Wang, Z.; Bain, J.; Newgard, C.B.; Muehlbauer, M.J.; Willis, M.S. Non-targeted metabolomics analysis of cardiac Muscle Ring Finger-1 (MuRF1), MuRF2, and MuRF3 in vivo reveals novel and redundant metabolic changes. Metabolomics 2015, 11, 312–322. [Google Scholar] [CrossRef]
- Attaix, D.; Ventadour, S.; Codran, A.; Béchet, D.; Taillandier, D.; Combaret, L. The ubiquitin-proteasome system and skeletal muscle wasting. Essays Biochem. 2005, 41, 173–186. [Google Scholar] [CrossRef]
- Jagoe, R.T.; Goldberg, A.L. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Eddins, M.J.; Marblestone, J.G.; Kumar, K.G.S.; Leach, C.A.; Sterner, D.E.; Mattern, M.R.; Nicholson, B. Targeting the ubiquitin E3 ligase MuRF1 to inhibit muscle atrophy. Cell Biochem. Biophys. 2011, 60, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Adams, V.; Bowen, T.S.; Werner, S.; Barthel, P.; Amberger, C.; Konzer, A.; Graumann, J.; Sehr, P.; Lewis, J.; Provaznik, J.; et al. Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J. Cachexia Sarcopenia Muscle 2019, 10, 1102–1115. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peris-Moreno, D.; Taillandier, D.; Polge, C. MuRF1/TRIM63, Master Regulator of Muscle Mass. Int. J. Mol. Sci. 2020, 21, 6663. https://doi.org/10.3390/ijms21186663
Peris-Moreno D, Taillandier D, Polge C. MuRF1/TRIM63, Master Regulator of Muscle Mass. International Journal of Molecular Sciences. 2020; 21(18):6663. https://doi.org/10.3390/ijms21186663
Chicago/Turabian StylePeris-Moreno, Dulce, Daniel Taillandier, and Cécile Polge. 2020. "MuRF1/TRIM63, Master Regulator of Muscle Mass" International Journal of Molecular Sciences 21, no. 18: 6663. https://doi.org/10.3390/ijms21186663
APA StylePeris-Moreno, D., Taillandier, D., & Polge, C. (2020). MuRF1/TRIM63, Master Regulator of Muscle Mass. International Journal of Molecular Sciences, 21(18), 6663. https://doi.org/10.3390/ijms21186663