Interaction between Cognitive Reserve and Biomarkers in Alzheimer Disease


Abstract
1. Introduction
2. Classification and the Use of New Biomarkers in Alzheimer Disease
- (1)
- Individuals with normal AD biomarkers.
- (2)
- Those in the continuum of AD (divided into “Alzheimer pathological change” and “AD”).
- (3)
- Those with a normal amyloid biomarker but with abnormal T, N, or both. This latter biomarker profile implies evidence of one or more neuropathological processes other than AD and has been labeled as “suspected non-Alzheimer pathophysiology”(SNAP) [21].
3. The Theory of Cognitive Reserve
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer disease |
| 18FDG-PET | 18F-fluorodesoxyglucose PET |
| MP4A-PET | Methylpiperidin-4-yl acetate PET |
| HE-AD | High education-Alzheimer disease |
| LE-AD | Low education-Alzheimer disease |
| CR | Cognitive reserve |
| CSFAβ1-42 | Cerebrospinal fluidΒ-Amyloid isoform made of 42 aminoacids |
References
- Dubois, B.; Feldman, H.H.; Jacova, C.; Cummings, J.L.; DeKosky, S.T.; Barberger-Gateau, P.; Gauthier, S. Revising the definition of Alzheimer’s disease: A new lexicon. Lancet Neurol. 2010, 9, 1118–1127. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol. Scand. 1996, 94, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Herholz, K. PET studies in dementia. Ann. Nucl. Med. 2003, 17, 79–89. [Google Scholar] [CrossRef]
- Ewers, M.; Inset, P.S.; Stern, Y.; Weiner, M.W. Alzheimer’s Disease Neuroimaging Initiative (ADNI). Cognitive reserve associated with FDG-PET in preclinical Alzheimer disease. Neurology 2013, 80, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Edison, P.; Archer, H.A.; Hinz, R.; Hammers, A.; Pavese, N.; Tai, Y.F.; Hotton, G.; Cutler, D.; Fox, N.C.; Kennedy, A.; et al. Amyloid, hypometabolism, and cognition in Alzheimer disease An [11C] P1B and [18F] FDG PET study. Neurology 2007, 68, 501–508. [Google Scholar] [CrossRef]
- Herholz, K.; Salmon, E.; Perani, D.; Baron, J.C.; Holthoff, V.; Frolich, L.; Schönknecht, P.; Ito, K.; Mielke, R.; Kalbe, E.; et al. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage 2002, 17, 302–316. [Google Scholar] [CrossRef]
- Tripathi, M.; Tripathi, M.; Damle, N.; Kushwaha, S.; Jaimini, A.; D’Souza, M.M.; Sharma, R.; Saw, S.; Mondal, A. Differential diagnosis of neurodegenerative dementias using metabolic phenotypes on F-18 FDG PET/CT. Neuroradiol. J. 2014, 27, 13–21. [Google Scholar] [CrossRef]
- Schoonenboom, N.S.M.; Pijnenburg, Y.A.L.; Mulder, C.; Rosso, S.M.; Van Elk, E.J.; Van Kamp, G.J.; Scheltens, P. Amyloid p (1–42) and phosphorylated tau in CSF as markers for early-onset Alzheimer disease. Neurology 2004, 62, 1580–1584. [Google Scholar] [CrossRef]
- Hansson, O.; Zetterberg, H.; Buchhave, P.; Londos, E.; Blennow, K.; Minthon, L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: A follow-up study. Lancet Neurol. 2006, 5, 228–234. [Google Scholar] [CrossRef]
- Clifford, R.J., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer Dement. 2018, 14, 535–562. [Google Scholar]
- Nelson, P.T.; Head, E.; Schmittm, F.A.; Davis, P.R.; Neltner, J.H.; Jicha, G.A.; Abner, E.L.; Smith, C.D.; Van Eldik, L.J.; Kryscio, R.J.; et al. Alzheimer’s disease is not “brain aging”: Neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011, 121, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Schmitt, F.A.; Lin, Y.; Abner, E.L.; Jicha, G.A.; Patel, E.; Sonnen, J.A. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain 2011, 134, 1506–1518. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, K.M.; Kennedy, K.M.; Devous, M.D., Sr.; Rieck, J.R.; Hebrank, A.C.; Diaz-Arrastia, R.; Park, D.C. beta-Amyloid burden in healthy aging: Regional distribution and cognitive consequences. Neurology 2012, 78, 387–395. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Qian, J.; Monsell, S.E.; Blacker, D.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Johnson, K.A.; Frosch, M.P.; Sperling, R.A.; et al. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann Neurol. 2014, 75, 597–601. [Google Scholar] [CrossRef]
- Fagan, A.M.; Roe, C.M.; Xiong, C.; Mintun, M.A.; Morris, J.C.; Holtzman, D.M. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007, 64, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Visser, P.J.; Verhey, F.; Knol, D.L.; Scheltens, P.; Wahlund, L.O.; Freund-Levi, Y.; Bürger, K. Prevalence and prognostic value of CSF markers of Alzheimer’s disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: A prospective cohort study. Lancet Neurol. 2009, 8, 619–627. [Google Scholar] [CrossRef]
- Buerger, K.; Ewers, M.; Pirttila, T.; Zinkowski, R.; Alafuzoff, I.; Teipel, S.J.; Hampel, H. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain 2006, 129 Pt 11, 3035–3041. [Google Scholar] [CrossRef]
- Clifford, R.J., Jr. PART and SNAP. Acta Neuropathol. 2014, 128, 773–776. [Google Scholar]
- Johnson, K.A.; Minoshima, S.; Bohnen, N.I.; Donohoe, K.J.; Foster, N.L.; Herscovitch, P.; Karlawish, J.H.; Rowe, C.C.; Hedrick, S.; Pappas, V.; et al. Update on appropriate use criteria for amyloid PET imaging: Dementia experts, mild cognitive impairment, and education. J. Nucl. Med. 2013, 54, 1011–1013. [Google Scholar] [CrossRef]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Blennow, K.; Vanmechelen, E.; Hampel, H. CSF total tau, AP42 and phosphorylated tauprotein as biomarkers for Alzheimer’s disease. Mol. Neurobiol. 2001, 24, 87–97. [Google Scholar] [CrossRef]
- Otto, M.; Wiltfang, J.; Tumani, H.; Zerr, I.; Lantsch, M.; Kornhuber, J.; Poser, S. Elevated levels of tau-protcin in cerebrospinal fluid of patients with Creutzfeldt—Jakob disease. Neurosci. Lett. 1997, 225, 210–212. [Google Scholar] [CrossRef]
- van Harter, A.C.; Kester, M.I.; Visser, P.J.; Blankenstein, M.A.; Pijnenburg, Y.A.; van der Flier, W.M.; Scheltens, P. Tau and P-tau as CSF biomarkers in dementia: A meta-analysis. Clin. Chem. Lab. Med. 2011, 49, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Nisticò, R. The β-secretase BACE1 in Alzheimer’s disease. Biol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef]
- Raz, N.; Lindenberger, U.; Rodrigue, K.M.; Kennedy, K.M.; Head, D.; Williamson, A.; Acker, J.D. Regional brain changes in aging healthy adults: General trends, individual differences and modifiers. Cereb. Cortex 2015, 15, 1676–1689. [Google Scholar] [CrossRef]
- Cabeza, R. Hemispheric asymmetry reduction in old adults: The HAROLD model. Psychol. Aging 2002, 17, 85–100. [Google Scholar] [CrossRef]
- Grady, C.L.; Springer, M.V.; Hongwanishkul, D.; McIntosh, A.R.; Winocur, G. Age-related changes in brain activity across the adult lifespan. J. Cogn. Neurosci. 2006, 18, 227–241. [Google Scholar] [CrossRef]
- Bäckman, L.; Lindenberger, U.; Li, S.-C.; Nyberg, L. Linking cognitive aging to alterations in dopamine neurotransmitter functioning: Recent data and future avenues. Neurosci. Biobehav. Rev. 2010, 34, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Cabeza, R.; Anderson, N.D.; Locantore, J.K.; McIntosh, A.R. Aging gracefully: Compensatory brain activity in high-performing older adults. Neuroimage 2002, 17, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Grady, C.; McIntosh, A.R.; Horwitz, B.; Maisog, J.; Ungerleider, L.; Mentis, M.; Pietrini, P.; Schapiro, M.; Haxby, J. Age-related reductions in human recognition memory due to impaired encoding. Science 1995, 269, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Fratiglioni, Grut, Forsell, Viitanen, Grafström, Holmén, Ericsson, Bäckman, Ahlbom e Winblad, Prevalence of Alzheimer’s disease and other dementias in an elderly urban population: Relationship with age, sex, and education. Neurology 1991, 41, 1886–1892. [CrossRef] [PubMed]
- Katzman, R.; Terry, R.; DeTeresa, R.; Brown, T.; Davies, P.; Fuld, P.; Peck, A. Clinical, pathological, and neurochemical changes in dementia: A subgroup with preserved mental status and numerous neocortical plaques. Ann. Neurol. 1988, 23, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Van Rossum, C.T.; Van Harskamp, F.; Van De Mheen, H.; Hofman, A.; Breteler, M.M.B. Education and the incidence of dementia in a large population-based study: The Rotterdam Study. Neurology 1999, 52, 663–666. [Google Scholar] [CrossRef]
- Schmand, B.; Smit, J.H.; Geerlings, M.I.; Lindeboom, J. The effects of intelligence and education on the development of dementia. A test of the brain reserve hypothesis. Psychol. Med. 1997, 27, 1337–1344. [Google Scholar] [CrossRef]
- Kesler, S.R.; Adams, H.F.; Blasey, C.M.; Bigler, E.D. Premorbid intellectual functioning, education, and brain size in traumatic brain injury: An investigation of the cognitive reserve hypothesis. Appl. Neuropsychol. 2003, 10, 153–162. [Google Scholar] [CrossRef]
- Evert, J.; Lawler, E.; Bogan, H.; Perls, T.T. Morbidity profiles of centenarians: Survivors, delayers, and escapers. Psychol. Sci. 2003, 58A, 232–237. [Google Scholar] [CrossRef]
- Perls, T.T. The different paths to 100. Am. J. Clin. Nutr. 2006, 83, 484S–487S. [Google Scholar] [CrossRef]
- Satz, P. Brain reserve capacity on symptom onset after brain injury: A formulation and review of evidence for threshold theory. Neuropsychology 1993, 7, 273–295. [Google Scholar] [CrossRef]
- Stern, Y. What is cognitive reserve? Theory and research application of the reserve concept. J. Int. Neuropsychol. Soc. 2002, 8, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Stern, Y. Cognitive reserve. Neuropsychologia 2009, 47, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Crystal, H.; Dickson, D.; Fuld, P.; Masur, D.; Scott, R.; Mehler, M.; Wolfson, L. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology 1988, 38, 1682. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.L.; Wolf, P.A.; Au, R.; White, R.; D’agostino, R.B. The effect of education on the incidence of dementia and Alzheimer’s disease in the Framingham Study. Neurology 1995, 45, 1707–1712. [Google Scholar] [CrossRef]
- Stern, Y.; Alexander, G.E.; Prohovnik, I.; Mayeux, R. Inverse relationship between education and parietotemporal perfusion deficit in Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1992, 32, 371–375. [Google Scholar] [CrossRef]
- Mortimer, J.A.; Borenstein, A.R.; Gosche, K.M.; Snowdon, D.A. Very early detection of Alzheimer neuropathology and the role of brain reserve in modifying its clinical expression. J. Geriatr. Psychiatry Neurol. 2005, 18, 218–223. [Google Scholar] [CrossRef]
- Mortimer, J.A.; Snowdon, D.A.; Markesbery, W.R. Head circumference. education and risk of dementia: Findings from the Nun Study. J. Clin. Exp. Neuropsychol. 2003, 25, 671–679. [Google Scholar] [CrossRef]
- Davis, D.G.; Schmitt, F.A.; Wekstein, D.R.; Markesbery, W.R. Alzheimer neuropathologic alterations in aged cognitively normal subjects. J. Neuropathol. Exp. Neurol. 1999, 58, 376–388. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Koberskaya, N.N.; Tabeeva, G.R. The modern concept of cognitive reserve. Neurol. Neuropsychiatry Psychosom. 2019, 11, 96–102. [Google Scholar] [CrossRef][Green Version]
- Stern, Y. Cognitive reserve and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2006, 20, S69–S74. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Parisi, J.E.; Salviati, A.; Floriach-Robert, M.; Boeve, B.F.; Ivnik, R.J.; McDonald, W.C. Neuropathology of cognitively normal elderly. J. Neuropathol. Exp. Neurol. 2003, 62, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.A.; Mann, D.M.A.; Sumpter, P.Q.; Yates, P.O. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J. Neurol. Sci. 1987, 78(2), 151–164. [Google Scholar] [CrossRef]
- Herholz, S.C.; Herholz, R.S.; Herholz, K. Non-pharmacological interventions and neuroplasticity in early stage Alzheimer’s disease. Expert Rev. Neurother. 2013, 13, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.K.; Tiwari, S.C. Cognitive functioning of community dwelling urban older adults with reference to socio-demographic variables. Indian J. Clin. Psychol. 2013, 40, 92–102. [Google Scholar]
- Fotenos, A.F.; Mintun, M.A.; Snyder, A.Z.; Morris, J.C.; Buckner, R.L. Brain volume decline in aging: Evidence for a relation between socioeconomic status, preclinical Alzheimer disease, and reserve. Arch. Neurol. 2008, 65, 113–120. [Google Scholar] [CrossRef]
- Bigio, E.H.; Hynan, L.S.; Sontag, E.; Satumtira, S.; White, C.L. Synapse loss is greater in presenile than senile onset Alzheimer disease: Implications for the cognitive reserve hypothesis. Neuropathol. Appl. Neurobiol. 2002, 28, 218–227. [Google Scholar] [CrossRef]
- Bruandet, A.; Richard, F.; Bombois, S.; Maurage, C.A.; Masse, I.; Amouyel, P.; Pasquier, F. Cognitive decline and survival in Alzheimer’s disease according to education l Level. Dement. Geriatr. Cogn. Disord. 2008, 25, 74–80. [Google Scholar] [CrossRef]
- Meng, X.; D’arcy, C. Education and dementia in the context of the cognitive reserve hypothesis: A systematic review with meta-analyses and qualitative analyses. PloS ONE 2012, 7, e38268. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yu, J.T.; Tan, M.S.; Tan, L. Cognitive reserve and Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 187–208. [Google Scholar] [CrossRef]
- Pascual-Leone, A.; Freitas, C.; Oberman, L.; Horvath, J.C.; Halko, M.; Eldaief, M.; Vahabzadeh-Hagh, A.M. Characterizing brain cortical plasticity and network dynamics across the age-span in health and disease with TMS-EEG and TMS-fMRI. Brain Topogr. 2011, 24, 302. [Google Scholar] [CrossRef] [PubMed]
- Altman, J. Are new neurons formed in the brains of adult mammals? Science 1962, 135, 1127–1128. [Google Scholar] [CrossRef] [PubMed]
- Lazarov, O.; Marr, R. Of mice and men: Neurogenesis. cognition and Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 43. [Google Scholar] [CrossRef]
- Lyness, S.A.; Zarow, C.; Chui, H.C. Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: A meta-analysis. Neurobiol. Aging 2003, 24, 1–23. [Google Scholar] [CrossRef]
- Insua, D.; Suarez, M.L.; Santamarina, G.; Sarasa, M.; Pesini, P. Dogs with canine counterpart of Alzheimer’s disease lose noradrenergic neurons. Neurobiol. Aging 2010, 31, 625–635. [Google Scholar] [CrossRef]
- Soldan, A.; Pettigrew, C.; Albert, M. Cognitive Reserve from the Perspective of Preclinical Alzheimer Disease: 2020 Update. Clin. Geriatr. Med. 2020, 36, 247–263. [Google Scholar] [CrossRef]
- Soldan, A.; Pettigrew, C.; Albert, M. Evaluating cognitive reserve through the prism of preclinical Alzheimer disease. Psychiatr. Clin. 2018, 41, 65–77. [Google Scholar] [CrossRef]
- Arenaza-Urquijo, E.M.; Vemuri, P. Resistance vs resilience to Alzheimer disease: Clarifying terminology for preclinical studies. Neurology 2018, 90, 695–703. [Google Scholar] [CrossRef]
- Cabeza, R.; Albert, M.; Belleville, S.; Craik, F.; Duarte, A.; Grady, C.; Rugg, M.D. Cognitive neuroscience of healthy aging: Maintenance, reserve, and compensation. Nat. Rev. Neurosci. 2018, 19, 701. [Google Scholar] [CrossRef]
- Carapelle, E.; Serra, L.; Modoni, S.; Falcone, M.; Caltagirone, C.; Bozzali, M.; Avolio, C. How the cognitive reserve interacts with β-amyloid deposition in mitigating FDG metabolism: An observational study. Medicine 2017, 96, e5876. [Google Scholar] [CrossRef] [PubMed]
- Petrie, E.C.; Cross, D.J.; Galasko, D.; Schellenberg, G.D.; Raskind, M.A.; Peskind, E.R.; Minoshima, S. Preclinical evidence of Alzheimer changes: Convergent cerebrospinal fluid biomarker and fluorodeoxyglucose positron emission tomography findings. Arch. Neurol. 2009, 66, 632–637. [Google Scholar] [CrossRef]
- Dowling, N.M.; Johnson, S.C.; Gleason, C.E.; Jagust, W.J.; Initiative, F.T.A.D.N. Alzheimer’s Disease Neuroimaging Initiative. The mediational effects of FDG hypometabolism on the association between cerebrospinal fluid biomarkers and neurocognitive function. NeuroImage 2015, 105, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Cappa, S.F. Cognitive reserve and successful ageing. J. Gerontol. Geriatr. 2017, 65, 296–298. [Google Scholar]
- Ingber, A.P.; Hassenstab, J.; Fagan, A.M.; Benzinger, T.L.; Grant, E.A.; Holtzman, D.M.; Morris, J.C.; Roe, C.M. Cerebrospinal Fluid Biomarkers and Reserve Variables as predictors of Future “Non Cognitive” Outcomes of Alzheimer’s Disease. J. Alzheimers Dis. 2016, 52, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Brayne, C.; Ince, P.G.; Keage, H.A.; McKeith, L.G.; Matthews, F.E.; Polvikoski, T.; Sulkava, R. Education, the brain and dementia: Neuroprotection or compensation? Brain 2010, 133, 2210–2216. [Google Scholar] [CrossRef]
- Serra, L.; Cercignani, M.; Petrosini, L.; Basile, B.; Perri, R.; Fadda, L.; Spano, B.; Marra, C.; Giubilei, F.; Carlesimo, G.A.; et al. Neuroanatomical correlates of cognitive reserve in Alzheimer disease. Rejuvenation Res. 2011, 14, 143–151. [Google Scholar] [CrossRef]


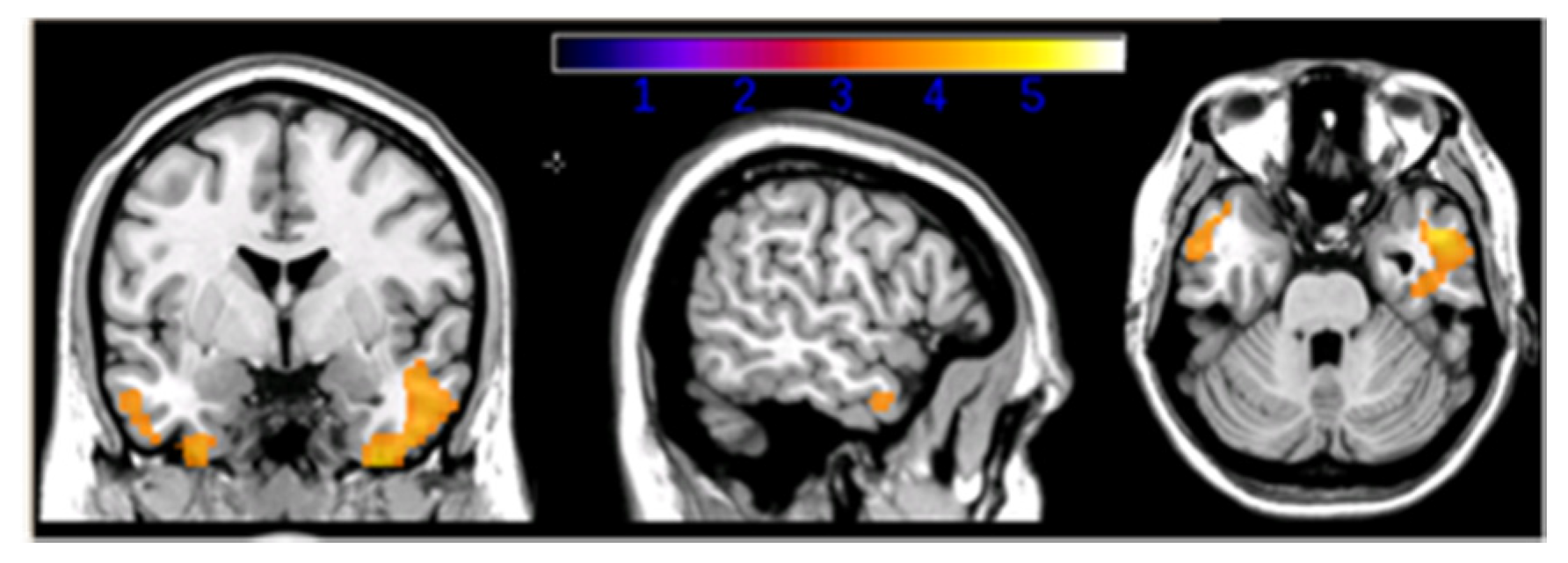{kind=link}
| Biomarkers | Changes in AD | |
|---|---|---|
| Aβ1-42 | Marked reduction in AD | Reduction CSF Aβ1-42 is gold standard for AD; low CSF Aβ1-42 is found in Lewy bodies dementia |
| 181p-tau | Marked increase in AD | High CSF 181P-tau is not specific for AD |
| T-tau | Marked increase in AD | High CSF T-tau is found in stroke, trauma and encephalities; very high CSF T-tau is found in Creutzfeld-Jakob |
| HE-AD (n = 12) | LE-AD (n = 15) | p Value | |
|---|---|---|---|
| Mean (SD) age (years) | 68.6 (7.2) | 73.9 (7.6) | n.s. |
| Sex (female|male) | 8|4 | 10|5 | n.s. |
| Mean (SD) MMSE score | 17.5 (6.2) | 17.9 (4.7) | n.s. |
| Mean (SD) ADL score | 4.3 (0.9) | 3.8 (1.3) | n.s. |
| Mean (SD) IADL score | 3.4 (1.8) | 3.8 (1.9) | n.s. |
| Mean (SD) GDS score | 5.25 (3.4) | 7.5 (4.5) | n.s. |
| Mean (SD) years of formal education | 10.4 (2.5) | 4.7 (0.7) | p < 0.001 |
| Mean (SD) of Aβ1-42 values (pg/ml) | 465.8 (140) | 534.8 (183.6) | n.s. |
| Mean (SD) of 181P-tau values (pg/ml) | 93.3 (65.6) | 62.7 (39.8) | n.s. |
| Mean (SD) of T-tau values (ng/ml) | 694.9 (570) | 616.3 (461.7) | n.s. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carapelle, E.; Mundi, C.; Cassano, T.; Avolio, C. Interaction between Cognitive Reserve and Biomarkers in Alzheimer Disease. Int. J. Mol. Sci. 2020, 21, 6279. https://doi.org/10.3390/ijms21176279
Carapelle E, Mundi C, Cassano T, Avolio C. Interaction between Cognitive Reserve and Biomarkers in Alzheimer Disease. International Journal of Molecular Sciences. 2020; 21(17):6279. https://doi.org/10.3390/ijms21176279
Chicago/Turabian StyleCarapelle, Elena, Ciro Mundi, Tommaso Cassano, and Carlo Avolio. 2020. "Interaction between Cognitive Reserve and Biomarkers in Alzheimer Disease" International Journal of Molecular Sciences 21, no. 17: 6279. https://doi.org/10.3390/ijms21176279
APA StyleCarapelle, E., Mundi, C., Cassano, T., & Avolio, C. (2020). Interaction between Cognitive Reserve and Biomarkers in Alzheimer Disease. International Journal of Molecular Sciences, 21(17), 6279. https://doi.org/10.3390/ijms21176279