Pathophysiology of Type 2 Diabetes Mellitus

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Type 2 Diabetes Mellitus: Background and Epidemiology
3. Risk Factors and Pathophysiology
3.1. Ethnicity and Family History/Genetic Predisposition
3.2. Obesity, Low Physical Activity and Unhealthy Diet
3.3. Pathophysiology
4. Mechanisms Leading to T2DM and Pathophysiology
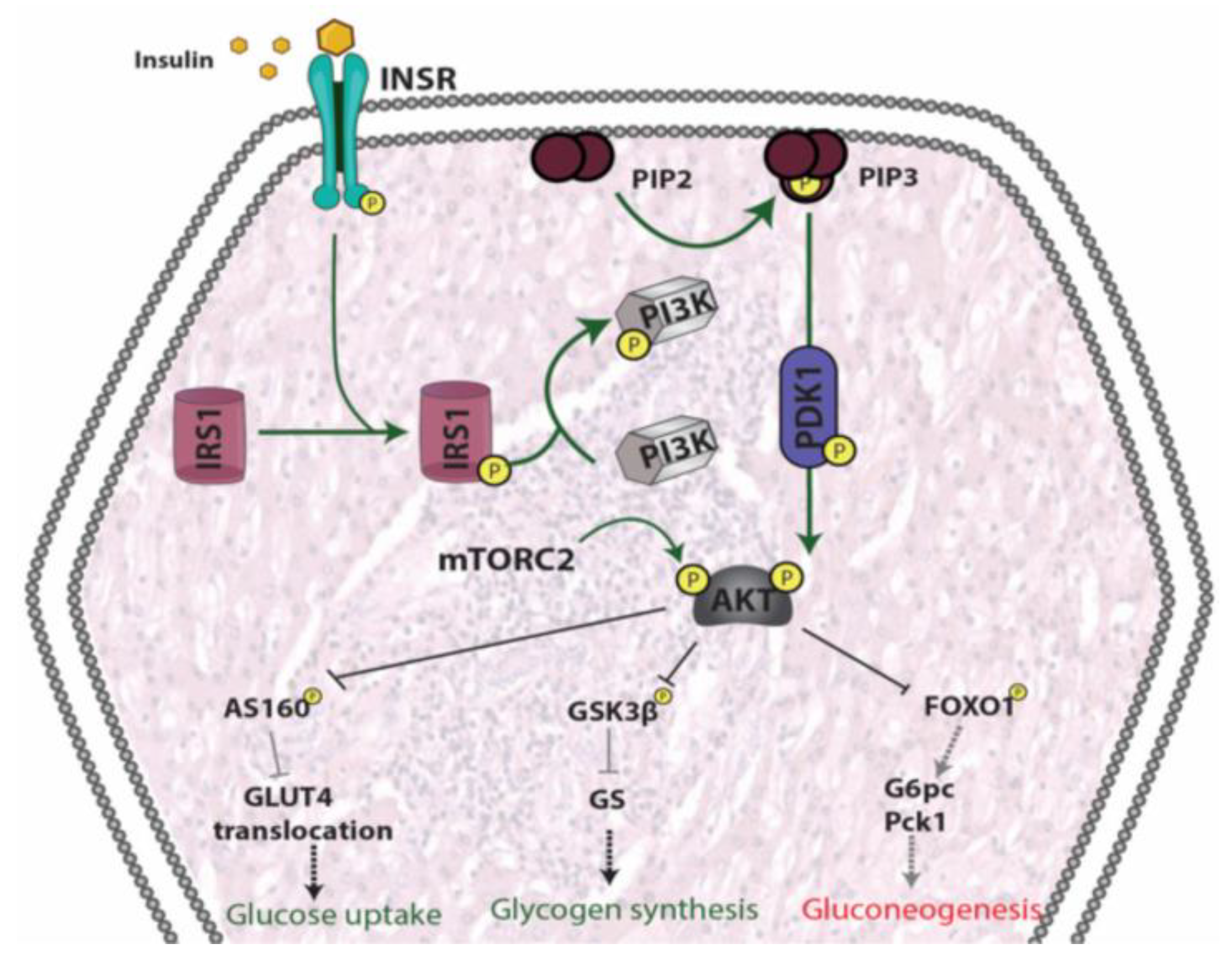
4.1. Insulin Secretion: Physiological and Dysfunctional Mechanisms Leading to T2DM
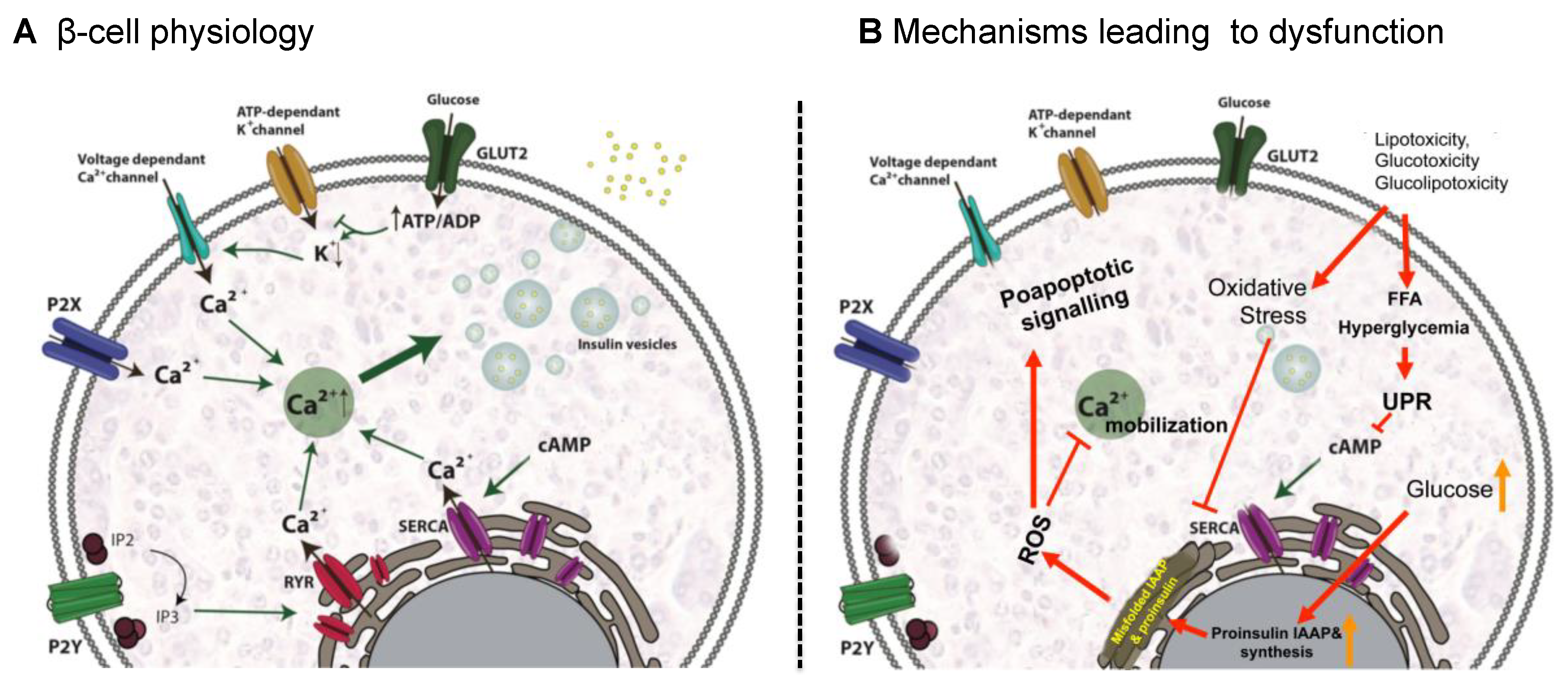
4.1.1. β-Cell Physiology
4.1.2. Mechanisms Leading to β-Cell Dysfunction
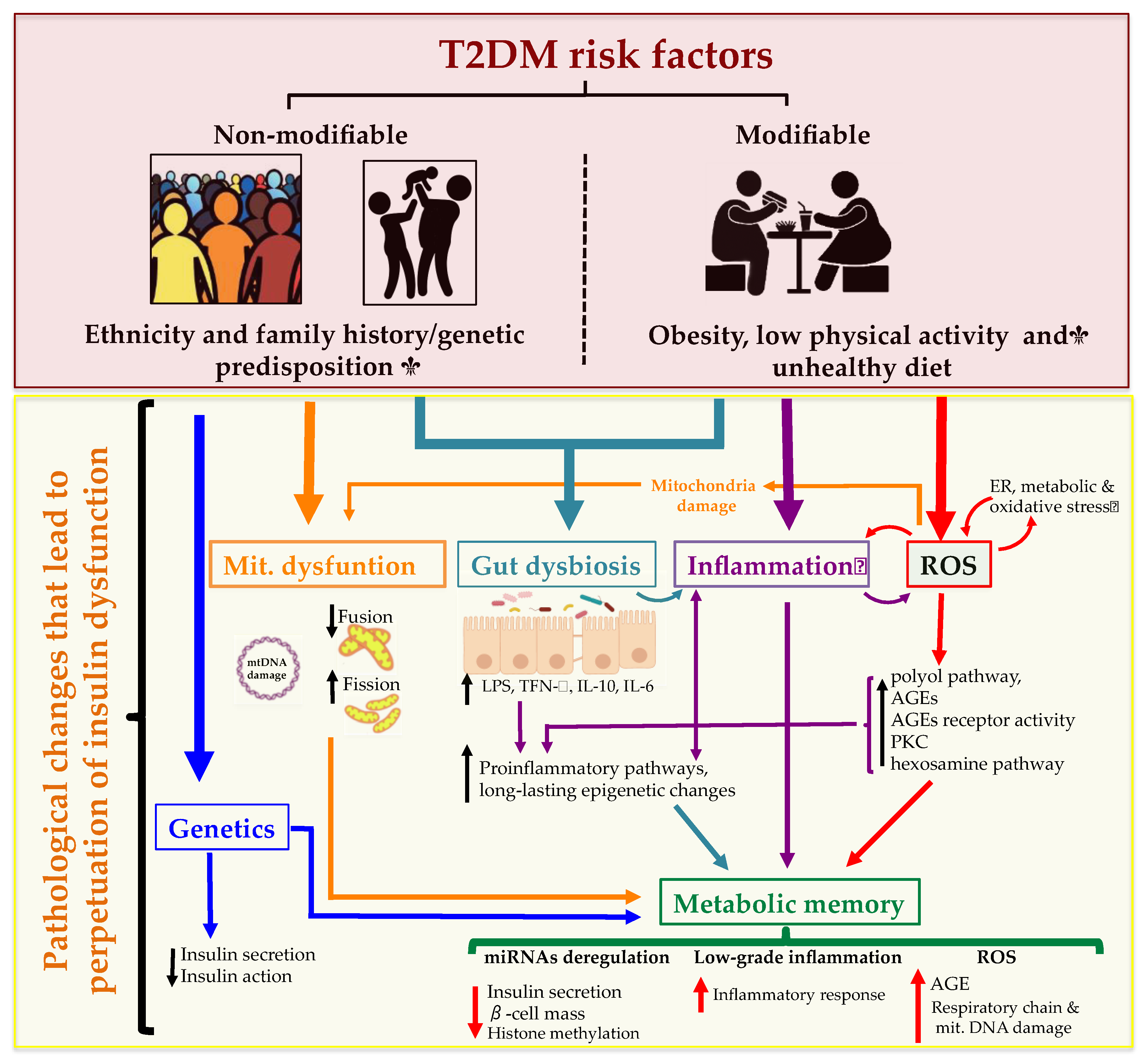
4.1.3. Pathological Conditions Perpetuating T2DM
Nutritional Factors
Physical Activity
Gut Dysbiosis
Metabolic Memory
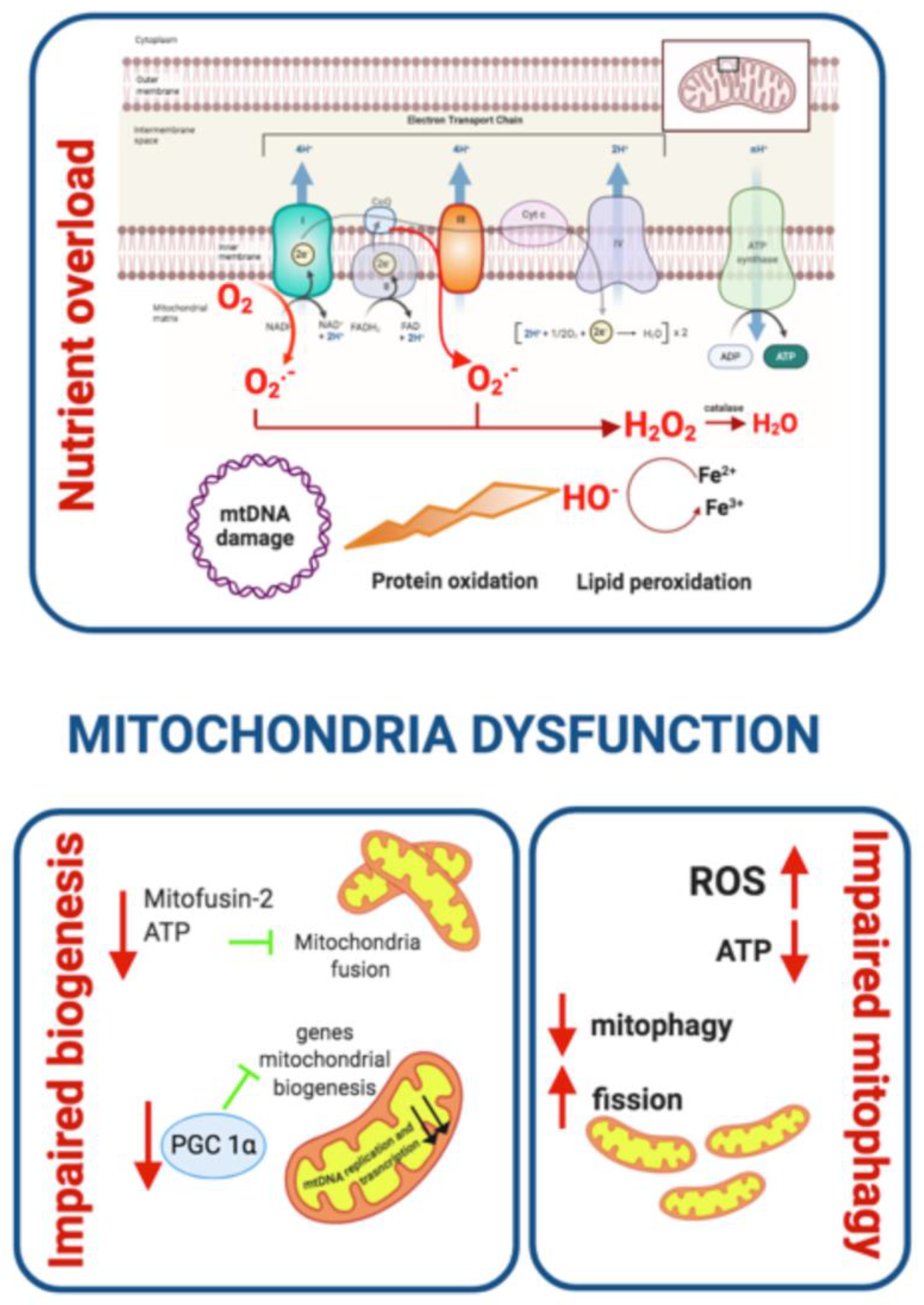
Mitochondrial Dysfunction
5. Insulin Resistance
5.1. Skeletal Muscle
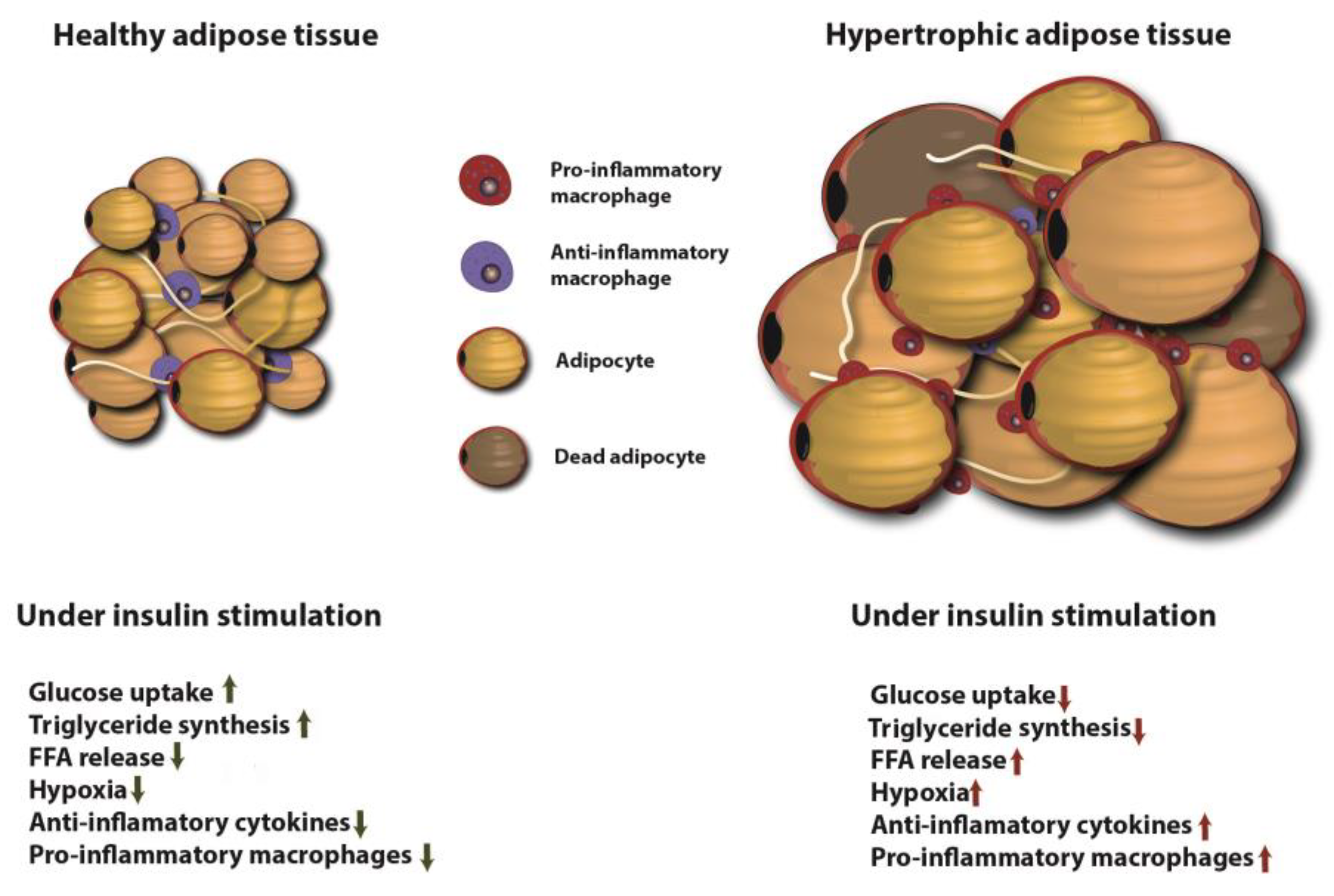
5.2. Adipose Tissue
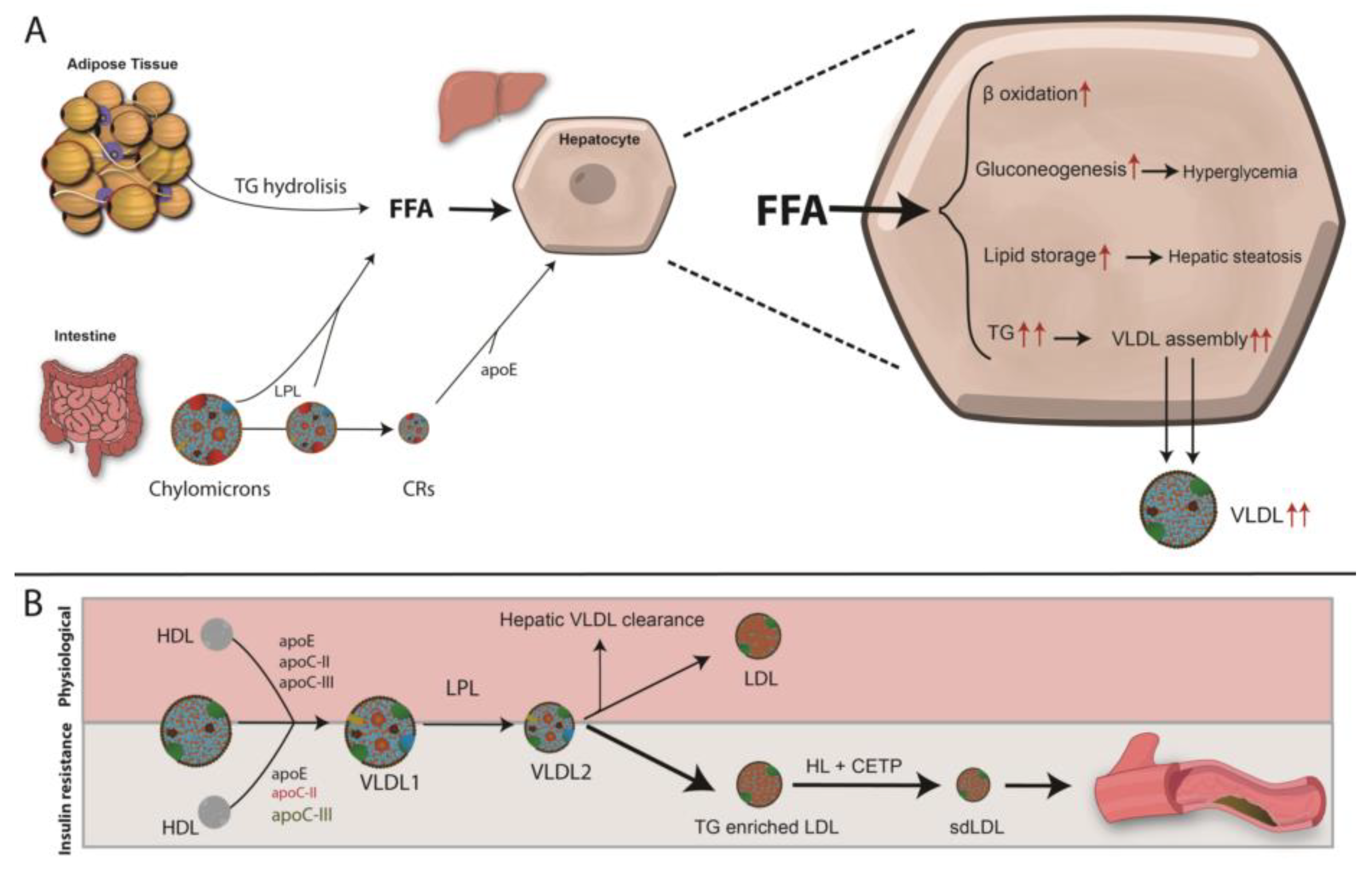
5.3. Liver
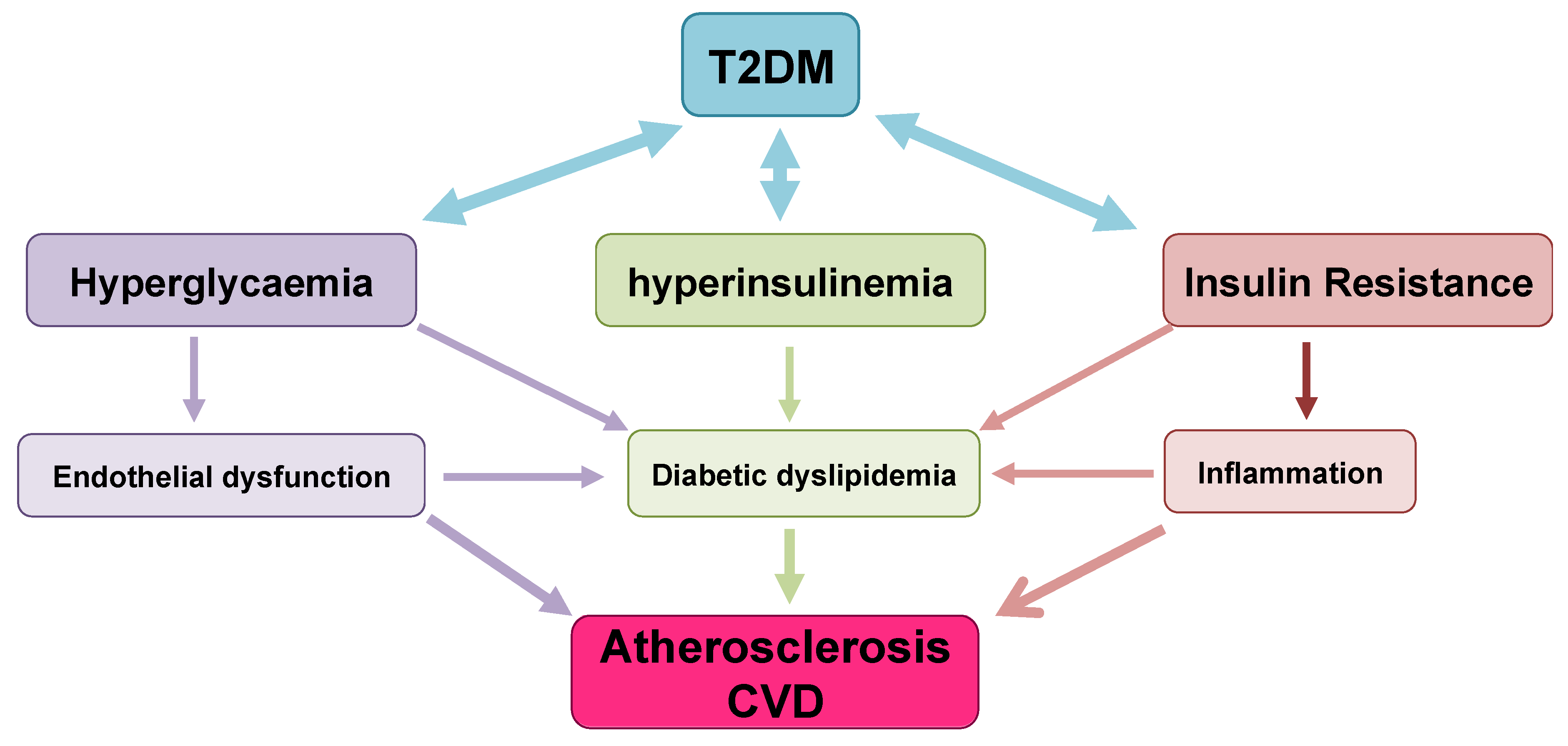
6. T2DM Outcomes/Complications: Cardiovascular Risk
6.1. Diabetic Dyslipidaemia and Atherosclerosis Development
Mechanisms Leading to T2DM Dyslipidemia and Atherosclerosis
6.2. Impaired Endothelial Function and Atherosclerosis Development
6.2.1. Mechanisms Leading to Endothelial Dysfunction in T2DM
6.2.2. Endothelial Dysfunction in T2DM and Atherosclerosis Development
6.3. Diabetes-Associated Chronic Inflammation and Atherosclerosis Progression
6.4. Adipokine Balance and CVD Risk
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Stumvoll, M.; Goldstein, B.J.; van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration. Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef]
- Defronzo, R.A. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef]
- Schwartz, S.S.; Epstein, S.; Corkey, B.E.; Grant, S.F.; Gavin, J.R., 3rd; Aguilar, R.B. The Time Is Right for a New Classification System for Diabetes: Rationale and Implications of the beta-Cell-Centric Classification Schema. Diabetes Care 2016, 39, 179–186. [Google Scholar] [CrossRef]
- Gaede, P.; Vedel, P.; Larsen, N.; Jensen, G.V.; Parving, H.H.; Pedersen, O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N. Engl. J. Med. 2003, 348, 383–393. [Google Scholar] [CrossRef]
- Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; Stampfer, M.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef]
- Grarup, N.; Sandholt, C.H.; Hansen, T.; Pedersen, O. Genetic susceptibility to type 2 diabetes and obesity: From genome-wide association studies to rare variants and beyond. Diabetologia 2014, 57, 1528–1541. [Google Scholar] [CrossRef]
- Wong, N.D.; Zhao, Y.; Patel, R.; Patao, C.; Malik, S.; Bertoni, A.G.; Correa, A.; Folsom, A.R.; Kachroo, S.; Mukherjee, J.; et al. Cardiovascular Risk Factor Targets and Cardiovascular Disease Event Risk in Diabetes: A Pooling Project of the Atherosclerosis Risk in Communities Study, Multi-Ethnic Study of Atherosclerosis, and Jackson Heart Study. Diabetes Care 2016, 39, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B.; Manson, J.E.; Stampfer, M.J.; Colditz, G.; Liu, S.; Solomon, C.G.; Willett, W.C. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N. Engl. J. Med. 2001, 345, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, E.S.; Dryden, D.M.; Vandermeer, B.; Ha, C.; Korownyk, C. Lifestyle interventions for patients with and at risk for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.; Cheung, C.K.; Swaminathan, R.; Nicholls, M.G.; Cockram, C.S. Obesity, albuminuria and hypertension among Hong Kong Chinese with non-insulin-dependent diabetes mellitus (NIDDM). Postgrad. Med. J. 1993, 69, 204–210. [Google Scholar] [CrossRef]
- Dabelea, D.; DeGroat, J.; Sorrelman, C.; Glass, M.; Percy, C.A.; Avery, C.; Hu, D.; D’Agostino, R.B., Jr.; Beyer, J.; Imperatore, G.; et al. Search for Diabetes in Navajo youth: Prevalence, incidence, and clinical characteristics: The Search for Diabetes in Youth Study. Diabetes Care 2009, 32 (Suppl. 2), S141–S147. [Google Scholar] [CrossRef]
- Liu, L.L.; Yi, J.P.; Beyer, J.; Mayer-Davis, E.J.; Dolan, L.M.; Dabelea, D.M.; Lawrence, J.M.; Rodriguez, B.L.; Marcovina, S.M.; Waitzfelder, B.E.; et al. Type 1 and Type 2 diabetes in Asian and Pacific Islander U.S. youth: The SEARCH for Diabetes in Youth Study. Diabetes Care 2009, 32 (Suppl. 2), S133–S140. [Google Scholar] [CrossRef]
- Karter, A.J.; Schillinger, D.; Adams, A.S.; Moffet, H.H.; Liu, J.; Adler, N.E.; Kanaya, A.M. Elevated rates of diabetes in Pacific Islanders and Asian subgroups: The Diabetes Study of Northern California (DISTANCE). Diabetes Care 2013, 36, 574–579. [Google Scholar] [CrossRef]
- Sattar, N.; Gill, J.M. Type 2 diabetes in migrant south Asians: Mechanisms, mitigation, and management. Lancet Diabetes Endocrinol. 2015, 3, 1004–1016. [Google Scholar] [CrossRef]
- McKeigue, P.M.; Shah, B.; Marmot, M.G. Relation of central obesity and insulin resistance with high diabetes prevalence and cardiovascular risk in South Asians. Lancet 1991, 337, 382–386. [Google Scholar] [CrossRef]
- Haines, L.; Wan, K.C.; Lynn, R.; Barrett, T.G.; Shield, J.P. Rising incidence of type 2 diabetes in children in the U.K. Diabetes Care 2007, 30, 1097–1101. [Google Scholar] [CrossRef]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41–47. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I. Genomics, type 2 diabetes, and obesity. N. Engl. J. Med. 2010, 363, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Dimas, A.S.; Lagou, V.; Barker, A.; Knowles, J.W.; Magi, R.; Hivert, M.F.; Benazzo, A.; Rybin, D.; Jackson, A.U.; Stringham, H.M.; et al. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes 2014, 63, 2158–2171. [Google Scholar] [CrossRef] [PubMed]
- Flannick, J.; Florez, J.C. Type 2 diabetes: Genetic data sharing to advance complex disease research. Nat. Rev. Genet. 2016, 17, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Franks, P.W.; Pearson, E.; Florez, J.C. Gene-environment and gene-treatment interactions in type 2 diabetes: Progress, pitfalls, and prospects. Diabetes Care 2013, 36, 1413–1421. [Google Scholar] [CrossRef]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E. Risk factors for type 2 diabetes mellitus: An exposure-wide umbrella review of meta-analyses. PLoS ONE 2018, 13, e0194127. [Google Scholar] [CrossRef]
- Carey, V.J.; Walters, E.E.; Colditz, G.A.; Solomon, C.G.; Willett, W.C.; Rosner, B.A.; Speizer, F.E.; Manson, J.E. Body fat distribution and risk of non-insulin-dependent diabetes mellitus in women. The Nurses’ Health Study. Am. J. Epidemiol. 1997, 145, 614–619. [Google Scholar] [CrossRef]
- Sinha, R.; Dufour, S.; Petersen, K.F.; LeBon, V.; Enoksson, S.; Ma, Y.Z.; Savoye, M.; Rothman, D.L.; Shulman, G.I.; Caprio, S. Assessment of skeletal muscle triglyceride content by (1)H nuclear magnetic resonance spectroscopy in lean and obese adolescents: Relationships to insulin sensitivity, total body fat, and central adiposity. Diabetes 2002, 51, 1022–1027. [Google Scholar] [CrossRef]
- Hillier, T.A.; Pedula, K.L. Complications in young adults with early-onset type 2 diabetes: Losing the relative protection of youth. Diabetes Care 2003, 26, 2999–3005. [Google Scholar] [CrossRef]
- Weinstein, A.R.; Sesso, H.D.; Lee, I.M.; Cook, N.R.; Manson, J.E.; Buring, J.E.; Gaziano, J.M. Relationship of physical activity vs body mass index with type 2 diabetes in women. JAMA 2004, 292, 1188–1194. [Google Scholar] [CrossRef]
- Lynch, J.; Helmrich, S.P.; Lakka, T.A.; Kaplan, G.A.; Cohen, R.D.; Salonen, R.; Salonen, J.T. Moderately intense physical activities and high levels of cardiorespiratory fitness reduce the risk of non-insulin-dependent diabetes mellitus in middle-aged men. Arch. Intern. Med. 1996, 156, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Venkatasamy, V.V.; Pericherla, S.; Manthuruthil, S.; Mishra, S.; Hanno, R. Effect of Physical activity on Insulin Resistance, Inflammation and Oxidative Stress in Diabetes Mellitus. J. Clin. Diagn. Res. 2013, 7, 1764–1766. [Google Scholar] [CrossRef] [PubMed]
- Strasser, B. Physical activity in obesity and metabolic syndrome. Ann. N. Y. Acad. Sci. 2013, 1281, 141–159. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Does exercise without weight loss improve insulin sensitivity? Diabetes Care 2003, 26, 944–945. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. (Lausanne) 2013, 4, 37. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Bunney, P.E.; Zink, A.N.; Holm, A.A.; Billington, C.J.; Kotz, C.M. Orexin activation counteracts decreases in nonexercise activity thermogenesis (NEAT) caused by high-fat diet. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef]
- Halban, P.A. Proinsulin processing in the regulated and the constitutive secretory pathway. Diabetologia 1994, 37 (Suppl. 2), S65–S72. [Google Scholar] [CrossRef]
- Boland, B.B.; Rhodes, C.J.; Grimsby, J.S. The dynamic plasticity of insulin production in beta-cells. Mol. Metab. 2017, 6, 958–973. [Google Scholar] [CrossRef]
- Rorsman, P.; Ashcroft, F.M. Pancreatic beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef] [PubMed]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Investig. 2011, 121, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S. The ryanodine receptor calcium channel of beta-cells: Molecular regulation and physiological significance. Diabetes 2002, 51, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Cuinas, A.; Garcia-Morales, V.; Vina, D.; Gil-Longo, J.; Campos-Toimil, M. Activation of PKA and Epac proteins by cyclic AMP depletes intracellular calcium stores and reduces calcium availability for vasoconstriction. Life Sci. 2016, 155, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Lustig, K.D.; Shiau, A.K.; Brake, A.J.; Julius, D. Expression cloning of an ATP receptor from mouse neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1993, 90, 5113–5117. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Webb, T.E.; King, B.F.; Burnstock, G.; Barnard, E.A. Characterisation of a recombinant P2Y purinoceptor. Eur. J. Pharmacol. 1995, 291, 281–289. [Google Scholar] [CrossRef]
- Valera, S.; Hussy, N.; Evans, R.J.; Adami, N.; North, R.A.; Surprenant, A.; Buell, G. A new class of ligand-gated ion channel defined by P2x receptor for extracellular ATP. Nature 1994, 371, 516–519. [Google Scholar] [CrossRef]
- Blachier, F.; Malaisse, W.J. Effect of exogenous ATP upon inositol phosphate production, cationic fluxes and insulin release in pancreatic islet cells. Biochim. Biophys. Acta 1988, 970, 222–229. [Google Scholar] [CrossRef]
- Li, G.D.; Milani, D.; Dunne, M.J.; Pralong, W.F.; Theler, J.M.; Petersen, O.H.; Wollheim, C.B. Extracellular ATP causes Ca2(+)− dependent and -independent insulin secretion in RINm5F cells. Phospholipase C mediates Ca2+ mobilization but not Ca2+ influx and membrane depolarization. J. Biol. Chem. 1991, 266, 3449–3457. [Google Scholar]
- Christensen, A.A.; Gannon, M. The Beta Cell in Type 2 Diabetes. Curr. Diabetes Rep. 2019, 19, 81. [Google Scholar] [CrossRef]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. beta-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, W.R.; Bone, R.N.; Sohn, P.; Syed, F.; Reissaus, C.A.; Mosley, A.L.; Wijeratne, A.B.; True, J.D.; Tong, X.; Kono, T.; et al. Endoplasmic reticulum stress alters ryanodine receptor function in the murine pancreatic beta cell. J. Biol. Chem. 2019, 294, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Hoang Do, O.; Thorn, P. Insulin secretion from beta cells within intact islets: Location matters. Clin. Exp. Pharmacol. Physiol. 2015, 42, 406–414. [Google Scholar] [CrossRef]
- Liu, M.; Weiss, M.A.; Arunagiri, A.; Yong, J.; Rege, N.; Sun, J.; Haataja, L.; Kaufman, R.J.; Arvan, P. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes. Metab. 2018, 20 (Suppl. 2), 28–50. [Google Scholar] [CrossRef] [PubMed]
- Dali-Youcef, N.; Mecili, M.; Ricci, R.; Andres, E. Metabolic inflammation: Connecting obesity and insulin resistance. Ann. Med. 2013, 45, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Hummasti, S.; Hotamisligil, G.S. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ. Res. 2010, 107, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Roca-Rivada, A.; Castelao, C.; Senin, L.L.; Landrove, M.O.; Baltar, J.; Belen Crujeiras, A.; Seoane, L.M.; Casanueva, F.F.; Pardo, M. FNDC5/irisin is not only a myokine but also an adipokine. PLoS ONE 2013, 8, e60563. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Graciano, M.F.; Valle, M.M.; Kowluru, A.; Curi, R.; Carpinelli, A.R. Regulation of insulin secretion and reactive oxygen species production by free fatty acids in pancreatic islets. Islets 2011, 3, 213–223. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Association, A.D. Prevention or Delay of Type 2 Diabetes: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019, 42, S29–S33. [Google Scholar] [CrossRef] [PubMed]
- Shamsuzzaman, A.S.; Winnicki, M.; Wolk, R.; Svatikova, A.; Phillips, B.G.; Davison, D.E.; Berger, P.B.; Somers, V.K. Independent association between plasma leptin and C-reactive protein in healthy humans. Circulation 2004, 109, 2181–2185. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenburgh, C.; Fiebig, R.; Chandwaney, R.; Ji, L.L. Aging and exercise training in skeletal muscle: Responses of glutathione and antioxidant enzyme systems. Am. J. Physiol. 1994, 267, R439–R445. [Google Scholar] [CrossRef]
- Polak, K.; Czyzyk, A.; Simoncini, T.; Meczekalski, B. New markers of insulin resistance in polycystic ovary syndrome. J. Endocrinol. Investig. 2017, 40, 1–8. [Google Scholar] [CrossRef]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 2008, 454, 463–469. [Google Scholar] [CrossRef]
- Park, K.H.; Zaichenko, L.; Brinkoetter, M.; Thakkar, B.; Sahin-Efe, A.; Joung, K.E.; Tsoukas, M.A.; Geladari, E.V.; Huh, J.Y.; Dincer, F.; et al. Circulating irisin in relation to insulin resistance and the metabolic syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 4899–4907. [Google Scholar] [CrossRef]
- El-Lebedy, D.H.; Ibrahim, A.A.; Ashmawy, I.O. Novel adipokines vaspin and irisin as risk biomarkers for cardiovascular diseases in type 2 diabetes mellitus. Diabetes Metab. Syndr. 2018, 12, 643–648. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Reparaz, J.; Kasper, L.H. Gut microbiome and the risk factors in central nervous system autoimmunity. FEBS Lett. 2014, 588, 4214–4222. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, E.; Tack, J. Obesity and metabolic syndrome: An inflammatory condition. Dig. Dis. 2012, 30, 148–153. [Google Scholar] [CrossRef]
- Biagi, E.; Candela, M.; Fairweather-Tait, S.; Franceschi, C.; Brigidi, P. Aging of the human metaorganism: The microbial counterpart. Age (Dordrecht) 2012, 34, 247–267. [Google Scholar] [CrossRef]
- Sircana, A.; Framarin, L.; Leone, N.; Berrutti, M.; Castellino, F.; Parente, R.; De Michieli, F.; Paschetta, E.; Musso, G. Altered Gut Microbiota in Type 2 Diabetes: Just a Coincidence? Curr. Diabetes Rep. 2018, 18, 98. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Watanabe, K.; Kimura, I. Gut Microbiota Dysbiosis Drives and Implies Novel Therapeutic Strategies for Diabetes Mellitus and Related Metabolic Diseases. Front. Immunol. 2017, 8, 1882. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The role of short-chain fatty acids in health and disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [CrossRef]
- Tang, C.; Ahmed, K.; Gille, A.; Lu, S.; Grone, H.J.; Tunaru, S.; Offermanns, S. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat. Med. 2015, 21, 173–177. [Google Scholar] [CrossRef]
- Neis, E.P.; Dejong, C.H.; Rensen, S.S. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef]
- Shan, Z.; Sun, T.; Huang, H.; Chen, S.; Chen, L.; Luo, C.; Yang, W.; Yang, X.; Yao, P.; Cheng, J.; et al. Association between microbiota-dependent metabolite trimethylamine-N-oxide and type 2 diabetes. Am. J. Clin. Nutr. 2017, 106, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.C.; Cull, C.A.; Frighi, V.; Holman, R.R. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: Progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA 1999, 281, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Gaede, P.H.; Jepsen, P.V.; Larsen, J.N.; Jensen, G.V.; Parving, H.H.; Pedersen, O.B. The Steno-2 study. Intensive multifactorial intervention reduces the occurrence of cardiovascular disease in patients with type 2 diabetes. Ugeskr. Laeger. 2003, 165, 2658–2661. [Google Scholar] [PubMed]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef]
- Ihnat, M.A.; Thorpe, J.E.; Kamat, C.D.; Szabo, C.; Green, D.E.; Warnke, L.A.; Lacza, Z.; Cselenyak, A.; Ross, K.; Shakir, S.; et al. Reactive oxygen species mediate a cellular ‘memory’ of high glucose stress signalling. Diabetologia 2007, 50, 1523–1531. [Google Scholar] [CrossRef]
- Ceriello, A.; Ihnat, M.A.; Thorpe, J.E. Clinical review 2: The “metabolic memory”: Is more than just tight glucose control necessary to prevent diabetic complications? J. Clin. Endocrinol. Metab. 2009, 94, 410–415. [Google Scholar] [CrossRef]
- Engerman, R.L. Pathogenesis of diabetic retinopathy. Diabetes 1989, 38, 1203–1206. [Google Scholar] [CrossRef]
- Olsen, A.S.; Sarras, M.P., Jr.; Leontovich, A.; Intine, R.V. Heritable transmission of diabetic metabolic memory in zebrafish correlates with DNA hypomethylation and aberrant gene expression. Diabetes 2012, 61, 485–491. [Google Scholar] [CrossRef]
- Simmons, D. Epigenetic Influences and Disease. Nat. Educ. 2008, 1, 6. [Google Scholar]
- Rosen, E.D.; Kaestner, K.H.; Natarajan, R.; Patti, M.E.; Sallari, R.; Sander, M.; Susztak, K. Epigenetics and Epigenomics: Implications for Diabetes and Obesity. Diabetes 2018, 67, 1923–1931. [Google Scholar] [CrossRef]
- Wahid, F.; Shehzad, A.; Khan, T.; Kim, Y.Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim. Biophys. Acta 2010, 1803, 1231–1243. [Google Scholar] [CrossRef]
- LaPierre, M.P.; Stoffel, M. MicroRNAs as stress regulators in pancreatic beta cells and diabetes. Mol. Metab. 2017, 6, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Esguerra, J.L.S.; Nagao, M.; Ofori, J.K.; Wendt, A.; Eliasson, L. MicroRNAs in islet hormone secretion. Diabetes Obes. Metab. 2018, 20 (Suppl. 2), 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ofori, J.K.; Salunkhe, V.A.; Bagge, A.; Vishnu, N.; Nagao, M.; Mulder, H.; Wollheim, C.B.; Eliasson, L.; Esguerra, J.L. Elevated miR-130a/miR130b/miR-152 expression reduces intracellular ATP levels in the pancreatic beta cell. Sci. Rep. 2017, 7, 44986. [Google Scholar] [CrossRef] [PubMed]
- Latreille, M.; Hausser, J.; Stutzer, I.; Zhang, Q.; Hastoy, B.; Gargani, S.; Kerr-Conte, J.; Pattou, F.; Zavolan, M.; Esguerra, J.L.; et al. MicroRNA-7a regulates pancreatic beta cell function. J. Clin. Investig. 2014, 124, 2722–2735. [Google Scholar] [CrossRef] [PubMed]
- Poy, M.N.; Hausser, J.; Trajkovski, M.; Braun, M.; Collins, S.; Rorsman, P.; Zavolan, M.; Stoffel, M. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc. Natl. Acad. Sci. USA 2009, 106, 5813–5818. [Google Scholar] [CrossRef] [PubMed]
- Latreille, M.; Herrmanns, K.; Renwick, N.; Tuschl, T.; Malecki, M.T.; McCarthy, M.I.; Owen, K.R.; Rulicke, T.; Stoffel, M. miR-375 gene dosage in pancreatic beta-cells: Implications for regulation of beta-cell mass and biomarker development. J. Mol. Med. (Berl.) 2015, 93, 1159–1169. [Google Scholar] [CrossRef]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015, 58, 443–455. [Google Scholar] [CrossRef]
- Blomen, V.A.; Boonstra, J. Stable transmission of reversible modifications: Maintenance of epigenetic information through the cell cycle. Cell. Mol. Life Sci. 2011, 68, 27–44. [Google Scholar] [CrossRef]
- Bogdanovic, O.; Veenstra, G.J. DNA methylation and methyl-CpG binding proteins: Developmental requirements and function. Chromosoma 2009, 118, 549–565. [Google Scholar] [CrossRef]
- Mosammaparast, N.; Shi, Y. Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. [Google Scholar] [CrossRef] [PubMed]
- Breving, K.; Esquela-Kerscher, A. The complexities of microRNA regulation: Mirandering around the rules. Int. J. Biochem. Cell. Biol. 2010, 42, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Gonzalo, I.G.; Lanting, L.; Natarajan, R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem. 2004, 279, 18091–18097. [Google Scholar] [CrossRef]
- Reddy, M.A.; Natarajan, R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc. Res. 2011, 90, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Al-Haddad, R.; Karnib, N.; Assaad, R.A.; Bilen, Y.; Emmanuel, N.; Ghanem, A.; Younes, J.; Zibara, V.; Stephan, J.S.; Sleiman, S.F. Epigenetic changes in diabetes. Neurosci. Lett. 2016, 625, 64–69. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kanwar, M.; Kennedy, A. Metabolic memory phenomenon and accumulation of peroxynitrite in retinal capillaries. Exp. Diabetes Res. 2007, 2007, 21976. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Abbas, S.N.; Odenbach, S. Reversal of hyperglycemia and diabetic nephropathy: Effect of reinstitution of good metabolic control on oxidative stress in the kidney of diabetic rats. J. Diabetes Complicat. 2004, 18, 282–288. [Google Scholar] [CrossRef]
- Thompson, J.A.; Webb, R.C. Potential role of Toll-like receptors in programming of vascular dysfunction. Clin. Sci. (Lond.) 2013, 125, 19–25. [Google Scholar] [CrossRef]
- Guarner, V.; Rubio-Ruiz, M.E. Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip. Top Gerontol. 2015, 40, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Stump, C.S.; Short, K.R.; Bigelow, M.L.; Schimke, J.M.; Nair, K.S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. USA 2003, 100, 7996–8001. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell. Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Focusing on mitochondrial form and function. Nat. Cell. Biol. 2018, 20, 735. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell. Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Kelley, D.E.; Goodpaster, B.; Wing, R.R.; Simoneau, J.A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol. 1999, 277, E1130–E1141. [Google Scholar] [CrossRef]
- Simoneau, J.A.; Veerkamp, J.H.; Turcotte, L.P.; Kelley, D.E. Markers of capacity to utilize fatty acids in human skeletal muscle: Relation to insulin resistance and obesity and effects of weight loss. FASEB J. 1999, 13, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Hickner, R.C.; Cortright, R.L.; Dohm, G.L.; Houmard, J.A. Lipid oxidation is reduced in obese human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1039–E1044. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef]
- Schrauwen-Hinderling, V.B.; Kooi, M.E.; Hesselink, M.K.; Jeneson, J.A.; Backes, W.H.; van Echteld, C.J.; van Engelshoven, J.M.; Mensink, M.; Schrauwen, P. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia 2007, 50, 113–120. [Google Scholar] [CrossRef]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.P.; Hesselink, M.K.; et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Ezraty, B.; Gennaris, A.; Barras, F.; Collet, J.F. Oxidative stress, protein damage and repair in bacteria. Nat. Rev. Microbiol. 2017, 15, 385–396. [Google Scholar] [CrossRef]
- Ceriello, A. The emerging challenge in diabetes: The “metabolic memory”. Vascul. Pharmacol. 2012, 57, 133–138. [Google Scholar] [CrossRef]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox. Signal 2020. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell. Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.; Naon, D.; Pich, S.; Soriano, F.X.; Vega, N.; Rieusset, J.; Laville, M.; Guillet, C.; Boirie, Y.; Wallberg-Henriksson, H.; et al. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: Effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 2005, 54, 2685–2693. [Google Scholar] [CrossRef]
- Novak, I. Mitophagy: A complex mechanism of mitochondrial removal. Antioxid. Redox. Signal 2012, 17, 794–802. [Google Scholar] [CrossRef]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta 2015, 1853, 2784–2790. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell. Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell. Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef]
- Quiros, P.M.; Ramsay, A.J.; Sala, D.; Fernandez-Vizarra, E.; Rodriguez, F.; Peinado, J.R.; Fernandez-Garcia, M.S.; Vega, J.A.; Enriquez, J.A.; Zorzano, A.; et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012, 31, 2117–2133. [Google Scholar] [CrossRef]
- Sebastian, D.; Hernandez-Alvarez, M.I.; Segales, J.; Sorianello, E.; Munoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Weng, S.W.; Chang, Y.H.; Su, Y.J.; Chang, C.M.; Tsai, C.J.; Shen, F.C.; Chuang, J.H.; Lin, T.K.; Liou, C.W.; et al. The Causal Role of Mitochondrial Dynamics in Regulating Insulin Resistance in Diabetes: Link through Mitochondrial Reactive Oxygen Species. Oxid. Med. Cell. Longev. 2018, 2018, 7514383. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, L.; Mollica, M.P.; Donizzetti, I.; Gifuni, G.; Sica, R.; Pignalosa, A.; Cavaliere, G.; Gaita, M.; De Filippo, C.; Zorzano, A.; et al. High-lard and high-fish-oil diets differ in their effects on function and dynamic behaviour of rat hepatic mitochondria. PLoS ONE 2014, 9, e92753. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Kadowaki, H.; Mori, Y.; Tobe, K.; Sakuta, R.; Suzuki, Y.; Tanabe, Y.; Sakura, H.; Awata, T.; Goto, Y.; et al. A subtype of diabetes mellitus associated with a mutation of mitochondrial DNA. N. Engl. J. Med. 1994, 330, 962–968. [Google Scholar] [CrossRef]
- Yang, B.T.; Dayeh, T.A.; Volkov, P.A.; Kirkpatrick, C.L.; Malmgren, S.; Jing, X.; Renstrom, E.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol. Endocrinol. 2012, 26, 1203–1212. [Google Scholar] [CrossRef]
- Tawata, M.; Hayashi, J.I.; Isobe, K.; Ohkubo, E.; Ohtaka, M.; Chen, J.; Aida, K.; Onaya, T. A new mitochondrial DNA mutation at 14577 T/C is probably a major pathogenic mutation for maternally inherited type 2 diabetes. Diabetes 2000, 49, 1269–1272. [Google Scholar] [CrossRef]
- Wang, D.; Taniyama, M.; Suzuki, Y.; Katagiri, T.; Ban, Y. Association of the mitochondrial DNA 5178A/C polymorphism with maternal inheritance and onset of type 2 diabetes in Japanese patients. Exp. Clin. Endocrinol. Diabetes 2001, 109, 361–364. [Google Scholar] [CrossRef]
- Tawata, M.; Ohtaka, M.; Iwase, E.; Ikegishi, Y.; Aida, K.; Onaya, T. New mitochondrial DNA homoplasmic mutations associated with Japanese patients with type 2 diabetes. Diabetes 1998, 47, 276–277. [Google Scholar] [CrossRef]
- Poulton, J.; Luan, J.; Macaulay, V.; Hennings, S.; Mitchell, J.; Wareham, N.J. Type 2 diabetes is associated with a common mitochondrial variant: Evidence from a population-based case-control study. Hum. Mol. Genet. 2002, 11, 1581–1583. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.L.; Zhou, X.; Li, X.; Zhao, L.; Liu, F. Variation of mitochondrial gene and the association with type 2 diabetes mellitus in a Chinese population. Diabetes Res. Clin. Pract. 2006, 73, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.; Wattis, J.A.; King, J.R.; MacDonald, I.A.; Mazzatti, D.J. The Effects of Insulin Resistance on Individual Tissues: An Application of a Mathematical Model of Metabolism in Humans. Bull. Math. Biol. 2016, 78, 1189–1217. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [PubMed]
- Nussey, S.; Whitehead, S. Endocrinology: An Integrated Approach; BIOS Scientific Publishers: Oxford, UK, 2001. [Google Scholar]
- Petersen, K.F.; Shulman, G.I. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 11G–18G. [Google Scholar] [CrossRef]
- Petersen, K.F.; Shulman, G.I. Cellular mechanism of insulin resistance in skeletal muscle. J. R Soc. Med. 2002, 95 (Suppl. 42), 8–13. [Google Scholar]
- Satoh, T. Molecular mechanisms for the regulation of insulin-stimulated glucose uptake by small guanosine triphosphatases in skeletal muscle and adipocytes. Int. J. Mol. Sci. 2014, 15, 18677–18692. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Lilly lecture 1987. The triumvirate: Beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988, 37, 667–687. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 2010, 476279. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Gaggini, M.; DeFronzo, R.A. Role of Adipose Tissue Insulin Resistance in the Natural History of Type 2 Diabetes: Results From the San Antonio Metabolism Study. Diabetes 2017, 66, 815–822. [Google Scholar] [CrossRef]
- Czech, M.P. Mechanisms of insulin resistance related to white, beige, and brown adipocytes. Mol. Metab. 2020, 34, 27–42. [Google Scholar] [CrossRef]
- Scherer, P.E. The many secret lives of adipocytes: Implications for diabetes. Diabetologia 2019, 62, 223–232. [Google Scholar] [CrossRef]
- Maki, K.C.; Kelley, K.M.; Lawless, A.L.; Hubacher, R.L.; Schild, A.L.; Dicklin, M.R.; Rains, T.M. Validation of insulin sensitivity and secretion indices derived from the liquid meal tolerance test. Diabetes Technol. Ther. 2011, 13, 661–666. [Google Scholar] [CrossRef]
- Titchenell, P.M.; Lazar, M.A.; Birnbaum, M.J. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol. Metab. 2017, 28, 497–505. [Google Scholar] [CrossRef]
- Cherrington, A.D.; Moore, M.C.; Sindelar, D.K.; Edgerton, D.S. Insulin action on the liver in vivo. Biochem. Soc. Trans. 2007, 35, 1171–1174. [Google Scholar] [CrossRef]
- Edgerton, D.S.; Lautz, M.; Scott, M.; Everett, C.A.; Stettler, K.M.; Neal, D.W.; Chu, C.A.; Cherrington, A.D. Insulin’s direct effects on the liver dominate the control of hepatic glucose production. J. Clin. Investig. 2006, 116, 521–527. [Google Scholar] [CrossRef]
- Van Schaftingen, E.; Gerin, I. The glucose-6-phosphatase system. Biochem. J. 2002, 362, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Han, H.S.; Kim, M.J.; Koo, S.H. CREB and FoxO1: Two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 2013, 46, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Montal, E.D.; Dewi, R.; Bhalla, K.; Ou, L.; Hwang, B.J.; Ropell, A.E.; Gordon, C.; Liu, W.J.; DeBerardinis, R.J.; Sudderth, J.; et al. PEPCK Coordinates the Regulation of Central Carbon Metabolism to Promote Cancer Cell Growth. Mol. Cell. 2015, 60, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, I.A.; Da Silva Morais, A.; Schroyen, B.; Van Hul, N.; Geerts, A. Insulin resistance in hepatocytes and sinusoidal liver cells: Mechanisms and consequences. J. Hepatol. 2007, 47, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Meshkani, R.; Adeli, K. Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clin. Biochem. 2009, 42, 1331–1346. [Google Scholar] [CrossRef]
- Gast, K.B.; Tjeerdema, N.; Stijnen, T.; Smit, J.W.; Dekkers, O.M. Insulin resistance and risk of incident cardiovascular events in adults without diabetes: Meta-analysis. PLoS ONE 2012, 7, e52036. [Google Scholar] [CrossRef]
- Haffner, S.M.; Lehto, S.; Ronnemaa, T.; Pyorala, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef]
- Beckman, J.A.; Creager, M.A.; Libby, P. Diabetes and atherosclerosis: Epidemiology, pathophysiology, and management. JAMA 2002, 287, 2570–2581. [Google Scholar] [CrossRef]
- Nesto, R.W. Correlation between cardiovascular disease and diabetes mellitus: Current concepts. Am. J. Med. 2004, 116 (Suppl. 5), 11S–22S. [Google Scholar] [CrossRef]
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002, 106, 3143–3421. [Google Scholar] [CrossRef]
- Reaven, G. Insulin resistance and coronary heart disease in nondiabetic individuals. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1754–1759. [Google Scholar] [CrossRef] [PubMed]
- Bornfeldt, K.E.; Tabas, I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell. Metab. 2011, 14, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.A.; Parkin, C.G. Is hyperglycemia a causal factor in cardiovascular disease? Does proving this relationship really matter? Yes. Diabetes Care 2009, 32 (Suppl. 2), S331–S333. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M.; Kuusisto, J. Insulin resistance and hyperglycaemia in cardiovascular disease development. Nat. Rev. Endocrinol. 2014, 10, 293–302. [Google Scholar] [CrossRef]
- Battisti, W.P.; Palmisano, J.; Keane, W.E. Dyslipidemia in patients with type 2 diabetes. relationships between lipids, kidney disease and cardiovascular disease. Clin. Chem. Lab. Med. 2003, 41, 1174–1181. [Google Scholar] [CrossRef]
- Lewis, G.F.; Steiner, G. Acute effects of insulin in the control of VLDL production in humans. Implications for the insulin-resistant state. Diabetes Care 1996, 19, 390–393. [Google Scholar] [CrossRef]
- Sparks, J.D.; Sparks, C.E.; Adeli, K. Selective hepatic insulin resistance, VLDL overproduction, and hypertriglyceridemia. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2104–2112. [Google Scholar] [CrossRef]
- Verges, B. Pathophysiology of diabetic dyslipidaemia: Where are we? Diabetologia 2015, 58, 886–899. [Google Scholar] [CrossRef]
- Taskinen, M.R. Diabetic dyslipidaemia: From basic research to clinical practice. Diabetologia 2003, 46, 733–749. [Google Scholar] [CrossRef]
- Chapman, M.J.; Ginsberg, H.N.; Amarenco, P.; Andreotti, F.; Boren, J.; Catapano, A.L.; Descamps, O.S.; Fisher, E.; Kovanen, P.T.; Kuivenhoven, J.A.; et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: Evidence and guidance for management. Eur. Heart J. 2011, 32, 1345–1361. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Varbo, A. Triglycerides and cardiovascular disease. Lancet 2014, 384, 626–635. [Google Scholar] [CrossRef]
- Boren, J.; Matikainen, N.; Adiels, M.; Taskinen, M.R. Postprandial hypertriglyceridemia as a coronary risk factor. Clin. Chim. Acta 2014, 431, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Dallinga-Thie, G.M.; Kroon, J.; Boren, J.; Chapman, M.J. Triglyceride-Rich Lipoproteins and Remnants: Targets for Therapy? Curr. Cardiol. Rep. 2016, 18, 67. [Google Scholar] [CrossRef] [PubMed]
- Varbo, A.; Freiberg, J.J.; Nordestgaard, B.G. Remnant Cholesterol and Myocardial Infarction in Normal Weight, Overweight, and Obese Individuals from the Copenhagen General Population Study. Clin. Chem. 2018, 64, 219–230. [Google Scholar] [CrossRef]
- Bernelot Moens, S.J.; Verweij, S.L.; Schnitzler, J.G.; Stiekema, L.C.A.; Bos, M.; Langsted, A.; Kuijk, C.; Bekkering, S.; Voermans, C.; Verberne, H.J.; et al. Remnant Cholesterol Elicits Arterial Wall Inflammation and a Multilevel Cellular Immune Response in Humans. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 969–975. [Google Scholar] [CrossRef]
- Steinberg, D.; Carew, T.E.; Fielding, C.; Fogelman, A.M.; Mahley, R.W.; Sniderman, A.D.; Zilversmit, D.B. Lipoproteins and the pathogenesis of atherosclerosis. Circulation 1989, 80, 719–723. [Google Scholar] [CrossRef]
- Cohn, J.S.; Marcoux, C.; Davignon, J. Detection, quantification, and characterization of potentially atherogenic triglyceride-rich remnant lipoproteins. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2474–2486. [Google Scholar] [CrossRef][Green Version]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C., Jr.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Curr. Opin. Cardiol. 2006, 21, 1–6. [Google Scholar] [CrossRef]
- Grundy, S.M. Metabolic syndrome: Connecting and reconciling cardiovascular and diabetes worlds. J. Am. Coll. Cardiol. 2006, 47, 1093–1100. [Google Scholar] [CrossRef]
- Ginsberg, H.N. Lipoprotein physiology in nondiabetic and diabetic states. Relationship to atherogenesis. Diabetes Care 1991, 14, 839–855. [Google Scholar] [CrossRef]
- Adiels, M.; Boren, J.; Caslake, M.J.; Stewart, P.; Soro, A.; Westerbacka, J.; Wennberg, B.; Olofsson, S.O.; Packard, C.; Taskinen, M.R. Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Verges, B. Abnormal hepatic apolipoprotein B metabolism in type 2 diabetes. Atherosclerosis 2010, 211, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.C.; Gordon, D.; Wetterau, J.R. Microsomal triglyceride transfer protein (MTP) regulation in HepG2 cells: Insulin negatively regulates MTP gene expression. J. Lipid. Res. 1995, 36, 1073–1081. [Google Scholar] [PubMed]
- Blasiole, D.A.; Davis, R.A.; Attie, A.D. The physiological and molecular regulation of lipoprotein assembly and secretion. Mol. Biosyst. 2007, 3, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.A.; Ginsberg, H.N. Complexity in the secretory pathway: The assembly and secretion of apolipoprotein B-containing lipoproteins. J. Biol. Chem. 2002, 277, 17377–17380. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Ginsberg, H.N. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol. Metab. 2011, 22, 353–363. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Kanter, J.E.; Shao, B.; Kramer, F.; Barnhart, S.; Shimizu-Albergine, M.; Vaisar, T.; Graham, M.J.; Crooke, R.M.; Manuel, C.R.; Haeusler, R.A.; et al. Increased apolipoprotein C3 drives cardiovascular risk in type 1 diabetes. J. Clin. Investig. 2019, 129, 4165–4179. [Google Scholar] [CrossRef]
- Haas, M.E.; Attie, A.D.; Biddinger, S.B. The regulation of ApoB metabolism by insulin. Trends Endocrinol. Metab. 2013, 24, 391–397. [Google Scholar] [CrossRef]
- Laatsch, A.; Merkel, M.; Talmud, P.J.; Grewal, T.; Beisiegel, U.; Heeren, J. Insulin stimulates hepatic low density lipoprotein receptor-related protein 1 (LRP1) to increase postprandial lipoprotein clearance. Atherosclerosis 2009, 204, 105–111. [Google Scholar] [CrossRef]
- Gordts, P.L.; Nock, R.; Son, N.H.; Ramms, B.; Lew, I.; Gonzales, J.C.; Thacker, B.E.; Basu, D.; Lee, R.G.; Mullick, A.E.; et al. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J. Clin. Investig. 2016, 126, 2855–2866. [Google Scholar] [CrossRef] [PubMed]
- Toth, P.P.; Barter, P.J.; Rosenson, R.S.; Boden, W.E.; Chapman, M.J.; Cuchel, M.; D’Agostino, R.B., Sr.; Davidson, M.H.; Davidson, W.S.; Heinecke, J.W.; et al. High-density lipoproteins: A consensus statement from the National Lipid Association. J. Clin. Lipidol. 2013, 7, 484–525. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Rader, D.J. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res. 2005, 96, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Van Lenten, B.J.; Fogelman, A.M.; Jackson, R.L.; Shapiro, S.; Haberland, M.E.; Edwards, P.A. Receptor-mediated uptake of remnant lipoproteins by cholesterol-loaded human monocyte-macrophages. J. Biol. Chem. 1985, 260, 8783–8788. [Google Scholar]
- Krauss, R.M. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care 2004, 27, 1496–1504. [Google Scholar] [CrossRef]
- Lamarche, B.; Uffelman, K.D.; Carpentier, A.; Cohn, J.S.; Steiner, G.; Barrett, P.H.; Lewis, G.F. Triglyceride enrichment of HDL enhances in vivo metabolic clearance of HDL apo A-I in healthy men. J. Clin. Investig. 1999, 103, 1191–1199. [Google Scholar] [CrossRef]
- Navab, M.; Reddy, S.T.; Van Lenten, B.J.; Fogelman, A.M. HDL and cardiovascular disease: Atherogenic and atheroprotective mechanisms. Nat. Rev. Cardiol. 2011, 8, 222–232. [Google Scholar] [CrossRef]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef]
- Zhou, H.; Shiu, S.W.; Wong, Y.; Tan, K.C. Impaired serum capacity to induce cholesterol efflux is associated with endothelial dysfunction in type 2 diabetes mellitus. Diabetes Vasc. Dis. Res. 2009, 6, 238–243. [Google Scholar] [CrossRef]
- Lathief, S.; Inzucchi, S.E. Approach to diabetes management in patients with CVD. Trends Cardiovasc. Med. 2016, 26, 165–179. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.R.; Small, D.M.; Li, Z.; Schaefer, E.J. Differences in LDL subspecies involve alterations in lipid composition and conformational changes in apolipoprotein B. J. Lipid. Res. 1996, 37, 1924–1935. [Google Scholar] [PubMed]
- Chait, A.; Brazg, R.L.; Tribble, D.L.; Krauss, R.M. Susceptibility of small, dense, low-density lipoproteins to oxidative modification in subjects with the atherogenic lipoprotein phenotype, pattern B. Am. J. Med. 1993, 94, 350–356. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Gaubatz, J.W.; Sun, W.; Dodge, R.C.; Crosby, J.R.; Jiang, J.; Couper, D.; Virani, S.S.; Kathiresan, S.; Boerwinkle, E.; et al. Small dense low-density lipoprotein-cholesterol concentrations predict risk for coronary heart disease: The Atherosclerosis Risk In Communities (ARIC) study. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1069–1077. [Google Scholar] [CrossRef]
- Witztum, J.L.; Mahoney, E.M.; Branks, M.J.; Fisher, M.; Elam, R.; Steinberg, D. Nonenzymatic glucosylation of low-density lipoprotein alters its biologic activity. Diabetes 1982, 31, 283–291. [Google Scholar] [CrossRef]
- Bays, H.E.; Toth, P.P.; Kris-Etherton, P.M.; Abate, N.; Aronne, L.J.; Brown, W.V.; Gonzalez-Campoy, J.M.; Jones, S.R.; Kumar, R.; La Forge, R.; et al. Obesity, adiposity, and dyslipidemia: A consensus statement from the National Lipid Association. J. Clin. Lipidol. 2013, 7, 304–383. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Haas, M.J.; Wong, N.C. Transcriptional control of apolipoprotein A-I gene expression in diabetes. Diabetes 2004, 53, 513–520. [Google Scholar] [CrossRef]
- Chung, S.; Sawyer, J.K.; Gebre, A.K.; Maeda, N.; Parks, J.S. Adipose tissue ATP binding cassette transporter A1 contributes to high-density lipoprotein biogenesis in vivo. Circulation 2011, 124, 1663–1672. [Google Scholar] [CrossRef]
- McGillicuddy, F.C.; Reilly, M.P.; Rader, D.J. Adipose modulation of high-density lipoprotein cholesterol: Implications for obesity, high-density lipoprotein metabolism, and cardiovascular disease. Circulation 2011, 124, 1602–1605. [Google Scholar] [CrossRef]
- Zhang, Y.; McGillicuddy, F.C.; Hinkle, C.C.; O’Neill, S.; Glick, J.M.; Rothblat, G.H.; Reilly, M.P. Adipocyte modulation of high-density lipoprotein cholesterol. Circulation 2010, 121, 1347–1355. [Google Scholar] [CrossRef]
- Farbstein, D.; Levy, A.P. HDL dysfunction in diabetes: Causes and possible treatments. Expert Rev. Cardiovasc. Ther. 2012, 10, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Ginsberg, H.N. Insulin resistance and cardiovascular disease. J. Clin. Investig. 2000, 106, 453–458. [Google Scholar] [CrossRef]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Guyton, J.R.; Slee, A.E.; Anderson, T.; Fleg, J.L.; Goldberg, R.B.; Kashyap, M.L.; Marcovina, S.M.; Nash, S.D.; O’Brien, K.D.; Weintraub, W.S.; et al. Relationship of lipoproteins to cardiovascular events: The AIM-HIGH Trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides and Impact on Global Health Outcomes). J. Am. Coll. Cardiol. 2013, 62, 1580–1584. [Google Scholar] [CrossRef]
- Athyros, V.G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. Dyslipidaemia of obesity, metabolic syndrome and type 2 diabetes mellitus: The case for residual risk reduction after statin treatment. Open Cardiovasc. Med. J. 2011, 5, 24–34. [Google Scholar] [CrossRef]
- Paneni, F.; Beckman, J.A.; Creager, M.A.; Cosentino, F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Eur. Heart J. 2013, 34, 2436–2443. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists, C.; Kearney, P.M.; Blackwell, L.; Collins, R.; Keech, A.; Simes, J.; Peto, R.; Armitage, J.; Baigent, C. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: A meta-analysis. Lancet 2008, 371, 117–125. [Google Scholar] [CrossRef]
- Goldberg, R.B. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J. Clin. Endocrinol. Metab. 2009, 94, 3171–3182. [Google Scholar] [CrossRef]
- Kessler, L.; Wiesel, M.L.; Attali, P.; Mossard, J.M.; Cazenave, J.P.; Pinget, M. Von Willebrand factor in diabetic angiopathy. Diabetes Metab. 1998, 24, 327–336. [Google Scholar]
- Giannini, C.; Mohn, A.; Chiarelli, F.; Kelnar, C.J. Macrovascular angiopathy in children and adolescents with type 1 diabetes. Diabetes Metab. Res. Rev. 2011, 27, 436–460. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxid. Med. Cell. Longev. 2010, 3, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Pereira, A.M.; Seica, R. Endothelial dysfunction—a major mediator of diabetic vascular disease. Biochim. Biophys. Acta 2013, 1832, 2216–2231. [Google Scholar] [CrossRef]
- Margetic, S. Inflammation and haemostasis. Biochem. Med. (Zagreb.) 2012, 22, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Guillausseau, P.J. Diabetes, advanced glycation endproducts and vascular disease. Vasc. Med. 1998, 3, 131–137. [Google Scholar] [CrossRef]
- Bakker, W.; Eringa, E.C.; Sipkema, P.; van Hinsbergh, V.W. Endothelial dysfunction and diabetes: Roles of hyperglycemia, impaired insulin signaling and obesity. Cell. Tissue Res. 2009, 335, 165–189. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef]
- Badimon, L.; Padro, T.; Vilahur, G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Lecube, A.; Pachon, G.; Petriz, J.; Hernandez, C.; Simo, R. Phagocytic activity is impaired in type 2 diabetes mellitus and increases after metabolic improvement. PLoS ONE 2011, 6, e23366. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.F.; Wang, X.L.; Tian, X.J.; Yang, Z.H.; Chu, G.P.; Zhang, J.; Li, M.; Shi, J.; Zhang, C. Nod-like receptor protein 1 inflammasome mediates neuron injury under high glucose. Mol. Neurobiol. 2014, 49, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Koenen, T.B.; Stienstra, R.; van Tits, L.J.; de Graaf, J.; Stalenhoef, A.F.; Joosten, L.A.; Tack, C.J.; Netea, M.G. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1beta transcription in human adipose tissue. Diabetes 2011, 60, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Zeadin, M.G.; Petlura, C.I.; Werstuck, G.H. Molecular mechanisms linking diabetes to the accelerated development of atherosclerosis. Can. J. Diabetes 2013, 37, 345–350. [Google Scholar] [CrossRef]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta. Diabetol. 2015, 52, 497–503. [Google Scholar] [CrossRef]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef]
- Northcott, J.M.; Yeganeh, A.; Taylor, C.G.; Zahradka, P.; Wigle, J.T. Adipokines and the cardiovascular system: Mechanisms mediating health and disease. Can. J. Physiol. Pharmacol. 2012, 90, 1029–1059. [Google Scholar] [CrossRef]
- Yamawaki, H. Vascular effects of novel adipocytokines: Focus on vascular contractility and inflammatory responses. Biol. Pharm. Bull. 2011, 34, 307–310. [Google Scholar] [CrossRef]
- Maeda, N.; Shimomura, I.; Kishida, K.; Nishizawa, H.; Matsuda, M.; Nagaretani, H.; Furuyama, N.; Kondo, H.; Takahashi, M.; Arita, Y.; et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat. Med. 2002, 8, 731–737. [Google Scholar] [CrossRef]
- Kim, J.Y.; van de Wall, E.; Laplante, M.; Azzara, A.; Trujillo, M.E.; Hofmann, S.M.; Schraw, T.; Durand, J.L.; Li, H.; Li, G.; et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Investig. 2007, 117, 2621–2637. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef]
- Fang, H.; Judd, R.L. Adiponectin Regulation and Function. Compr. Physiol. 2018, 8, 1031–1063. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Wang, M.; Wang, Q.A.; Scherer, P.E. Adiponectin-mediated antilipotoxic effects in regenerating pancreatic islets. Endocrinology 2015, 156, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Pratt, B.T.; Barnes, M.; McMullen, M.R.; Nagy, L.E. Molecular mechanism for adiponectin-dependent M2 macrophage polarization: Link between the metabolic and innate immune activity of full-length adiponectin. J. Biol. Chem. 2011, 286, 13460–13469. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Bertolani, C.; Guerra, C.T.; Aleffi, S.; Galastri, S.; Trappoliere, M.; Vizzutti, F.; Gelmini, S.; Laffi, G.; Pinzani, M.; et al. Adenosine monophosphate-activated protein kinase modulates the activated phenotype of hepatic stellate cells. Hepatology 2008, 47, 668–676. [Google Scholar] [CrossRef]
- Ezenwaka, C.E.; Kalloo, R. Caribbean female patients with type 2 diabetes mellitus have lower serum levels of adiponectin than nondiabetic subjects. Neth. J. Med. 2005, 63, 64–69. [Google Scholar]
- Dullaart, R.P.; Kappelle, P.J.; Dallinga-Thie, G.M. Carotid intima media thickness is associated with plasma adiponectin but not with the leptin:adiponectin ratio independently of metabolic syndrome. Atherosclerosis 2010, 211, 393–396. [Google Scholar] [CrossRef]
- Iwashima, Y.; Katsuya, T.; Ishikawa, K.; Ouchi, N.; Ohishi, M.; Sugimoto, K.; Fu, Y.; Motone, M.; Yamamoto, K.; Matsuo, A.; et al. Hypoadiponectinemia is an independent risk factor for hypertension. Hypertension 2004, 43, 1318–1323. [Google Scholar] [CrossRef]
- Pilz, S.; Horejsi, R.; Moller, R.; Almer, G.; Scharnagl, H.; Stojakovic, T.; Dimitrova, R.; Weihrauch, G.; Borkenstein, M.; Maerz, W.; et al. Early atherosclerosis in obese juveniles is associated with low serum levels of adiponectin. J. Clin. Endocrinol. Metab. 2005, 90, 4792–4796. [Google Scholar] [CrossRef]
- Sattar, N.; Wannamethee, G.; Sarwar, N.; Tchernova, J.; Cherry, L.; Wallace, A.M.; Danesh, J.; Whincup, P.H. Adiponectin and coronary heart disease: A prospective study and meta-analysis. Circulation 2006, 114, 623–629. [Google Scholar] [CrossRef]
- Ouchi, N.; Kihara, S.; Arita, Y.; Nishida, M.; Matsuyama, A.; Okamoto, Y.; Ishigami, M.; Kuriyama, H.; Kishida, K.; Nishizawa, H.; et al. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation 2001, 103, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Z.; Lee, M.J.; Hu, H.; Pray, J.; Wu, H.B.; Hansen, B.C.; Shuldiner, A.R.; Fried, S.K.; McLenithan, J.C.; Gong, D.W. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: Possible role in modulating insulin action. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1253–E1261. [Google Scholar] [CrossRef] [PubMed]
- Landecho, M.F.; Tuero, C.; Valenti, V.; Bilbao, I.; de la Higuera, M.; Fruhbeck, G. Relevance of Leptin and Other Adipokines in Obesity-Associated Cardiovascular Risk. Nutrients 2019, 11, 2664. [Google Scholar] [CrossRef] [PubMed]
- Schaffler, A.; Neumeier, M.; Herfarth, H.; Furst, A.; Scholmerich, J.; Buchler, C. Genomic structure of human omentin, a new adipocytokine expressed in omental adipose tissue. Biochim. Biophys. Acta 2005, 1732, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Auguet, T.; Quintero, Y.; Riesco, D.; Morancho, B.; Terra, X.; Crescenti, A.; Broch, M.; Aguilar, C.; Olona, M.; Porras, J.A.; et al. New adipokines vaspin and omentin. Circulating levels and gene expression in adipose tissue from morbidly obese women. BMC Med. Genet. 2011, 12, 60. [Google Scholar] [CrossRef]
- Pan, H.Y.; Guo, L.; Li, Q. Changes of serum omentin-1 levels in normal subjects and in patients with impaired glucose regulation and with newly diagnosed and untreated type 2 diabetes. Diabetes Res. Clin. Pract. 2010, 88, 29–33. [Google Scholar] [CrossRef]
- Tan, B.K.; Adya, R.; Farhatullah, S.; Lewandowski, K.C.; O’Hare, P.; Lehnert, H.; Randeva, H.S. Omentin-1, a novel adipokine, is decreased in overweight insulin-resistant women with polycystic ovary syndrome: Ex vivo and in vivo regulation of omentin-1 by insulin and glucose. Diabetes 2008, 57, 801–808. [Google Scholar] [CrossRef]
- Cai, R.C.; Wei, L.; Di, J.Z.; Yu, H.Y.; Bao, Y.Q.; Jia, W.P. Expression of omentin in adipose tissues in obese and type 2 diabetic patients. Zhonghua Yi Xue Za Zhi 2009, 89, 381–384. [Google Scholar]
- Elsaid, N.H.; Sadik, N.A.; Ahmed, N.R.; Fayez, S.E.; Mohammed, N.A.E. Serum omentin-1 levels in type 2 diabetic obese women in relation to glycemic control, insulin resistance and metabolic parameters. J. Clin. Transl. Endocrinol. 2018, 13, 14–19. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Lin, F.; Han, K.; Wang, X. Omentin-1 attenuates lipopolysaccharide (LPS)-induced U937 macrophages activation by inhibiting the TLR4/MyD88/NF-kappaB signaling. Arch. Biochem. Biophys. 2020, 679, 108187. [Google Scholar] [CrossRef]
- Fernandez-Trasancos, A.; Agra, R.M.; Garcia-Acuna, J.M.; Fernandez, A.L.; Gonzalez-Juanatey, J.R.; Eiras, S. Omentin treatment of epicardial fat improves its anti-inflammatory activity and paracrine benefit on smooth muscle cells. Obesity (Silver Spring) 2017, 25, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Zabetian-Targhi, F.; Mirzaei, K.; Keshavarz, S.A.; Hossein-Nezhad, A. Modulatory Role of Omentin-1 in Inflammation: Cytokines and Dietary Intake. J. Am. Coll. Nutr. 2016, 35, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Dimova, R.; Tankova, T. The role of vaspin in the development of metabolic and glucose tolerance disorders and atherosclerosis. Biomed. Res. Int. 2015, 2015, 823481. [Google Scholar] [CrossRef] [PubMed]
- Esteghamati, A.; Noshad, S.; Mousavizadeh, M.; Zandieh, A.; Nakhjavani, M. Association of vaspin with metabolic syndrome: The pivotal role of insulin resistance. Diabetes Metab. J. 2014, 38, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kwak, S.H.; Lee, Y.; Moon, M.K.; Lim, S.; Park, Y.J.; Jang, H.C.; Kim, M.S. Plasma vaspin concentrations are elevated in metabolic syndrome in men and are correlated with coronary atherosclerosis in women. Clin. Endocrinol. (Oxf.) 2011, 75, 628–635. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. https://doi.org/10.3390/ijms21176275
Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, Ostolaza H, Martín C. Pathophysiology of Type 2 Diabetes Mellitus. International Journal of Molecular Sciences. 2020; 21(17):6275. https://doi.org/10.3390/ijms21176275
Chicago/Turabian StyleGalicia-Garcia, Unai, Asier Benito-Vicente, Shifa Jebari, Asier Larrea-Sebal, Haziq Siddiqi, Kepa B. Uribe, Helena Ostolaza, and César Martín. 2020. "Pathophysiology of Type 2 Diabetes Mellitus" International Journal of Molecular Sciences 21, no. 17: 6275. https://doi.org/10.3390/ijms21176275
APA StyleGalicia-Garcia, U., Benito-Vicente, A., Jebari, S., Larrea-Sebal, A., Siddiqi, H., Uribe, K. B., Ostolaza, H., & Martín, C. (2020). Pathophysiology of Type 2 Diabetes Mellitus. International Journal of Molecular Sciences, 21(17), 6275. https://doi.org/10.3390/ijms21176275