Measuring Extracellular Vesicles by Conventional Flow Cytometry: Dream or Reality?

 , ,
, ,
Abstract

1. Introduction
2. Results
2.1. EVs Generation and Flow Cytometer Set Up
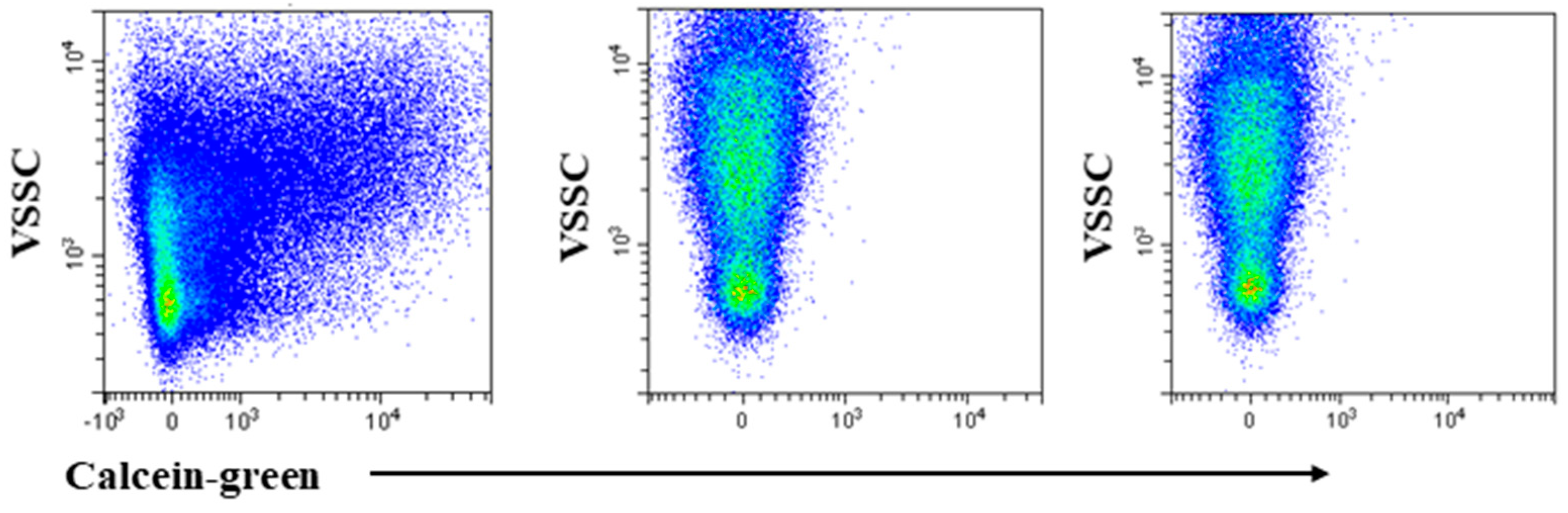
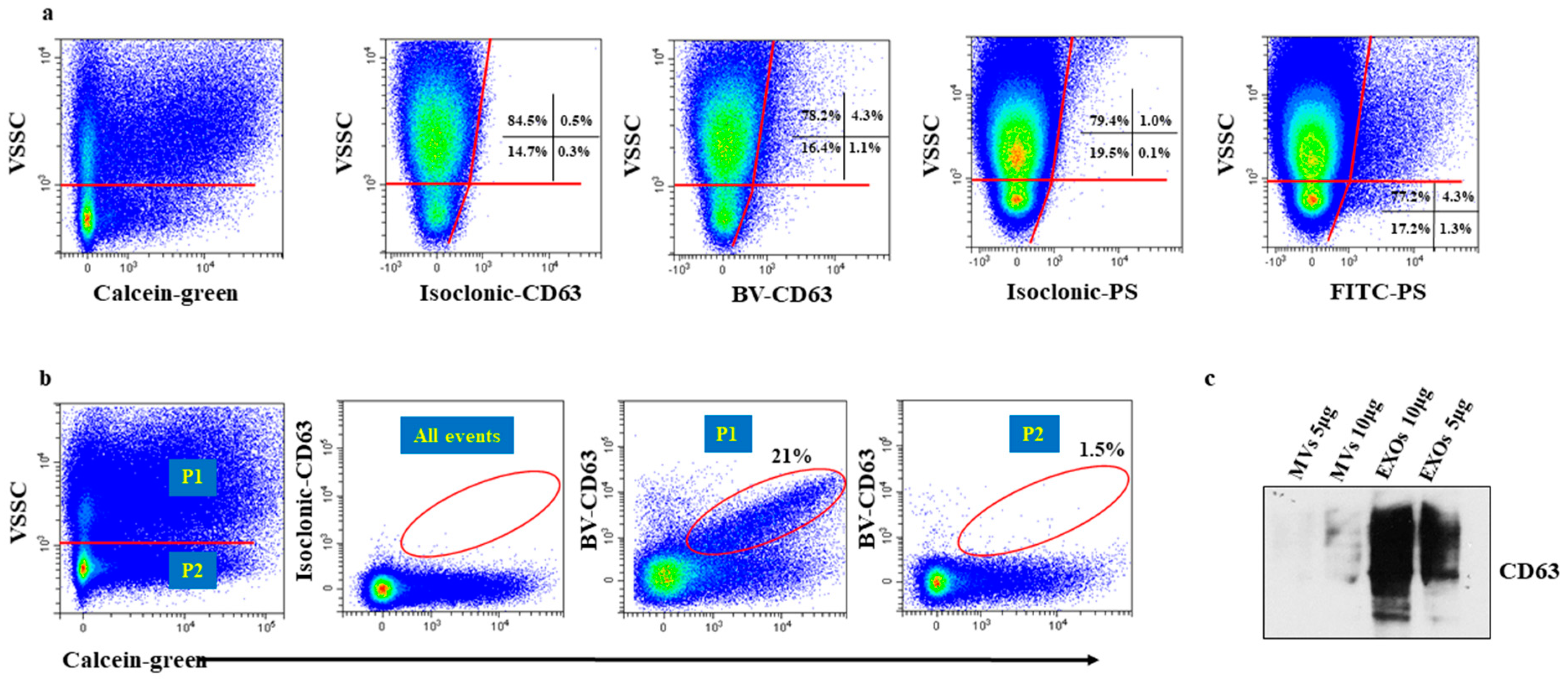
2.2. Identification of Small and Medium Sized EVs by VSSC and Calceins
2.3. Particles in the Low VSSC Signal Region Do Not Stain for Tetraspanins Because of an Insufficient Number of Binding Sites
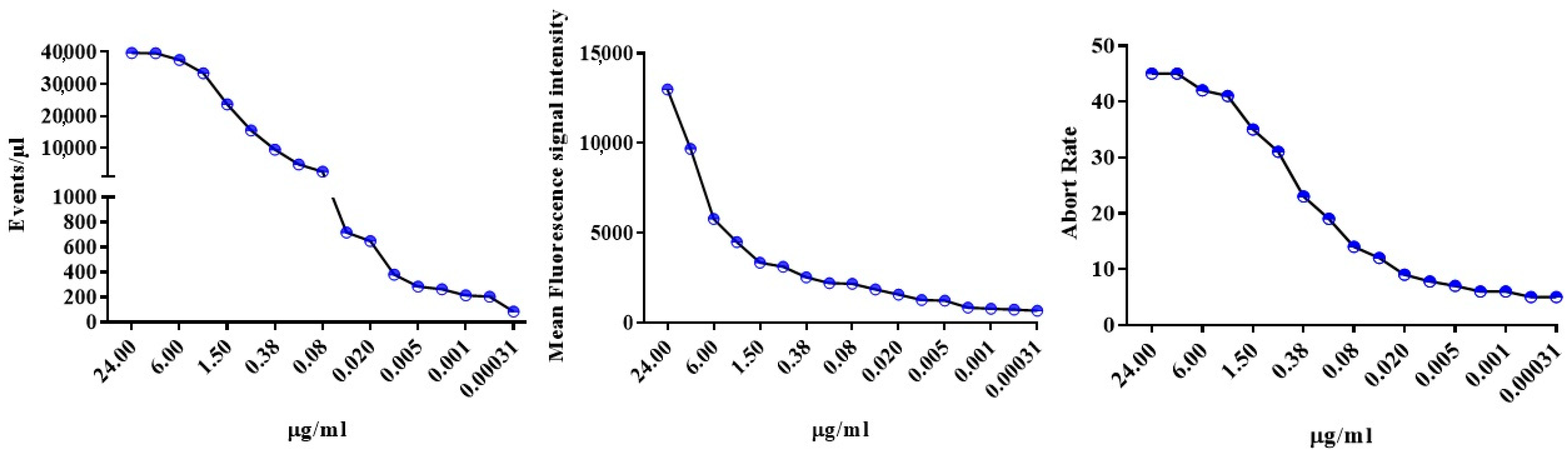
2.4. Swarming and Abort Rate Affect EVs Detection
3. Discussion
4. Methods
4.1. Reagents
4.2. EVs Staining by Calcein-Green and Calcein-Violet, and Anti-Tetraspanins moAbs
4.3. Microbead Experiments
4.4. Flow Cytometry
4.5. Western Blot Analysis
4.6. Fluorescence Microscopy
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lane, R.E.; Korbie, D.; Hill, M.M.; Trau, M. Extracellular vesicles as circulating cancer biomarkers: Opportunities and challenges. Clin. Transl. Med. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Extracellular vesicles, news about their role in immune cells: Physiology, pathology and diseases. Clin. Exp. Immunol. 2019, 196, 318–327. [Google Scholar] [CrossRef]
- Lucchetti, D.; Fattorossi, A.; Sgambato, A. Extracellular Vesicles in Oncology: Progress and Pitfalls in the Methods of Isolation and Analysis. Biotechnol. J. 2019, 14, e1700716. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ma, L.; Wang, S.; Chen, C.; Zhang, W.; Yang, L.; Hang, W.; Nolan, J.P.; Wu, L.; Yan, X. Light-scattering detection below the level of single fluorescent molecules for high-resolution characterization of functional nanoparticles. ACS Nano 2014, 8, 10998–11006. [Google Scholar] [CrossRef]
- Morales-Kastresana, A.; Musich, T.A.; Welsh, J.A.; Telford, W.; Demberg, T.; Wood, J.; Bigos, M.; Ross, C.D.; Kachynski, A.; Dean, A.; et al. High-fidelity detection and sorting of nanoscale vesicles in viral disease and cancer. J. Extracell. Vesicles 2019, 8, 1597603. [Google Scholar] [CrossRef] [PubMed]
- Chýlek, P. Absorption and scattering of light by small particles. By C. F. Bohren and d. R. Huffman. Appl. Opt. 1986, 25, 3166. [Google Scholar] [PubMed]
- McVey, M.J.; Spring, C.M.; Kuebler, W.M. Improved resolution in extracellular vesicle populations using 405 instead of 488 nm side scatter. J. Extracell. Vesicles 2018, 7. [Google Scholar] [CrossRef]
- Brittain, G.C.; Chen, Y.Q.; Martinez, E.; Tang, V.A.; Renner, T.M.; Langlois, M.A.; Gulnik, S. A Novel Semiconductor-Based Flow Cytometer with Enhanced Light-Scatter Sensitivity for the Analysis of Biological Nanoparticles. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Lucchetti, D.; Calapà, F.; Palmieri, V.; Fanali, C.; Carbone, F.; Papa, A.; De Maria, R.; De Spirito, M.; Sgambato, A. Differentiation Affects the Release of Exosomes from Colon Cancer Cells and Their Ability to Modulate the Behavior of Recipient Cells. Am. J. Pathol. 2017, 187, 1633–1647. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, J.M.; Kim, J.; Hur, J.; Park, S.; Kim, K.; Shin, H.-J.; Chwae, Y.J. Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc. Natl. Acad. Sci. USA 2018, 11, E11721–E11730. [Google Scholar] [CrossRef]
- Tsien, R.Y. Fluorescent probes of cell signaling. Annu. Rev. Neurosci. 1989, 12, 227–253. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.D.; Mitchell, A.J.; Searles, C.D. An accurate, precise method for general labeling of extracellular vesicles. MethodsX 2015, 2, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Nordin, J.Z.; Lee, Y.; Vader, P.; Mäger, I.; Johansson, H.J.; Heusermann, W.; Wiklander, O.P.; Hällbrink, M.; Seow, Y.; Bultema, J.; et al. Ultrafiltration with size-exclusion liquid chromatography for high yieldisolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine 2015, 11, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Roederer, M. Spectral compensation for flow cytometry: Visualization artifacts, limitations, and caveats. Cytometry 2001, 45, 194–205. [Google Scholar] [CrossRef]
- de Rond, L.; Libregts, S.; Rikkert, L.G.; Hau, C.M.; van der Pol, E.; Nieuwland, R.; van Leeuwen, T.G.; Coumans, F. Refractive index to evaluate staining specificity of extracellular vesicles by flow cytometry. J. Extracell. Vesicles 2019, 8, 1643671. [Google Scholar] [CrossRef]
- Coumans, F.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef]
- Escola, J.-M.; Kleijmeer, M.J.; Stoorvogel, W.; Griffith, J.; Yoshie, O.; Geuze, H.J. Selective enrichment of tetraspan proteins on the internal vesiclesof multivesicular endosomes and exosomes secreted by humanB-lymphocytes. J. Biol. Chem. 1998, 273, 20121–20127. [Google Scholar] [CrossRef]
- Chutipongtanate, S.; Greis, K.D. Multiplex Biomarker Screening Assay for Urinary Extracellular Vesicles Study: A Targeted Label-Free Proteomic Approach. Sci. Rep. 2018, 9, 15039. [Google Scholar] [CrossRef]
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.M.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627. [Google Scholar] [CrossRef]
- Linares, R.; Tan, S.; Gounou, C.; Arraud, N.; Brisson, A.R. High-speed centrifugation induces aggregation of extracellular vesicles. J. Extracell. Vesicles 2015, 4, 29509. [Google Scholar] [CrossRef]
- Maas, S.L.; de Vrij, J.; van der Vlist, E.J.; Geragousian, B.; van Bloois, L.; Mastrobattista, E.; Schiffelers, R.M.; Wauben, M.H.; Broekman, M.L.; Nolte-’t Hoen, E.N. Possibilities and limitations of current technologies for quantification of biological extracellular vesicles and synthetic mimics. J. Control. Release 2015, 200, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.P.; Jones, J.C. Detection of platelet vesicles by flow cytometry. Platelets 2017, 28, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Holloway, J.A.; Wilkinson, J.S.; Englyst, N.A. Extracellular Vesicle Flow Cytometry Analysis and Standardization. Front. Cell Dev. Biol. 2017, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; van Gemert, M.J.; Sturk, A.; Nieuwland, R.; van Leeuwen, T.G. Single vs swarm detection of microparticles and exosomes by flow cytometry. J. Thromb. Haemost. 2012, 10, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Kormelink, T.G.; Arkesteijn, G.J.; Nauwelaers, F.A.; van den Engh, G.; Nolte-’t Hoen, E.N.; Wauben, M.H. Prerequisites for the analysis and sorting of extracellular vesicle subpopulations by high-resolution flow cytometry. Cytom. Part A 2016, 89, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Libregts, S.F.W.M.; Arkesteijn, G.J.A.; Németh, A.; Nolte-’t Hoen, E.N.M.; Wauben, M.H.M. Flow cytometric analysis of extracellular vesicle subsets in plasma: Impact of swarm by particles of non-interest. J. Thromb. Haemost. 2018, 16, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.P.; Ryan, K.H.; Siegel, A.L. Steric hindrance as a factor in the reaction of labeled antibody with cell surface antigenic determinants. J. Histochem. Cytochem. 1978, 26, 618–621. [Google Scholar] [CrossRef] [PubMed]
- De Vita, M.; Catzola, V.; Buzzonetti, A.; Fossati, M.; Battaglia, A.; Zamai, L.; Fattorossi, A. Unexpected interference in cell surface staining by monoclonal antibodies to unrelated antigens. Cytom. Part B Clin. Cytom. 2015, 88, 352–354. [Google Scholar] [CrossRef]
- Kibria, G.; Ramos, E.K.; Lee, K.E.; Bedoyan, S.; Huang, S.; Samaeekia, R.; Athman, J.J.; Harding, C.V.; Lötvall, J.; Harris, L.; et al. A rapid, automated surface protein profiling of single circulating exosomes in human blood. Sci. Rep. 2016, 6, 36502. [Google Scholar] [CrossRef]
- Van der Pol, E.; Coumans, F.A.; Grootemaat, A.E.; Gardiner, C.; Sargent, I.L.; Harrison, P.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J. Thromb. Haemost. 2014, 12, 1182–1192. [Google Scholar] [CrossRef]
- Welsh, J.A.; Horak, P.; Wilkinson, J.S.; Ford, V.J.; Jones, J.C.; Smith, D. FCMPASS Software Aids Extracellular Vesicle Light Scatter Standardization. Cytom. Part A 2000, 97, 569–581. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic[s] Measured | Analyte | Analyte Detector | Reporter | Isotype | Clone | Final Concentration | Manufacturer | Catalogue. Number | Lot Number |
|---|---|---|---|---|---|---|---|---|---|
| Intracellular Esterase activity | Vesicles esterases | Calcein-green | Green-fluorescent calcein | NA | NA | 1 μM | Life Technologies | C3100MP | 1837717 |
| Calcein-violet | Violet-fluorescent calcein | NA | NA | 1 μM | Life Technologies | C34858 | 2018203 | ||
| Cell surface protein | Human CD63 | Anti-human CD63 antibody | Brilliant Violet 421™ | Mouse IgG1k | H5C6 | 1.25 μg/mL−1 | BioLegend | 353030 | B275650 |
| Human CD63 | Anti-human CD63 antibody | NA | Mouse IgG1k | H5C6 | 5 μg/mL−1 | BD Pharmingen | 556019 | VP036 | |
| Membrane Phospholipids | Phosphatidylserine | Anti-Phosphatidylserine Antibody | Alexa Fluor 488 | Mouse IgG1 | 1H6 | 1.25 μg/mL−1 | Millipore | 16-256 | 2926493 |
| Anti-Phosphatidylserine Antibody | NA | Mouse IgG1 | 1H6 | 5 μg/mL−1 | Millipore | 05-719 | 2867579 | ||
| Mouse IgG1 Fluorescein-conjugated Antibody | Miltenyi Microbeads | 50 nm-microbeads | Fluorescein | Mouse IgG1k | N/A | Dilution 1/20 | R&D system | IC002F | 1171 |
| Anti-mouse IgG MicroBeads, human | NA | NA | NA | Mouse IgG1 | NA | 5 µl from the bottle | Miltenyi Biotec | 130-057-501 | 5171201227 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucchetti, D.; Battaglia, A.; Ricciardi-Tenore, C.; Colella, F.; Perelli, L.; De Maria, R.; Scambia, G.; Sgambato, A.; Fattorossi, A. Measuring Extracellular Vesicles by Conventional Flow Cytometry: Dream or Reality? Int. J. Mol. Sci. 2020, 21, 6257. https://doi.org/10.3390/ijms21176257
Lucchetti D, Battaglia A, Ricciardi-Tenore C, Colella F, Perelli L, De Maria R, Scambia G, Sgambato A, Fattorossi A. Measuring Extracellular Vesicles by Conventional Flow Cytometry: Dream or Reality? International Journal of Molecular Sciences. 2020; 21(17):6257. https://doi.org/10.3390/ijms21176257
Chicago/Turabian StyleLucchetti, Donatella, Alessandra Battaglia, Claudio Ricciardi-Tenore, Filomena Colella, Luigi Perelli, Ruggero De Maria, Giovanni Scambia, Alessandro Sgambato, and Andrea Fattorossi. 2020. "Measuring Extracellular Vesicles by Conventional Flow Cytometry: Dream or Reality?" International Journal of Molecular Sciences 21, no. 17: 6257. https://doi.org/10.3390/ijms21176257
APA StyleLucchetti, D., Battaglia, A., Ricciardi-Tenore, C., Colella, F., Perelli, L., De Maria, R., Scambia, G., Sgambato, A., & Fattorossi, A. (2020). Measuring Extracellular Vesicles by Conventional Flow Cytometry: Dream or Reality? International Journal of Molecular Sciences, 21(17), 6257. https://doi.org/10.3390/ijms21176257