Distinctive Evidence Involved in the Role of Endocannabinoid Signalling in Parkinson’s Disease: A Perspective on Associated Therapeutic Interventions



 ,
, 
 and
and
Abstract
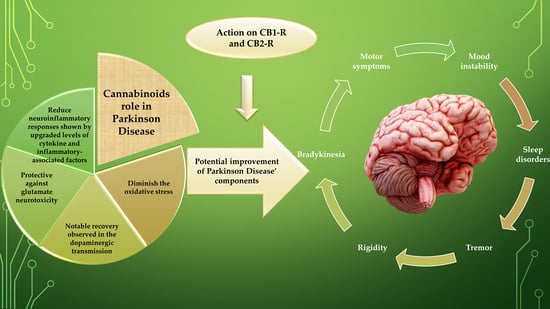
1. Introduction
2. The Endocannabinoid System
2.1. Cannabinoid Receptors (CB1 and CB2 Receptors) and Other ECS Associated Receptors
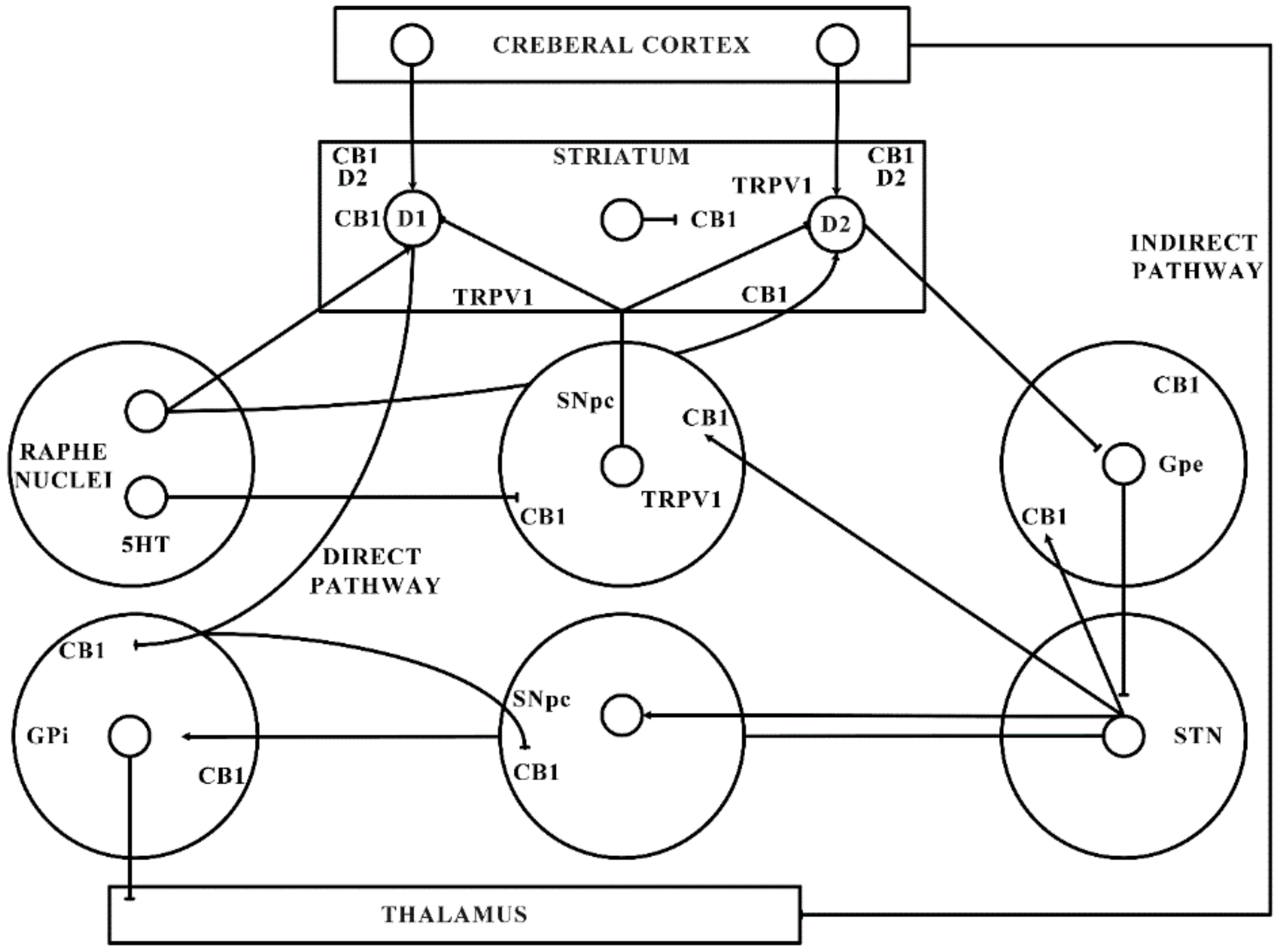
2.2. Dopamine and Cannabinoid Interactions
- Directly interacts and regulates the expression of dopaminergic neurons (by forming heteromers with dopaminergic receptors, as shown in Figure 1).
- Impedes the transduction of dopamine (DA) signals in CB-co-localized postsynaptic DA receptors.
3. Alterations Observed in ECS and Basal Ganglia in PD
4. Consequences of Basal Ganglia and Cortico-Striatal Plasticity in PD Associated Long Term Depression (LTD)
5. Experimental Studies Implying Potential Interventions of Cannabinoids as “Multi-Targeted” Compounds in Neuroprotection and Treatment of Motor and Non-Motor Complications
- Firstly, they diminish the augmented oxidative stress (OS) in PD, the process that appears to be irrespective of CB receptor participation.
- Firstly, there is indeed a strong involvement of TRPV1, CB1, and CB2 receptors linked with the cerebellum and basal ganglia eCBs that form the basis of regulating centres of movement.
- Secondly, there is proof of a potent inhibitory activity on the motor function of these endogenous, plant derivatives, and synthetic cannabinoids by optimizing the performance of numerous conventional neurotransmitters.
- Thirdly, there are significant alterations in the eCB transmission observed in human and PD animal models within basal ganglia.
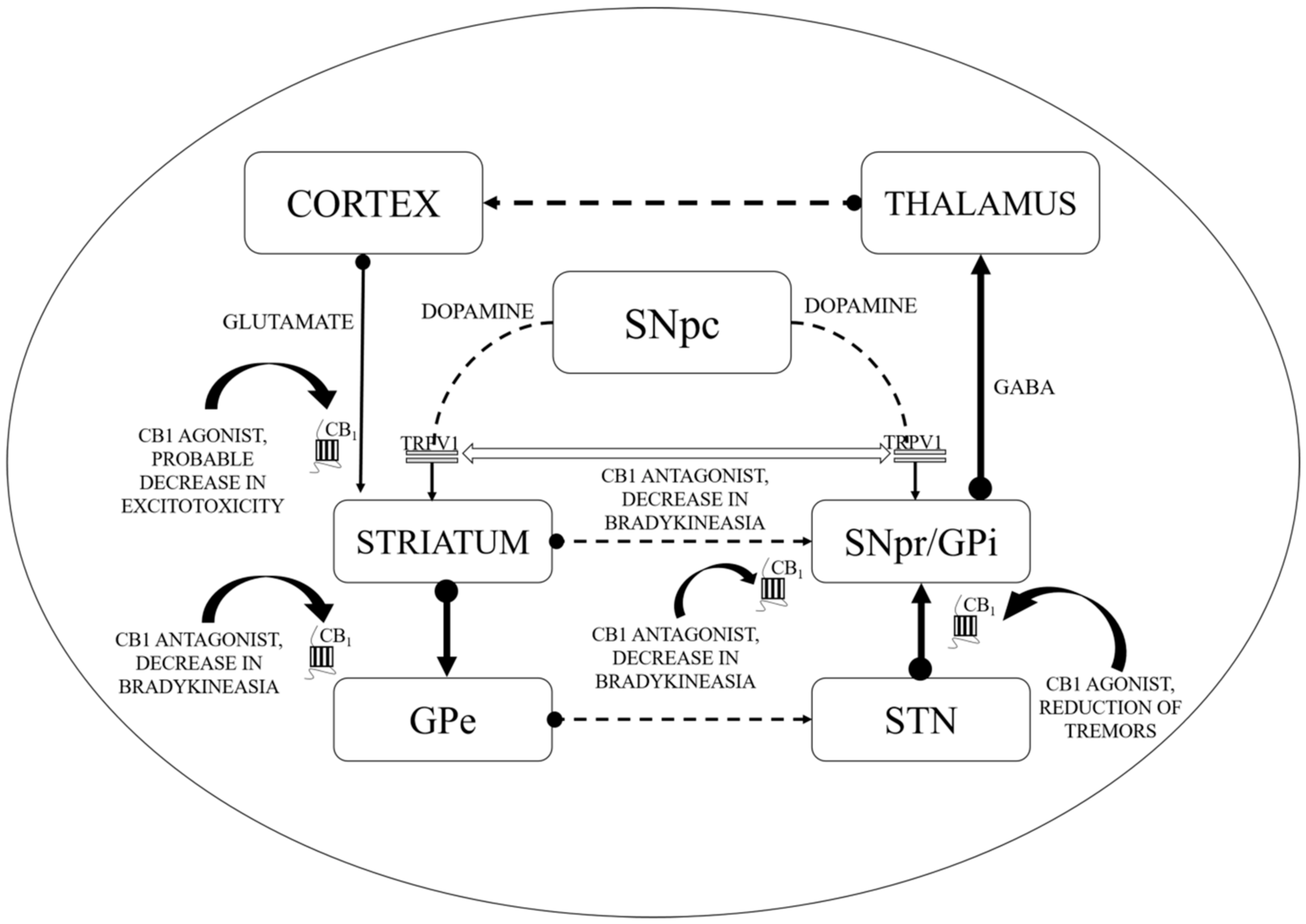
5.1. Use of Cannabinoids in Reduction of LID: Implications of CB1 Agonists and Antagonists
5.2. Therapeutic Implications of Cannabinoids in Oxidative Stress during PD
5.3. Antidepressant and Analgesic Effect of Cannabinoids in PD
5.4. Therapeutic Implications of Cannabinoids in Excitotoxicity in PD
5.5. Therapeutic Implications of Cannabinoids in Sleep Disturbances Associated with PD
6. Δ9-THCV as a Promising Cannabinoid in the PD Treatment
7. Conclusions and Future Approach
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Marzo, V. The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 2009, 60, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Melck, D.; Bisogno, T.; De Petrocellis, L. Endocannabinoids: Endogenous cannabinoid receptor ligands with neuromodulatory action. Trends. Neurosci. 1998, 21, 521–528. [Google Scholar] [CrossRef]
- Lu, H.C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Hanus, L.O.; Mechoulam, R. Novel natural and synthetic ligands of the endocannabinoid system. Curr. Med. Chem. 2010, 17, 1341–1359. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M. The Endocannabinoid System and its Manifold Central Actions. In Handbook of Neurochemistry and Molecular Neurobiology: Neural Lipids; Lajtha, A., Tettamanti, G., Goracci, G., Eds.; Springer: Boston, MA, USA, 2009; pp. 385–405. [Google Scholar] [CrossRef]
- Grosu, F.; Ungureanu, A.; Bianchi, E.; Moscu, B.; Coldea, L.; Stupariu, A.L.; Pirici, I.; Roman-Filip, C.C. Multifocal and multicentric low-grade oligoastrocytoma in a young patient. Rom. J. Morphol. Embryol. 2017, 58, 207–210. [Google Scholar]
- Buhmann, C.; Mainka, T.; Ebersbach, G.; Gandor, F. Evidence for the use of cannabinoids in Parkinson’s disease. J. Neural. Transm. (Vienna) 2019, 126, 913–924. [Google Scholar] [CrossRef]
- Mainka, T.; Stork, J.; Hidding, U.; Buhmann, C. Cannabis bei Parkinson—Hype oder Heilmittel? [Cannabis in Parkinson’s Disease: Hype or help? Fortschr. Neurol. Psychiatr. 2018, 86, 106–116. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; El-Tawil, O.S.; Bungau, S.G.; Atanasov, A.G. Applications of Antioxidants in Metabolic Disorders and Degenerative Diseases: Mechanistic Approach. Oxidative Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Di Marzo, V.; Hill, M.P.; Bisogno, T.; Crossman, A.R.; Brotchie, J.M. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson’s disease. FASEB J. 2000, 14, 1432–1438. [Google Scholar] [CrossRef]
- Gubellini, P.; Picconi, B.; Bari, M.; Battista, N.; Calabresi, P.; Centonze, D.; Bernardi, G.; Finazzi-Agrò, A.; Maccarrone, M. Experimental parkinsonism alters endocannabinoid degradation: Implications for striatal glutamatergic transmission. J. Neurosci. 2002, 22, 6900–6907. [Google Scholar] [CrossRef]
- Hernandes, M.S.; Café-Mendes, C.C.; Britto, L.R. NADPH oxidase and the degeneration of dopaminergic neurons in parkinsonian mice. Oxid. Med. Cell Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Fezza, F.; Galati, S.; Battista, N.; Napolitano, S.; Finazzi-Agrò, A.; Bernardi, G.; Brusa, L.; Pierantozzi, M.; Stanzione, P.; et al. High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson’s disease patients. Ann. Neurol. 2005, 57, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, D.M.; Goyal, V. Parkinson’s disease: A review. Neurol. India 2018, 66, S26–S35. [Google Scholar] [CrossRef]
- Zeng, X.S.; Geng, W.S.; Jia, J.J.; Chen, L.; Zhang, P.P. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Daim, M.M.; Zakhary, N.I.; Aleya, L.; Bungau, S.G.; Bohara, R.A.; Siddiqi, N.J. Aging, Metabolic, and Degenerative Disorders: Biomedical Value of Antioxidants. Oxidative Med. Cell. Longev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Behl, T.; Bungau, S.; Kumar, A.; Uddin, M.S.; Mehta, V.; Zengin, G.; Mathew, B.; Shah, M.A.; Arora, S. Dysregulation of the Gut-Brain Axis, Dysbiosis and Influence of numerous factors on Gut Microbiota associated Parkinson’s Disease. Curr. Neuropharmacol. 2020, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Desai Bradaric, B.; Patel, A.; Schneider, J.A.; Carvey, P.M.; Hendey, B. Evidence for angiogenesis in Parkinson’s disease, incidental Lewy body disease, and progressive supranuclear palsy. J. Neural. Transm. (Vienna) 2012, 119, 59–71. [Google Scholar] [CrossRef]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects [Internet]; Codon Publications: Brisbane, Australia, 2018. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Schapira, A.H. Etiology of Parkinson’s disease. Neurology 2006, 66, S10–S23. [Google Scholar] [CrossRef]
- Gupta, M.; Kant, K.; Sharma, R.; Kumar, A. Evaluation of In Silico Anti-parkinson Potential of β-asarone. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 128–135. [Google Scholar] [CrossRef]
- Rascol, O.; Payoux, P.; Ory, F.; Ferreira, J.J.; Brefel-Courbon, C.; Montastruc, J.L. Limitations of current Parkinson’s disease therapy. Ann. Neurol. 2003, 53, S3–S15. [Google Scholar] [CrossRef] [PubMed]
- Utsumi, H.; Okuma, Y.; Kano, O.; Suzuki, Y.; Iijima, M.; Tomimitsu, H.; Hashida, H.; Kubo, S.; Suzuki, M.; Nanri, K.; et al. Evaluation of the efficacy of pramipexole for treating levodopa-induced dyskinesia in patients with Parkinson’s disease. Intern. Med. 2013, 52, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Moreno-Martet, M.; Rodríguez-Cueto, C.; Palomo-Garo, C.; Gómez-Cañas, M.; Valdeolivas, S.; Guaza, C.; Romero, J.; Guzmán, M.; Mechoulam, R.; et al. Prospects for cannabinoid therapies in basal ganglia disorders. Br. J. Pharmacol. 2011, 163, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef]
- ElSohly, M.A.; Gul, W. Constituents of Cannabis Sativa. In Handbook of Cannabis; Oxford University Press: Oxford, UK, 2014; pp. 3–22. [Google Scholar] [CrossRef]
- García, M.C.; Cinquina, V.; Palomo-Garo, C.; Rábano, A.; Fernández-Ruiz, J. Identification of CB2 receptors in human nigral neurons that degenerate in Parkinson’s disease. Neurosci. Lett. 2015, 587, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kluger, B.; Triolo, P.; Jones, W.; Jankovic, J. The therapeutic potential of cannabinoids for movement disorders. Mov. Disord. 2015, 30, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, I.; Kotwani, A. Role of endocannabinoids in the progression of diabetic retinopathy. Diabetes Metab. Res. Rev. 2016, 32, 251–259. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Di Marzo, V. Non-CB1, non-CB2 receptors for endocannabinoids, plant cannabinoids, and synthetic cannabimimetics: Focus on G-protein-coupled receptors and transient receptor potential channels. J. Neuroimmune Pharmacol. 2010, 5, 103–121. [Google Scholar] [CrossRef]
- Gasperi, V.; Fezza, F.; Pasquariello, N.; Bari, M.; Oddi, S.; Agrò, A.F.; Maccarrone, M. Endocannabinoids in adipocytes during differentiation and their role in glucose uptake. Cell Mol. Life Sci. 2007, 64, 219–229. [Google Scholar] [CrossRef]
- Cascio, M.G.; Marini, P. Biosynthesis and Fate of Endocannabinoids. In Endocannabinoids; Pertwee, R.G., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 39–58. [Google Scholar] [CrossRef]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef]
- Di Marzo, V.; Bisogno, T.; De Petrocellis, L. Anandamide: Some like it hot. Trends Pharmacol. Sci. 2001, 22, 346–349. [Google Scholar] [CrossRef]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Kano, M. Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J. Neurosci. 2007, 27, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Borchert, P.; Hinz, B. A simple method for simultaneous determination of N-arachidonoylethanolamine, N-oleoylethanolamine, N-palmitoylethanolamine and 2-arachidonoylglycerol in human cells. Anal. Bioanal. Chem. 2015, 407, 1781–1787. [Google Scholar] [CrossRef]
- Liu, J.; Batkai, S.; Pacher, P.; Harvey-White, J.; Wagner, J.A.; Cravatt, B.F.; Gao, B.; Kunos, G. Lipopolysaccharide induces anandamide synthesis in macrophages via CD14/MAPK/phosphoinositide 3-kinase/NF-kappaB independently of platelet-activating factor. J. Biol. Chem. 2003, 278, 45034–45039. [Google Scholar] [CrossRef]
- Pacher, P.; Kunos, G. Modulating the endocannabinoid system in human health and disease: Successes and failures. FEBS J. 2013, 280, 1918–1943. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, A.; Martinez, A. The Endocannabinoid System and Parkinson Disease. In The Endocannabinoid System; Murillo-Rodríguez, E., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 63–81. [Google Scholar] [CrossRef]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef]
- Wilson, R.I.; Nicoll, R.A. Endocannabinoid signaling in the brain. Science 2002, 296, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Y.; Chen, C. Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist 2015, 21, 152–168. [Google Scholar] [CrossRef]
- Hegde, V.L.; Nagarkatti, M.; Nagarkatti, P.S. Cannabinoid receptor activation leads to massive mobilization of myeloid-derived suppressor cells with potent immunosuppressive properties. Eur. J. Immunol. 2010, 40, 3358–3371. [Google Scholar] [CrossRef]
- Di Marzo, V.; Berrendero, F.; Bisogno, T.; González, S.; Cavaliere, P.; Romero, J.; Cebeira, M.; Ramos, J.A.; Fernández-Ruiz, J.J. Enhancement of anandamide formation in the limbic forebrain and reduction of endocannabinoid contents in the striatum of delta9-tetrahydrocannabinol-tolerant rats. J. Neurochem. 2000, 74, 1627–1635. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Toczek, M.; Malinowska, B. Enhanced endocannabinoid tone as a potential target of pharmacotherapy. Life Sci. 2018, 204, 20–45. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, D.G. A Personal Retrospective: Elevating Anandamide (AEA) by Targeting Fatty Acid Amide Hydrolase (FAAH) and the Fatty Acid Binding Proteins (FABPs). Front. Pharmacol. 2016, 7, 370. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Deutsch, D.G. Unique pathway for anandamide synthesis and liver regeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 6339–6340. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Tsuboi, K.; Uyama, T. Metabolic Enzymes for Endocannabinoids and Endocannabinoid-Like Mediators. In The Endocannabinoidome; Di Marzo, V., Wang, J., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 111–135. [Google Scholar] [CrossRef]
- Murataeva, N.; Straiker, A.; Mackie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Ternianov, A.; Pérez-Ortiz, J.M.; Solesio, M.E.; García-Gutiérrez, M.S.; Ortega-Álvaro, A.; Navarrete, F.; Leiva, C.; Galindo, M.F.; Manzanares, J. Overexpression of CB2 cannabinoid receptors results in neuroprotection against behavioral and neurochemical alterations induced by intracaudate administration of 6-hydroxydopamine. Neurobiol. Aging 2012, 33, 421.e1-16. [Google Scholar] [CrossRef] [PubMed]
- Mailleux, P.; Vanderhaeghen, J.J. Distribution of neuronal cannabinoid receptor in the adult rat brain: A comparative receptor binding radioautography and in situ hybridization histochemistry. Neuroscience 1992, 48, 655–668. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [CrossRef]
- Mezey, E.; Tóth, Z.E.; Cortright, D.N.; Arzubi, M.K.; Krause, J.E.; Elde, R.; Guo, A.; Blumberg, P.M.; Szallasi, A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc. Natl. Acad. Sci. USA 2000, 97, 3655–3660. [Google Scholar] [CrossRef]
- Carta, A.R.; Pisanu, A.; Carboni, E. Do PPAR-Gamma Agonists Have a Future in Parkinson’s Disease Therapy? Parkinsons. Dis. 2011, 2011. [Google Scholar] [CrossRef]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Price, D.A.; Martinez, A.A.; Seillier, A.; Koek, W.; Acosta, Y.; Fernandez, E.; Strong, J.R.; Lutz, B.; Marsicano, G.; Roberts, J.L.; et al. WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Eur. J. Neurosci. 2009, 29, 2177–2186. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; García, C.; Sagredo, O.; Gómez-Ruiz, M.; de Lago, E. The endocannabinoid system as a target for the treatment of neuronal damage. Expert Opin. Ther. Targets 2010, 14, 387–404. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Romero, J.; Velasco, G.; Tolón, R.M.; Ramos, J.A.; Guzmán, M. Cannabinoid CB2 receptor: A new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007, 28, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lanciego, J.L.; Barroso-Chinea, P.; Rico, A.J.; Conte-Perales, L.; Callén, L.; Roda, E.; Gómez-Bautista, V.; López, I.P.; Lluis, C.; Labandeira-García, J.L.; et al. Expression of the mRNA coding the cannabinoid receptor 2 in the pallidal complex of Macaca fascicularis. J. Psychopharmacol. 2011, 25, 97–104. [Google Scholar] [CrossRef]
- López-Sendón Moreno, J.L.; García Caldentey, J.; Trigo Cubillo, P.; Ruiz Romero, C.; García Ribas, G.; Alonso Arias, M.A.; García de Yébenes, M.J.; Tolón, R.M.; Galve-Roperh, I.; Sagredo, O.; et al. A double-blind, randomized, cross-over, placebo-controlled, pilot trial with Sativex in Huntington’s disease. J. Neurol. 2016, 263, 1390–1400. [Google Scholar] [CrossRef]
- Molina-Holgado, F.; Pinteaux, E.; Moore, J.D.; Molina-Holgado, E.; Guaza, C.; Gibson, R.M.; Rothwell, N.J. Endogenous interleukin-1 receptor antagonist mediates anti-inflammatory and neuroprotective actions of cannabinoids in neurons and glia. J. Neurosci. 2003, 23, 6470–6474. [Google Scholar] [CrossRef]
- Rodríguez-Cueto, C.; Benito, C.; Fernández-Ruiz, J.; Romero, J.; Hernández-Gálvez, M.; Gómez-Ruiz, M. Changes in CB1 and CB2 receptors in the post-mortem cerebellum of humans affected by spinocerebellar ataxias. Br. J. Pharmacol. 2014, 171, 1472–1489. [Google Scholar] [CrossRef]
- Smith, S.R.; Denhardt, G.; Terminelli, C. The anti-inflammatory activities of cannabinoid receptor ligands in mouse peritonitis models. Eur. J. Pharmacol. 2001, 432, 107–119. [Google Scholar] [CrossRef]
- Babayeva, M.; Assefa, H.; Basu, P.; Chumki, S.; Loewy, Z. Marijuana Compounds: A Nonconventional Approach to Parkinson’s Disease Therapy. Parkinsons Dis. 2016, 2016. [Google Scholar] [CrossRef]
- Batista, L.A.; Gobira, P.H.; Viana, T.G.; Aguiar, D.C.; Moreira, F.A. Inhibition of endocannabinoid neuronal uptake and hydrolysis as strategies for developing anxiolytic drugs. Behav. Pharmacol. 2014, 25, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Dos Anjos-Garcia, T.; Ullah, F.; Falconi-Sobrinho, L.L.; Coimbra, N.C. CB 1 cannabinoid receptor-mediated anandamide signalling reduces the defensive behaviour evoked through GABA A receptor blockade in the dorsomedial division of the ventromedial hypothalamus. Neuropharmacology 2017, 113, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, E.; Rossi, F.; Maione, S. Role of TRPV1 receptors in descending modulation of pain. Mol. Cell Endocrinol. 2008, 286, S79–S83. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Pinilla, E.; Aguinaga, D.; Navarro, G.; Rico, A.J.; Oyarzábal, J.; Sánchez-Arias, J.A.; Lanciego, J.L.; Franco, R. Targeting CB 1 and GPR55 Endocannabinoid Receptors as a Potential Neuroprotective Approach for Parkinson’s Disease. Mol. Neurobiol. 2019, 56, 5900–5910. [Google Scholar] [CrossRef] [PubMed]
- Celorrio, M.; Rojo-Bustamante, E.; Fernández-Suárez, D.; Sáez, E.; Estella-Hermoso de Mendoza, A.; Müller, C.E.; Ramírez, M.J.; Oyarzábal, J.; Franco, R.; Aymerich, M.S. GPR55: A therapeutic target for Parkinson’s disease? Neuropharmacology 2017, 125, 319–332. [Google Scholar] [CrossRef]
- Kotsikorou, E.; Madrigal, K.E.; Hurst, D.P.; Sharir, H.; Lynch, D.L.; Heynen-Genel, S.; Milan, L.B.; Chung, T.D.; Seltzman, H.H.; Bai, Y.; et al. Identification of the GPR55 agonist binding site using a novel set of high-potency GPR55 selective ligands. Biochemistry 2011, 50, 5633–5647. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Baker, D.; Pryce, G.; Davies, W.L.; Hiley, C.R. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol. Sci. 2006, 27, 1–4. [Google Scholar] [CrossRef]
- Saliba, S.W.; Jauch, H.; Gargouri, B.; Keil, A.; Hurrle, T.; Volz, N.; Mohr, F.; van der Stelt, M.; Bräse, S.; Fiebich, B.L. Anti-neuroinflammatory effects of GPR55 antagonists in LPS-activated primary microglial cells. J. Neuroinflamm. 2018, 15, 322. [Google Scholar] [CrossRef]
- Wu, C.S.; Chen, H.; Sun, H.; Zhu, J.; Jew, C.P.; Wager-Miller, J.; Straiker, A.; Spencer, C.; Bradshaw, H.; Mackie, K.; et al. GPR55, a G-Protein Coupled Receptor for Lysophosphatidylinositol, Plays a Role in Motor Coordination. PLoS ONE 2013, 8, e60314. [Google Scholar] [CrossRef]
- Mátyás, F.; Yanovsky, Y.; Mackie, K.; Kelsch, W.; Misgeld, U.; Freund, T.F. Subcellular localization of type 1 cannabinoid receptors in the rat basal ganglia. Neuroscience 2006, 137, 337–361. [Google Scholar] [CrossRef] [PubMed]
- Gerdeman, G.; Lovinger, D.M. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J. Neurophysiol. 2001, 85, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez De Fonseca, F.; Gorriti, M.A.; Bilbao, A.; Escuredo, L.; García-Segura, L.M.; Piomelli, D.; Navarro, M. Role of the endogenous cannabinoid system as a modulator of dopamine transmission: Implications for Parkinson’s disease and schizophrenia. Neurotox. Res. 2001, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- García, C.; Palomo-Garo, C.; Gómez-Gálvez, Y.; Fernández-Ruiz, J. Cannabinoid–dopamine interactions in the physiology and physiopathology of the basal ganglia. Br. J. Pharmacol. 2016, 173, 2069–2079. [Google Scholar] [CrossRef]
- Kearn, C.S.; Blake-Palmer, K.; Daniel, E.; Mackie, K.; Glass, M. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: A mechanism for receptor cross-talk? Mol. Pharmacol. 2005, 67, 1697–1704. [Google Scholar] [CrossRef]
- Glass, M.; Felder, C.C. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: Evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997, 17, 5327–5333. [Google Scholar] [CrossRef]
- Jarrahian, A.; Watts, V.J.; Barker, E.L. D2 dopamine receptors modulate Galpha-subunit coupling of the CB1 cannabinoid receptor. J. Pharmacol. Exp. Ther. 2004, 308, 880–886. [Google Scholar] [CrossRef]
- Bamford, N.S.; Robinson, S.; Palmiter, R.D.; Joyce, J.A.; Moore, C.; Meshul, C.K. Dopamine modulates release from corticostriatal terminals. J. Neurosci. 2004, 24, 9541–9552. [Google Scholar] [CrossRef]
- Centonze, D.; Battista, N.; Rossi, S.; Mercuri, N.B.; Finazzi-Agrò, A.; Bernardi, G.; Calabresi, P.; Maccarrone, M. A critical interaction between dopamine D2 receptors and endocannabinoids mediates the effects of cocaine on striatal gabaergic Transmission. Neuropsychopharmacology 2004, 29, 1488–1497. [Google Scholar] [CrossRef]
- Usiello, A.; Baik, J.H.; Rougé-Pont, F.; Picetti, R.; Dierich, A.; LeMeur, M.; Piazza, P.V.; Borrelli, E. Distinct functions of the two isoforms of dopamine D2 receptors. Nature 2000, 408, 199–203. [Google Scholar] [CrossRef]
- Pickel, V.M.; Chan, J.; Kash, T.L.; Rodríguez, J.J.; MacKie, K. Compartment-specific localization of cannabinoid 1 (CB1) and mu-opioid receptors in rat nucleus accumbens. Neuroscience 2004, 127, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Agnati, L.F.; Ferré, S.; Lluis, C.; Franco, R.; Fuxe, K. Molecular mechanisms and therapeutical implications of intramembrane receptor/receptor interactions among heptahelical receptors with examples from the striatopallidal GABA neurons. Pharmacol. Rev. 2003, 55, 509–550. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; Ciruela, F.; Woods, A.S.; Lluis, C.; Franco, R. Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci. 2007, 30, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Casadó, V.; Cortés, A.; Ferrada, C.; Mallol, J.; Woods, A.; Lluis, C.; Canela, E.I.; Ferré, S. Basic concepts in G-protein-coupled receptor homo- and heterodimerization. Sci. World J. 2007, 7, 48–57. [Google Scholar] [CrossRef]
- Martín, A.B.; Fernandez-Espejo, E.; Ferrer, B.; Gorriti, M.A.; Bilbao, A.; Navarro, M.; Rodriguez de Fonseca, F.; Moratalla, R. Expression and function of CB1 receptor in the rat striatum: Localization and effects on D1 and D2 dopamine receptor-mediated motor behaviors. Neuropsychopharmacology 2008, 33, 1667–1679. [Google Scholar] [CrossRef]
- Maneuf, Y.P.; Crossman, A.R.; Brotchie, J.M. The cannabinoid receptor agonist WIN 55,212-2 reduces D2, but not D1, dopamine receptor-mediated alleviation of akinesia in the reserpine-treated rat model of Parkinson’s disease. Exp. Neurol. 1997, 148, 265–270. [Google Scholar] [CrossRef]
- Andersson, M.; Usiello, A.; Borgkvist, A.; Pozzi, L.; Dominguez, C.; Fienberg, A.A.; Svenningsson, P.; Fredholm, B.B.; Borrelli, E.; Greengard, P.; et al. Cannabinoid action depends on phosphorylation of dopamine- and cAMP-regulated phosphoprotein of 32 kDa at the protein kinase A site in striatal projection neurons. J. Neurosci. 2005, 25, 8432–8438. [Google Scholar] [CrossRef][Green Version]
- Ferré, S.; Goldberg, S.R.; Lluis, C.; Franco, R. Looking for the role of cannabinoid receptor heteromers in striatal function. Neuropharmacology 2009, 56, 226–234. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J. The endocannabinoid system as a target for the treatment of motor dysfunction. Br. J. Pharmacol. 2009, 156, 1029–1040. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Lastres-Becker, I.; Cabranes, A.; González, S.; Ramos, J.A. Endocannabinoids and basal ganglia functionality. Prostaglandins Leukot. Essent Fatty Acids 2002, 66, 257–267. [Google Scholar] [CrossRef]
- Stampanoni Bassi, M.; Sancesario, A.; Morace, R.; Centonze, D.; Iezzi, E. Cannabinoids in Parkinson’s Disease. Cannabis Cannabinoid Res. 2017, 2, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Voutsinos, B.; Fournier, M.; Labie, C.; Steinberg, R.; Souilhac, J.; Le Fur, G.; Soubrié, P. Blockade of cannabinoid receptors by SR141716 selectively increases Fos expression in rat mesocorticolimbic areas via reduced dopamine D2 function. Neuroscience 1999, 91, 607–620. [Google Scholar] [CrossRef]
- Meschler, J.P.; Conley, T.J.; Howlett, A.C. Cannabinoid and dopamine interaction in rodent brain: Effects on locomotor activity. Pharmacol. Biochem. Behav. 2000, 67, 567–573. [Google Scholar] [CrossRef]
- Nava, F.; Carta, G.; Battasi, A.M.; Gessa, G.L. D2 dopamine receptors enable Δ9-tetrahydrocannabinol induced memory impairment and reduction of hippocampal extracellular acetylcholine concentration. Br. J. Pharmacol. 2000, 130, 1201–1210. [Google Scholar] [CrossRef]
- Comelli, F.; Bettoni, I.; Colleoni, M.; Giagnoni, G.; Costa, B. Beneficial effects of a Cannabis sativa extract treatment on diabetes-induced neuropathy and oxidative stress. Phytother. Res. 2009, 23, 1678–1684. [Google Scholar] [CrossRef]
- Bezard, E.; Brotchie, J.M.; Gross, C.E. Pathophysiology of levodopa-induced dyskinesia: Potential for new therapies. Nat. Rev. Neurosci. 2001, 2, 577–588. [Google Scholar] [CrossRef]
- Reddy, V.; Grogan, D.; Ahluwalia, M.; Salles, É.L.; Ahluwalia, P.; Khodadadi, H.; Alverson, K.; Nguyen, A.; Raju, S.P.; Gaur, P.; et al. Targeting the endocannabinoid system: A predictive, preventive, and personalized medicine-directed approach to the management of brain pathologies. EPMA J. 2020, 11, 217–250. [Google Scholar] [CrossRef]
- García-Arencibia, M.; García, C.; Fernández-Ruiz, J. Cannabinoids and Parkinson’s disease. CNS Neurol Disord. Drug Targets 2009, 8, 432–439. [Google Scholar] [CrossRef]
- DeLong, M.R.; Wichmann, T. Circuits and circuit disorders of the basal ganglia. Arch. Neurol. 2007, 64, 20–24. [Google Scholar] [CrossRef]
- Kawaguchi, Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J. Neurosci. 1993, 13, 4908–4923. [Google Scholar] [CrossRef]
- Calabresi, P.; Picconi, B.; Tozzi, A.; Di Filippo, M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007, 30, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Hsu, K.S. Progress in understanding the factors regulating reversibility of long-term potentiation. Rev. Neurosci. 2001, 12, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Raymond, C.R. LTP forms 1, 2 and 3: Different mechanisms for the “long” in long-term potentiation. Trends Neurosci. 2007, 30, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Bernardi, G.; Koch, G. Mechanisms of disease: Basic-research-driven investigations in humans—The case of hyperkinetic disorders. Nat. Clin. Pract. Neurol. 2007, 3, 572–580. [Google Scholar] [CrossRef]
- Chen, Z.; Xiong, C.; Pancyr, C.; Stockwell, J.; Walz, W.; Cayabyab, F.S. Prolonged adenosine A1 receptor activation in hypoxia and pial vessel disruption focal cortical ischemia facilitates clathrin-mediated AMPA receptor endocytosis and long-lasting synaptic inhibition in rat hippocampal CA3-CA1 synapses: Differential regulation of GluA2 and GluA1 subunits by p38 MAPK and JNK. J. Neurosci. 2014, 34, 9621–9643. [Google Scholar] [CrossRef]
- Chung, H.J.; Ge, W.P.; Qian, X.; Wiser, O.; Jan, Y.N.; Jan, L.Y. G protein-activated inwardly rectifying potassium channels mediate depotentiation of long-term potentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 635–640. [Google Scholar] [CrossRef]
- Izumi, Y.; Zorumski, C.F. GABA and Endocannabinoids Mediate Depotentiation of Schaffer Collateral Synapses Induced by Stimulation of Temperoammonic Inputs. PLoS ONE 2016, 11, e0149034. [Google Scholar] [CrossRef]
- Chagas, M.H.; Eckeli, A.L.; Zuardi, A.W.; Pena-Pereira, M.A.; Sobreira-Neto, M.A.; Sobreira, E.T.; Camilo, M.R.; Bergamaschi, M.M.; Schenck, C.H.; Hallak, J.E.; et al. Cannabidiol can improve complex sleep-related behaviours associated with rapid eye movement sleep behaviour disorder in Parkinson’s disease patients: A case series. J. Clin. Pharm. Ther. 2014, 39, 564–566. [Google Scholar] [CrossRef]
- Espay, A.J.; Morgante, F.; Merola, A.; Fasano, A.; Marsili, L.; Fox, S.H.; Bezard, E.; Picconi, B.; Calabresi, P.; Lang, A.E. Levodopa-induced dyskinesia in Parkinson disease: Current and evolving concepts. Ann. Neurol. 2018, 84, 797–811. [Google Scholar] [CrossRef]
- Marrs, W.R.; Blankman, J.L.; Horne, E.A.; Thomazeau, A.; Lin, Y.H.; Coy, J.; Bodor, A.L.; Muccioli, G.G.; Hu, S.S.J.; Woodruff, G.; et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat. Neurosci. 2010, 13, 951–957. [Google Scholar] [CrossRef]
- Pan, J.P.; Zhang, H.Q.; Guo, Y.F.; Cao, X.H.; Liu, L.J. Some subtypes of endocannabinoid/endovanilloid receptors mediate docosahexaenoic acid-induced enhanced spatial memory in rats. Brain Res. 2011, 1412, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Pan, B.; Gao, X.; Blankman, J.L.; Cravatt, B.F.; Liu, Q. Genetic deletion of monoacylglycerol lipase alters endocannabinoid-mediated retrograde synaptic depression in the cerebellum. J. Physiol. 2011, 589, 4847–4855. [Google Scholar] [CrossRef] [PubMed]
- Sagredo, O.; García-Arencibia, M.; de Lago, E.; Finetti, S.; Decio, A.; Fernández-Ruiz, J. Cannabinoids and neuroprotection in basal ganglia disorders. Mol. Neurobiol. 2007, 36, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Makkar, R.; Behl, T.; Bungau, S.; Zengin, G.; Mehta, V.; Kumar, A.; Uddin, M.S.; Ashraf, G.M.; Abdel-Daim, M.M.; Arora, S.; et al. Nutraceuticals in Neurological Disorders. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Kaur, I.; Behl, T.; Bungau, S.; Zengin, G.; Kumar, A.; El-Esawi, M.A.; Khullar, G.; Venkatachalam, T.; Arora, S. The endocannabinoid signaling pathway as an emerging target in pharmacotherapy, earmarking mitigation of destructive events in rheumatoid arthritis. Life Sci. 2020, 118109. [Google Scholar] [CrossRef] [PubMed]
- García, C.; Palomo-Garo, C.; García-Arencibia, M.; Ramos, J.; Pertwee, R.; Fernández-Ruiz, J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Δ9-THCV in animal models of Parkinson’s disease. Br. J. Pharmacol. 2011, 163, 1495–1506. [Google Scholar] [CrossRef]
- Pan, H.; Mukhopadhyay, P.; Rajesh, M.; Patel, V.; Mukhopadhyay, B.; Gao, B.; Haskó, G.; Pacher, P. Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J. Pharmacol. Exp. Ther. 2009, 328, 708–714. [Google Scholar] [CrossRef]
- Hebert-Chatelain, E.; Reguero, L.; Puente, N.; Lutz, B.; Chaouloff, F.; Rossignol, R.; Piazza, P.V.; Benard, G.; Grandes, P.; Marsicano, G. Cannabinoid control of brain bioenergetics: Exploring the subcellular localization of the CB1 receptor. Mol. Metab. 2014, 3, 495–504. [Google Scholar] [CrossRef]
- Yamaori, S.; Ebisawa, J.; Okushima, Y.; Yamamoto, I.; Watanabe, K. Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: Role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci. 2011, 88, 730–736. [Google Scholar] [CrossRef]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Di Filippo, M.; Picconi, B.; Tozzi, A.; Ghiglieri, V.; Rossi, A.; Calabresi, P. The endocannabinoid system in Parkinson’s disease. Curr. Pharm. Des. 2008, 14, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Uivarosan, D.; Abdel-Daim, M.M.; Endres, L.; Purza, L.; Iovan, C.; Bungau, S.; Furau, C.G.; Tit, D.M. Effects of a proteic swine extract associated to recovery treatment on functional independence and quality of life in patients post stroke. Farmacia 2018, 66, 826–830. [Google Scholar] [CrossRef]
- Uivarosan, D.; Tit, D.M.; Iovan, C.; Nistor-Cseppento, D.C.; Endres, L.; Lazar, L.; Sava, C.; Sabau, A.M.; Buhas, C.; Moleriu, L.C.; et al. Effects of combining modern recovery techniques with neurotrophic medication and standard treatment in stroke patients. Sci. Total Environ. 2019, 679, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Sagredo, O.; Pazos, M.R.; García, C.; Pertwee, R.; Mechoulam, R.; Martínez-Orgado, J. Cannabidiol for neurodegenerative disorders: Important new clinical applications for this phytocannabinoid? Br. J. Clin. Pharmacol. 2013, 75, 323–333. [Google Scholar] [CrossRef]
- McPartland, J.M.; Duncan, M.; Di Marzo, V.; Pertwee, R.G. Are cannabidiol and Δ9-tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br. J. Pharmacol. 2015, 172, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Junior, N.C.; Campos, A.C.; Guimarães, F.S.; Del-Bel, E.; Zimmermann, P.R.; Brum, L.; Hallak, J.E.; Crippa, J.A.; Zuardi, A.W. Biological bases for a possible effect of cannabidiol in Parkinson’s disease. Braz. J. Psychiatry 2020, 42, 218–224. [Google Scholar] [CrossRef]
- Janefjord, E.; Mååg, J.L.; Harvey, B.S.; Smid, S.D. Cannabinoid effects on β amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cell Mol. Neurobiol. 2014, 34, 31–42. [Google Scholar] [CrossRef]
- Martín-Moreno, A.M.; Reigada, D.; Ramírez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to Alzheimer’s disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef]
- Santos, N.A.; Martins, N.M.; Sisti, F.M.; Fernandes, L.S.; Ferreira, R.S.; Queiroz, R.H.; Santos, A.C. The neuroprotection of cannabidiol against MPP⁺-induced toxicity in PC12 cells involves trkA receptors, upregulation of axonal and synaptic proteins, neuritogenesis, and might be relevant to Parkinson’s disease. Toxicol. In Vitro 2015, 30, 231–240. [Google Scholar] [CrossRef]
- Moldzio, R.; Pacher, T.; Krewenka, C.; Kranner, B.; Novak, J.; Duvigneau, J.C.; Rausch, W.D. Effects of cannabinoids Δ(9)-tetrahydrocannabinol, Δ(9)-tetrahydrocannabinolic acid and cannabidiol in MPP+ affected murine mesencephalic cultures. Phytomedicine 2012, 19, 819–824. [Google Scholar] [CrossRef]
- Lotan, I.; Treves, T.A.; Roditi, Y.; Djaldetti, R. Cannabis (medical marijuana) treatment for motor and non-motor symptoms of Parkinson disease: An open-label observational study. Clin. Neuropharmacol. 2014, 37, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Chagas, M.H.; Zuardi, A.W.; Tumas, V.; Pena-Pereira, M.A.; Sobreira, E.T.; Bergamaschi, M.M.; dos Santos, A.C.; Teixeira, A.L.; Hallak, J.E.; Crippa, J.A. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: An exploratory double-blind trial. J. Psychopharmacol. 2014, 28, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.B.; Bain, P.G.; Teare, L.; Liu, X.; Joint, C.; Wroath, C.; Parkin, S.G.; Fox, P.; Wright, D.; Hobart, J.; et al. Cannabis for dyskinesia in Parkinson disease: A randomized double-blind crossover study. Neurology 2004, 63, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Koppel, B.S. Cannabis in the Treatment of Dystonia, Dyskinesias, and Tics. Neurotherapeutics 2015, 12, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Venderová, K.; Růzicka, E.; Vorísek, V.; Visnovský, P. Survey on cannabis use in Parkinson’s disease: Subjective improvement of motor symptoms. Mov. Disord. 2004, 19, 1102–1106. [Google Scholar] [CrossRef]
- Zuardi, A.W.; Crippa, J.A.; Hallak, J.E.; Pinto, J.P.; Chagas, M.H.; Rodrigues, G.G.; Dursun, S.M.; Tumas, V. Cannabidiol for the treatment of psychosis in Parkinson’s disease. J. Psychopharmacol. 2009, 23, 979–983. [Google Scholar] [CrossRef]
- Finseth, T.A.; Hedeman, J.L.; Brown, R.P.; Johnson, K.I.; Binder, M.S.; Kluger, B.M. Self-reported efficacy of cannabis and other complementary medicine modalities by Parkinson’s disease patients in Colorado. Evid. Based Complement. Alternat. Med. 2015, 2015. [Google Scholar] [CrossRef]
- Ahlskog, J.E.; Muenter, M.D. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov. Disord. 2001, 16, 448–458. [Google Scholar] [CrossRef]
- Brotchie, J.M. Adjuncts to dopamine replacement: A pragmatic approach to reducing the problem of dyskinesia in Parkinson’s disease. Mov. Disord. 1998, 13, 871–876. [Google Scholar] [CrossRef]
- Morgese, M.G.; Cassano, T.; Cuomo, V.; Giuffrida, A. Anti-dyskinetic effects of cannabinoids in a rat model of Parkinson’s disease: Role of CB1 and TRPV1 receptors. Exp. Neurol. 2007, 208, 110–119. [Google Scholar] [CrossRef]
- Sieradzan, K.A.; Fox, S.H.; Hill, M.; Dick, J.P.; Crossman, A.R.; Brotchie, J.M. Cannabinoids reduce levodopa-induced dyskinesia in Parkinson’s disease: A pilot study. Neurology 2001, 57, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Morgese, M.G.; Cassano, T.; Gaetani, S.; Macheda, T.; Laconca, L.; Dipasquale, P.; Ferraro, L.; Antonelli, T.; Cuomo, V.; Giuffrida, A. Neurochemical changes in the striatum of dyskinetic rats afteradministration of the cannabinoid agonist WIN55,212-2. Neurochem. Int. 2009, 54, 56–64. [Google Scholar] [CrossRef]
- Alex, M.; Teresa, M.; Grazia, M.M.; Trabace, L.; Andrea, G. The cannabinoid agonist WIN55212-2 decreases L-DOPA-induced PKA activation and dyskinetic behavior in 6-OHDA-treated rats. Neurosci. Res. 2012, 72, 236–242. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Cebeira, M.; de Ceballos, M.L.; Zeng, B.Y.; Jenner, P.; Ramos, J.A.; Fernández-Ruiz, J.J. Increased cannabinoid CB1 receptor binding and activation of GTP-binding proteins in the basal ganglia of patients with Parkinson’s syndrome and of MPTP-treated marmosets. Eur. J. Neurosci. 2001, 14, 1827–1832. [Google Scholar] [CrossRef]
- Lee, J.; Di Marzo, V.; Brotchie, J.M. A role for vanilloid receptor 1 (TRPV1) and endocannabinnoid signalling in the regulation of spontaneous and L-DOPA induced locomotion in normal and reserpine-treated rats. Neuropharmacology 2006, 51, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, L.; Bonasera, S.J.; Hopf, F.W.; O’Dell, L.; Giorgetti, M.; Jongsma, M.; Carra, S.; Pierucci, M.; Di Giovanni, G.; Esposito, E.; et al. Impact of serotonin 2C receptor null mutation on physiology and behavior associated with nigrostriatal dopamine pathway function. J. Neurosci. 2009, 29, 8156–8165. [Google Scholar] [CrossRef] [PubMed]
- Hermann, H.; Marsicano, G.; Lutz, B. Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience 2002, 109, 451–460. [Google Scholar] [CrossRef]
- Mathur, B.N.; Capik, N.A.; Alvarez, V.A.; Lovinger, D.M. Serotonin induces long-term depression at corticostriatal synapses. J. Neurosci. 2011, 31, 7402–7411. [Google Scholar] [CrossRef]
- Häring, M.; Grieb, M.; Monory, K.; Lutz, B.; Moreira, F.A. Cannabinoid CB₁ receptor in the modulation of stress coping behavior in mice: The role of serotonin and different forebrain neuronal subpopulations. Neuropharmacology 2013, 65, 83–89. [Google Scholar] [CrossRef]
- Meschler, J.P.; Howlett, A.C.; Madras, B.K. Cannabinoid receptor agonist and antagonist effects on motor function in normal and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP)-treated non-human primates. Psychopharmacology 2001, 156, 79–85. [Google Scholar] [CrossRef]
- Segovia, G.; Mora, F.; Crossman, A.R.; Brotchie, J.M. Effects of CB1 cannabinoid receptor modulating compounds on the hyperkinesia induced by high-dose levodopa in the reserpine-treated rat model of Parkinson’s disease. Mov. Disord. 2003, 18, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Espejo, E.; Caraballo, I.; Rodriguez de Fonseca, F.; Ferrer, B.; El Banoua, F.; Flores, J.A.; Galan-Rodriguez, B. Experimental parkinsonism alters anandamide precursor synthesis, and functional deficits are improved by AM404: A modulator of endocannabinoid function. Neuropsychopharmacology 2004, 29, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Kreitzer, A.C.; Malenka, R.C. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 2007, 445, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Espejo, E.; Caraballo, I.; de Fonseca, F.R.; El Banoua, F.; Ferrer, B.; Flores, J.A.; Galan-Rodriguez, B. Cannabinoid CB1 antagonists possess antiparkinsonian efficacy only in rats with very severe nigral lesion in experimental parkinsonism. Neurobiol. Dis. 2005, 18, 591–601. [Google Scholar] [CrossRef]
- González, S.; Scorticati, C.; García-Arencibia, M.; de Miguel, R.; Ramos, J.A.; Fernández-Ruiz, J. Effects of rimonabant, a selective cannabinoid CB1 receptor antagonist, in a rat model of Parkinson’s disease. Brain Res. 2006, 1073–1074, 209–219. [Google Scholar] [CrossRef]
- Kelsey, J.E.; Harris, O.; Cassin, J. The CB(1) antagonist rimonabant is adjunctively therapeutic as well as monotherapeutic in an animal model of Parkinson’s disease. Behav. Brain Res. 2009, 203, 304–307. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Kyles, P.; Tyagi, S.C.; Tyagi, N. Autophagy of Mitochondria: A Promising Therapeutic Target for Neurodegenerative Disease. Cell Biochem. Biophys. 2014, 70, 707–719. [Google Scholar] [CrossRef]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef]
- More, S.V.; Choi, D.K. Promising cannabinoid-based therapies for Parkinson’s disease: Motor symptoms to neuroprotection. Mol. Neurodegener. 2015, 10, 17. [Google Scholar] [CrossRef]
- Cassol, O.J.; Comim, C.M.; Silva, B.R.; Hermani, F.V.; Constantino, L.S.; Felisberto, F.; Petronilho, F.; Hallak, J.E.; De Martinis, B.S.; Zuardi, A.W.; et al. Treatment with cannabidiol reverses oxidative stress parameters, cognitive impairment and mortality in rats submitted to sepsis by cecal ligation and puncture. Brain Res. 2010, 1348, 128–138. [Google Scholar] [CrossRef]
- Velez-Pardo, C.; Jimenez-Del-Rio, M.; Lores-Arnaiz, S.; Bustamante, J. Protective effects of the synthetic cannabinoids CP55,940 and JWH-015 on rat brain mitochondria upon paraquat exposure. Neurochem. Res. 2010, 35, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Juknat, A.; Pietr, M.; Kozela, E.; Rimmerman, N.; Levy, R.; Gao, F.; Coppola, G.; Geschwind, D.; Vogel, Z. Microarray and pathway analysis reveal distinct mechanisms underlying cannabinoid-mediated modulation of LPS-induced activation of BV-2 microglial cells. PLoS ONE 2013, 8, e61462. [Google Scholar] [CrossRef] [PubMed]
- Juknat, A.; Pietr, M.; Kozela, E.; Rimmerman, N.; Levy, R.; Coppola, G.; Geschwind, D.; Vogel, Z. Differential transcriptional profiles mediated by exposure to the cannabinoids cannabidiol and Δ9-tetrahydrocannabinol in BV-2 microglial cells. Br. J. Pharmacol. 2012, 165, 2512–2528. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, L.; Tegeder, I.; Barhum, Y.; Melamed, E.; Roditi, Y.; Djaldetti, R. Contribution of genetic variants to pain susceptibility in Parkinson disease. Eur. J. Pain 2012, 16, 1243–1250. [Google Scholar] [CrossRef]
- Ellis, R.J.; Toperoff, W.; Vaida, F.; van den Brande, G.; Gonzales, J.; Gouaux, B.; Bentley, H.; Atkinson, J.H. Smoked medicinal cannabis for neuropathic pain in HIV: A randomized, crossover clinical trial. Neuropsychopharmacology 2009, 34, 672–680. [Google Scholar] [CrossRef]
- Wilsey, B.; Marcotte, T.; Tsodikov, A.; Millman, J.; Bentley, H.; Gouaux, B.; Fishman, S. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J. Pain 2008, 9, 506–521. [Google Scholar] [CrossRef]
- Hill, K.P. Medical Marijuana for Treatment of Chronic Pain and Other Medical and Psychiatric Problems: A Clinical Review. JAMA 2015, 313, 2474–2483. [Google Scholar] [CrossRef]
- Lynch, M.E.; Campbell, F. Cannabinoids for treatment of chronic non-cancer pain; a systematic review of randomized trials. Br. J. Clin. Pharmacol. 2011, 72, 735–744. [Google Scholar] [CrossRef]
- Gorzalka, B.B.; Hill, M.N. Putative role of endocannabinoid signaling in the etiology of depression and actions of antidepressants. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 1575–1585. [Google Scholar] [CrossRef]
- Bambico, F.R.; Hattan, P.R.; Garant, J.P.; Gobbi, G. Effect of delta-9-tetrahydrocannabinol on behavioral despair and on pre- and postsynaptic serotonergic transmission. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 38, 88–96. [Google Scholar] [CrossRef]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Vicente-Sánchez, A.; Garzón, J. Cannabinoid receptors couple to NMDA receptors to reduce the production of NO and the mobilization of zinc induced by glutamate. Antioxid Redox. Signal. 2013, 19, 1766–1782. [Google Scholar] [CrossRef] [PubMed]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Gutiérrez, S.O.; van der Stelt, M.; et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef]
- Naidoo, V.; Nikas, S.P.; Karanian, D.A.; Hwang, J.; Zhao, J.; Wood, J.T.; Alapafuja, S.O.; Vadivel, S.K.; Butler, D.; Makriyannis, A.; et al. A new generation fatty acid amide hydrolase inhibitor protects against kainate-induced excitotoxicity. J. Mol. Neurosci. 2011, 43, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Leonoudakis, D.; Abood, M.E.; Beattie, E.C. Cannabinoid receptor activation reduces TNFalpha-induced surface localization of AMPAR-type glutamate receptors and excitotoxicity. Neuropharmacology 2010, 58, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Chiarlone, A.; Bellocchio, L.; Blázquez, C.; Resel, E.; Soria-Gómez, E.; Cannich, A.; Ferrero, J.J.; Sagredo, O.; Benito, C.; Romero, J.; et al. A restricted population of CB1 cannabinoid receptors with neuroprotective activity. Proc. Natl. Acad. Sci. USA 2014, 111, 8257–8262. [Google Scholar] [CrossRef]
- Porter, B.; Macfarlane, R.; Walker, R. The frequency and nature of sleep disorders in a community-based population of patients with Parkinson’s disease. Eur. J. Neurol. 2008, 15, 50–54. [Google Scholar] [CrossRef]
- Videnovic, A.; Golombek, D. Circadian and sleep disorders in Parkinson’s disease. Exp. Neurol. 2013, 243, 45–56. [Google Scholar] [CrossRef]
- Trotti, L.M.; Bliwise, D.L. Treatment of the sleep disorders associated with Parkinson’s disease. Neurotherapeutics 2014, 11, 68–77. [Google Scholar] [CrossRef]
- Stacy, M. Sleep disorders in Parkinson’s disease: Epidemiology and management. Drugs Aging 2002, 19, 733–739. [Google Scholar] [CrossRef]
- Suraev, A.S.; Marshall, N.S.; Vandrey, R.; McCartney, D.; Benson, M.J.; McGregor, I.S.; Grunstein, R.R.; Hoyos, C.M. Cannabinoid therapies in the management of sleep disorders: A systematic review of preclinical and clinical studies. Sleep Med. Rev. 2020, 53, 101339. [Google Scholar] [CrossRef]
- Russo, E.B.; Guy, G.W.; Robson, P.J. Cannabis, pain, and sleep: Lessons from therapeutic clinical trials of Sativex, a cannabis-based medicine. Chem. Biodivers 2007, 4, 1729–1743. [Google Scholar] [CrossRef] [PubMed]
- Koppel, B.S.; Brust, J.C.; Fife, T.; Bronstein, J.; Youssof, S.; Gronseth, G.; Gloss, D. Systematic review: Efficacy and safety of medical marijuana in selected neurologic disorders: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2014, 82, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Yang, X.; Ma, Y.; Wu, N.; Liu, Z. The CB1 cannabinoid receptor agonist reduces L-DOPA-induced motor fluctuation and ERK1/2 phosphorylation in 6-OHDA-lesioned rats. Drug Des. Devel. Ther. 2014, 8, 2173–2180. [Google Scholar] [CrossRef] [PubMed][Green Version]
- González-Aparicio, R.; Moratalla, R. Oleoylethanolamide reduces L-DOPA-induced dyskinesia via TRPV1 receptor in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2014, 62, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Bok, E.; Chung, Y.C.; Baik, H.H.; Jin, B.K. Cannabinoids prevent lipopolysaccharide-induced neurodegeneration in the rat substantia nigra in vivo through inhibition of microglial activation and NADPH oxidase. Brain Res. 2012, 1451, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.B.; Zeissler, M.L.; Hanemann, C.O.; Zajicek, J.P. Δ⁹-tetrahydrocannabinol (Δ⁹-THC) exerts a direct neuroprotective effect in a human cell culture model of Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2012, 38, 535–547. [Google Scholar] [CrossRef]
- Jeon, P.; Yang, S.; Jeong, H.; Kim, H. Cannabinoid receptor agonist protects cultured dopaminergic neurons from the death by the proteasomal dysfunction. Anat. Cell Biol. 2011, 44, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Gorman, A.M.; Finn, D.P.; Dowd, E. The effects of cannabinoid drugs on abnormal involuntary movements in dyskinetic and non-dyskinetic 6-hydroxydopamine lesioned rats. Brain Res. 2010, 1363, 40–48. [Google Scholar] [CrossRef]
- Espadas, I.; Keifman, E.; Palomo-Garo, C.; Burgaz, S.; García, C.; Fernández-Ruiz, J.; Moratalla, R. Beneficial effects of the phytocannabinoid Δ 9-THCV in L-DOPA-induced dyskinesia in Parkinson’s disease. Neurobiol. Dis. 2020, 141, 104892. [Google Scholar] [CrossRef]
- García-Arencibia, M.; González, S.; de Lago, E.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: Importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 2007, 1134, 162–170. [Google Scholar] [CrossRef]
- Gómez-Gálvez, Y.; Palomo-Garo, C.; Fernández-Ruiz, J.; García, C. Potential of the cannabinoid CB(2) receptor as a pharmacological target against inflammation in Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Molina-Holgado, F.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: Relevance to Parkinson’s disease. Neurobiol. Dis. 2005, 19, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Aymerich, M.S.; Aso, E.; Abellanas, M.A.; Tolon, R.M.; Ramos, J.A.; Ferrer, I.; Romero, J.; Fernández-Ruiz, J. Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem. Pharmacol. 2018, 157, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Nunes, I.; Tovmasian, L.T.; Silva, R.M.; Burke, R.E.; Goff, S.P. Pitx3 is required for development of substantia nigra dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2003, 100, 4245–4250. [Google Scholar] [CrossRef]
- Hwang, D.Y.; Fleming, S.M.; Ardayfio, P.; Moran-Gates, T.; Kim, H.; Tarazi, F.I.; Chesselet, M.F.; Kim, K.S. 3,4-dihydroxyphenylalanine reverses the motor deficits in Pitx3-deficient aphakia mice: Behavioral characterization of a novel genetic model of Parkinson’s disease. J. Neurosci. 2005, 25, 2132–2137. [Google Scholar] [CrossRef]
- Fuchs, J.; Mueller, J.C.; Lichtner, P.; Schulte, C.; Munz, M.; Berg, D.; Wüllner, U.; Illig, T.; Sharma, M.; Gasser, T. The transcription factor PITX3 is associated with sporadic Parkinson’s disease. Neurobiol. Aging 2009, 30, 731–738. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Design | Cannabinoids | Patients | Observations | References |
|---|---|---|---|---|
| Case series | Smoked cannabis, 1 g cannabis (2–9% THC) | 5 | No relief in tremors after one administration | [97,136] |
| Patient survey | Smoked cannabis | 84 | 46% patients elaborate few benefits, 45% improvement of bradykinesia, 14% LID, 31% of rest tremor | [97,141] |
| Four-week open label | CBD up to 400 mg/day | 6 | Brief psychiatric rating scale improvement along with PD psychosis questionnaire | [97,142] |
| Patient survey | Cannabis | 9 | 7 patients (78%) reported mood and sleep improvements, 2 patients improved motor symptoms | [97,143] |
| Randomized, double-blind, placebo-controlled | CBD 75 or 300 mg/day | 21 | Improvement for total PD psychosis questionnaire—39 score and daily activity sub-scores | [97,138] |
| Case series | CBD 75 or 300 mg/day | 4 | Improvements in rapid eye movement sleep behaviour disorder | [97,114] |
| Open label | Smoked cannabis, 0.5 g cannabis | 22 | Patients reported benefits for tremor, rigidity, pain, sleep, and bradykinesia (30 min after smoking cannabis) | [97,137] |
| Four weeks randomized, double-blind, placebo-controlled crossover | Cannador (1.25 mg CBD and 2.5 mg THC) | 17 | No benefits for LIDs on multiple outcomes | [97,139] |
| Compound | Model Involved | Activity Profile | References |
|---|---|---|---|
| OCE | A double-blind crossover study in dyskinetic patients | In PD patients, OCE was ineffective for treating levodopa-induced dyskinesia | [188] |
| WIN-55,212-2 | PD model of L-DOPA—induced motor fluctuation | WIN-55,212-2 reduced AIMs to L-DOPA significantly by enhancing DARPP-32 and ERK1/2 phosphorylation in striatal neurons | [189] |
| OEA | 6-OHDA PD mice model | OEA reduces symptoms and molecular markers of dyskinesia including the striatal overexpression of FosB. | [190] |
| HU-210 and WIN-55,212-2 | LPS injection in rats intra-nigral | HU-210 and WIN-55,212-2 elevated the survival of the nigral neurons, inhibited the ROS generation, NADPH oxidase as well as pro-inflammatory mediators | [191] |
| THC | Lactacystin, MPP+, paraquat-induced neurotoxicity in SH-SY5Y cells | The neuroprotective effect is exhibited by THC by PPAR-γ receptor activation against all toxins | [192] |
| WIN-55,212-2 | Proteasome inhibitors PSI-induced cytotoxicity in PC12 cells | PC12 cells are protected by WIN-55,212-2 and impede the cytoplasmic aggregation of α-synuclein and parkin | [193] |
| HU210 and AM251 | L-DOPA-induced dyskinesia in the rat model | Subtypes of AIMs are significantly reduced by HU210 while no effect shown on AIMs by AM251 | [194] |
| WIN-55,212-2 | In 6-OHDA injected rats, AIMs induced by L-DOPA | WIN-55,212-2 mitigated L-DOPA induced AIMs | [149] |
| CBD, THC, THCA, | Cytotoxicity in mice mesencephalic cultures induced by MPP+ | All exhibited antioxidative effects. Dopaminergic neurons protected by THCA and THC | [136] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behl, T.; Kaur, G.; Bungau, S.; Jhanji, R.; Kumar, A.; Mehta, V.; Zengin, G.; Brata, R.; Hassan, S.S.u.; Fratila, O. Distinctive Evidence Involved in the Role of Endocannabinoid Signalling in Parkinson’s Disease: A Perspective on Associated Therapeutic Interventions. Int. J. Mol. Sci. 2020, 21, 6235. https://doi.org/10.3390/ijms21176235
Behl T, Kaur G, Bungau S, Jhanji R, Kumar A, Mehta V, Zengin G, Brata R, Hassan SSu, Fratila O. Distinctive Evidence Involved in the Role of Endocannabinoid Signalling in Parkinson’s Disease: A Perspective on Associated Therapeutic Interventions. International Journal of Molecular Sciences. 2020; 21(17):6235. https://doi.org/10.3390/ijms21176235
Chicago/Turabian StyleBehl, Tapan, Gagandeep Kaur, Simona Bungau, Rishabh Jhanji, Arun Kumar, Vineet Mehta, Gokhan Zengin, Roxana Brata, Syed Shams ul Hassan, and Ovidiu Fratila. 2020. "Distinctive Evidence Involved in the Role of Endocannabinoid Signalling in Parkinson’s Disease: A Perspective on Associated Therapeutic Interventions" International Journal of Molecular Sciences 21, no. 17: 6235. https://doi.org/10.3390/ijms21176235
APA StyleBehl, T., Kaur, G., Bungau, S., Jhanji, R., Kumar, A., Mehta, V., Zengin, G., Brata, R., Hassan, S. S. u., & Fratila, O. (2020). Distinctive Evidence Involved in the Role of Endocannabinoid Signalling in Parkinson’s Disease: A Perspective on Associated Therapeutic Interventions. International Journal of Molecular Sciences, 21(17), 6235. https://doi.org/10.3390/ijms21176235