Abstract
The endoplasmic reticulum (ER) is an important organelle involved in protein quality control and cellular homeostasis. The accumulation of unfolded proteins leads to an ER stress, followed by an adaptive response via the activation of the unfolded protein response (UPR), PKR-like ER kinase (PERK), inositol-requiring transmembrane kinase/endoribonuclease 1α (IRE1α) and activating transcription factor 6 (ATF6) pathways. However, prolonged cell stress activates apoptosis signaling leading to cell death. Neuronal cells are particularly sensitive to protein misfolding, consequently ER and UPR dysfunctions were found to be involved in many neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis and prions diseases, among others characterized by the accumulation and aggregation of misfolded proteins. Pharmacological UPR modulation in affected tissues may contribute to the treatment and prevention of neurodegeneration. The association between ER stress, UPR and neuropathology is well established. In this review, we provide up-to-date evidence of UPR activation in neurodegenerative disorders followed by therapeutic strategies targeting the UPR and ameliorating the toxic effects of protein unfolding and aggregation.
1. ER Functions and Connections with Other Organelles
The endoplasmic reticulum (ER) is a large organelle spread throughout the cytoplasm and divided into three morphologies that include the nuclear envelope (NE), peripheral ER cisternae, and an interconnected tubular network [1]. ER is considered multifunctional since it is specialized in lipid and steroid synthesis, Ca2+ homeostasis and storage, carbohydrate metabolism, and protein synthesis [2]. This multi-functional nature requires, in addition to a myriad of proteins and a unique physical structure, interconnections and coordination with other organelles. Thus, ER has numerous contact sites with all membrane-bound organelles, including the plasma membrane (PM), mitochondria, Golgi, endosomes, and peroxisomes [1]. Structural connectivity at the level of ER-PM and ER-mitochondria junctions is the best studied.
1.1. ER and Plasma Membrane
The ER is the cell’s major Ca2+ store and forms an extensive and dynamic network of contacts with the PM. It is becoming increasingly evident that contact sites between the ER and the PM play a role in Ca2+ exchange. Influx of Ca2+ across the plasma membrane in many cells is achieved by store operated Ca2+ entry (SOCE). At ER-PM junctions, stromal-interacting molecule (STIM) proteins sense a drop in ER Ca2+ levels, undergo a conformational change repositioning tubular structures throughout the ER to ER-PM and directly activate Orai, the pore-forming component of the Ca2+- release-activated Ca2+(CRAC) channel, triggering channel opening and Ca2+ influx [3]. In addition, ER-PM contact sites are important for the phosphatidylinositol metabolism specifically for the regulation of the lipid signaling molecule phosphatidylinositol 4-phosphate (PI4P). This is controlled by the oxysterol-binding homology (Osh) protein family and the integral ER membrane proteins vesicle-associated membrane protein- associated protein (VAP) after the activation of Sac1 phosphatase [1]. Deletion of Osh proteins in yeast cells resulted in a six to seven-fold increase in PI4P levels; furthermore, the addition of recombinant Osh3 to a microsome fraction depleted peripherally bound proteins able to stimulate Sac1 phosphatase activity, suggesting that Osh proteins control PI4P levels at ER-PM contact sites [4].
1.2. ER and Mitochondria
The sites of ER in contact with mitochondria have been referred to as mitochondria-associated ER membrane (MAM). This communication is vital for the cell fate; it operates as a structural allocation for multiple scaffold proteins and regulatory factors and it is associated with multiple functions including lipid transfer, autophagosome formation, mitochondrial fission, Ca2+ homeostasis and apoptosis [5]. At ER contact sites, there is an influx of Ca2+ into the intermembrane space and matrix of the mitochondria. ER channels release Ca2+ directly to the mitochondrial membrane and this requires IP3 receptor (IP3R) interaction with the voltage-dependent anion selective channel protein 1 (VDAC1). Changes in Ca2+ levels have been shown to affect apoptosis, mitochondrial division and motility, and to regulate the activity of mitochondrial Ca2+-binding proteins [1,6]. MAM is also necessary for lipid flipping during lipid biosynthesis. During this process, phosphatidylserine (PS) is synthesized from phosphatidylalanine on the ER membrane; then, PS conversion to phosphatidylethanolamine (PE) uses proteins on the mitochondria and PE conversion to phosphatidylcholine (PC) uses ER localized enzymes. Therefore, the phospholipid moves between the two membranes before each conversion step. Furthermore, ER tubules were shown to be involved in mitochondrial biogenesis by defining the position of the mitochondrial division machinery recruitment in yeast and mammalian cells [7]. Several types of molecular bridges mediate the contacts between these two organelles, such as the ER-mitochondria encounter structures (ERMES) complex identified in yeast and the mitochondrial fusion protein mitofusin 2 (Mfn2) in mammalian cells, which suggest that contact site formation may be highly regulated during different ER-mitochondria functions [7]. Alterations in ER-mitochondria signaling have pleiotropic effects on a variety of intracellular events resulting in mitochondrial damage, Ca2+ dyshomeostasis, ER stress, defects in lipid metabolism, autophagy, reduced respiratory chain activity and oxidative phosphorylation; mitochondria and ER play a central role in the regulation of neurological activities. Multiple studies revealed that these alterations are common in neurodegenerative disease [8,9].
2. ER Stress and the Unfolded Protein Response (UPR)
The ER is the principal site for biosynthesis of proteins, post-translational modification, folding and assembly of newly synthesized proteins. As a membranous compartment, the ER is extremely sensitive to changes that affect its structure, integrity and function leading to a disruption of proteins’ folding. If the final tertiary structure cannot be attained, unfolded proteins are translocated to the cytosol and subjected to ubiquitination and proteasome-dependent degradation, known as ER-associated degradation (ERAD). The accumulation of unfolded or misfolded proteins inside the cell results in the failure of the ER to cope with the excess of protein load leading to “ER stress” and to several pathological conditions [10].
Eukaryotic cells can adapt by attenuating the rate of protein synthesis, upregulating the expression of genes encoding chaperones and other proteins that prevent polypeptide aggregation and degrading accumulated misfolded proteins. This set of cellular responses is obtained after the activation of an integrated intracellular signaling cascade: the “Unfolded Protein Response” (UPR).
UPR is controlled by three sensor proteins: PERK (PKR-like ER kinase), IRE1α (inositol-requiring transmembrane kinase/endoribonuclease 1α) and ATF6 (activating transcription factor 6). Under normal conditions, these proteins are associated with BiP or GRP78 (78 KDa glucose-regulated protein) and thus remain inactive. Although the mechanism behind the detection of the unfolded protein accumulation in the ER by PERK and IRE1α remains obscure, Carrara et al. [11] proposed that the ATPase domain of BiP interacts with PERK an IRE1α, which dissociates when an unfolded protein binds to the canonical substrate binding domain of BiP. Therefore, under ER stress, BiP is released, and UPR cascade is activated after further dimerization and autophosphorylation of PERK and IRE1α, as well as regulated intramembrane proteolysis of ATF6 (Figure 1).
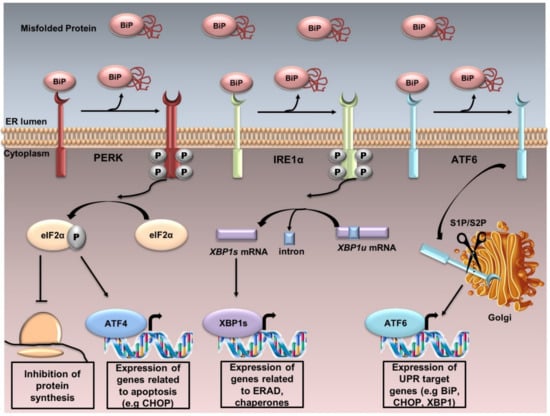
Figure 1.
The UPR. UPR is controlled by three sensor proteins: PERK, IRE1α and ATF6. Under normal conditions, these proteins are associated with BiP and thus remain inactive. Under ER stress, BiP is released from these sensor proteins, and UPR cascade is activated after further dimerization and autophosphorylation of PERK and IRE1α, and regulated intramembrane proteolysis of ATF6. PERK activation induces the phosphorylation of eIF2α. Phosphorylated eIF2α reduces the global protein synthesis and promotes the translation of the activating transcription factor 4 (ATF4) which upregulates the genes related to apoptosis. IRE1α dimerizes, autotransphosphorylates, and its endoribonuclease activity enables the splicing of the unspliced X-box binding protein 1 XBP1u to the spliced XBP1s. XBP1s upregulates the transcription of several genes involved in UPR and ERAD. ATF6 is localized at the ER in unstressed cells. Under ER stress, ATF6 translocates from the ER to the Golgi, where it is cleaved sequentially by the enzymes site 1 protease (S1P) and site 2 protease (S2P). Active ATF6 is transferred into the nucleus, where it binds the promoters of several genes involved in UPR (such as C/EBPα-homologous protein (GADD153 or CHOP), BiP, XBP1).
PERK is a type I ER transmembrane protein, containing a cytoplasmic serine/threonine protein kinase domain. PERK activation induces the phosphorylation of the α subunit of eukaryotic translation initiation factor (eIF2α) at the residue serine 51. Phosphorylated eIF2α (p-eIF2α) reduces the global protein synthesis and promotes the translation of ATF4 because of a longer resident time on its first uORF (upstream open reading frame) of the translation initiation complex. ATF4 upregulates the genes related to apoptosis, including the pro-apoptotic factor GADD153 or CHOP. The PERK/ATF4/CHOP signaling pathway plays a pivotal function in inducing cell apoptosis; PERK−/−, ATF4−/− cells and eIF2α (Ser51Ala)−/− cells fail to induce CHOP during ER stress [12].
IRE1 is also a type I ER transmembrane protein which senses ER stress and has serine/threonine kinase and endonuclease activities. The mammalian genome encodes for two isoforms: IRE1α and IRE1β [13]. Most mammalian UPR research is conducted by IRE1α. The accumulation of unfolded proteins in the ER stimulates IRE1α oligomerization and autophosphorylation, activating the endoribonuclease activity enabling the splicing of Xbp1 mRNA through the cleavage of a 26-nucleotide sequence. This results in a frame shift of the unspliced form (XBP1u), producing eventually an active transcription factor, the spliced form (XBP1s). XBP1s upregulates the transcription of several genes involved in UPR and ERAD. Under prolonged stress, IRE1α promotes cell death by activating the apoptotic-signaling kinase-1 (ASK1), followed by the activation of downstream kinases, Jun-N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38 MAPK), leading to apoptosis [14]. Furthermore, IRE1 RNase activity is involved in the regulated IRE1-dependent decay (RIDD), known to selectively degrade ER-associated mRNAs coding secretory or membrane proteins in order to unburden the protein load of the ER [15,16].
ATF6 is the third transmembrane mediator of UPR. It is an ER-associated type II transmembrane protein. Two isoforms have been described: ATF6α and ATF6β [17]. ATF6α has a higher transcriptional activity than ATF6β. Under ER stress, ATF6 translocates from the ER to the Golgi, where it is cleaved sequentially by S1P and S2P. Active ATF6 is transferred into the nucleus, where it binds the promoters of several genes involved in UPR (such as CHOP, BiP, XBP1) and induces target-gene transcription [18].
Overall, UPR mediators are involved in maintaining ER homeostasis. Excessive or long-term accumulation of misfolded proteins leads to the failure of UPR’s adaptive responses and the initiation of the regulated cell death.
2.1. Interaction between UPR, Protein Aggregation and Neurodegeneration
Misfolded proteins may lose their physiological activity, acquire neurotoxicity and lead to chronic brain inflammation [19]. Neuronal cells are particularly sensitive to protein misfolding, thus, excessive misfolding and aggregation lead to disrupted function of synapses, apoptosis and selective neuronal death [20] (Figure 2).
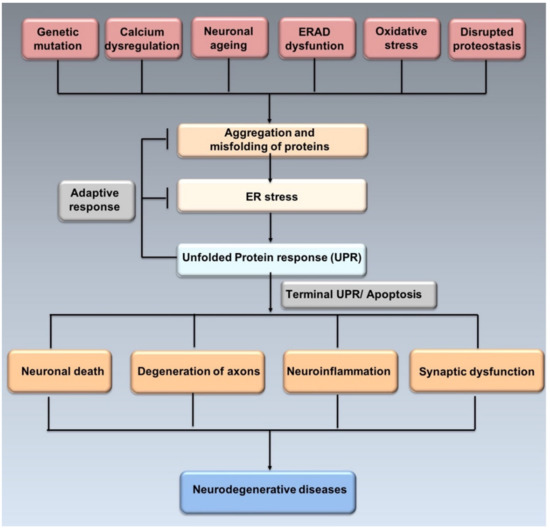
Figure 2.
ER stress, UPR and neurodegeneration. Genetic mutations, ageing, oxidative stress, disrupted proteostasis and other stimuli may induce the misfolding and aggregation of proteins leading to ER stress. In order to resolve ER stress, adaptive response through UPR is activated. However, prolonged ER stress induces apoptosis affecting neurons and synaptic funcion, thus leading to neurogenerative diseases (adapted from Hetz et al., 2017 [21]).
ER stress mediates abnormal neuronal death in psychiatric diseases, such as schizophrenia, depression and post-traumatic stress disorder (PTSD) [21]. Evidence from transgenic animal models, neuropathological and genetic studies suggests that many neurodegenerative diseases are caused by the misfolding, aggregation and accumulation in the brain of an underlying protein (Figure 3) [22].
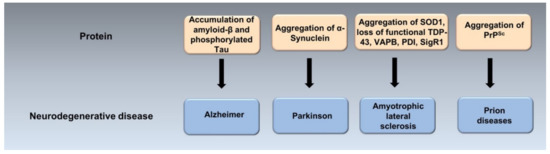
Figure 3.
Protein misfolding and aggregation in the pathogenesis of neurodegenerative diseases. The accumulation of misfolded and aggregated particular proteins may play a crucial role in the pathogenesis of neurodegenrative diseases called protein misfolding disoders (PMDs). PMDs include Alzheimer’s (AD), Parkinson’s (PD), Amyotrophic lateral sclerosis (ALS) and prion diseases, among others. They are caused by misfolding and aggregation of amyloid-β and p-tau in AD, α-Synuclein in PD, SOD-1, TDP-43, VAPB, PDI, SigR1 in ALS and PrPSc in prion diseases.
Accumulating evidence suggests that UPR mediators are directly involved in the physiopathology of protein misfolding disorders (PMDs) [23], thus ER stress markers have been described in neurodegenerative diseases. Interestingly, in patients’ brains affected with PMDs, ER stress markers often co-localize with protein aggregates [24,25]. Different UPR markers such as p-IRE1, active ATF6α, p-PERK and p-eIF2α, BiP and CHOP were found in patients with neurodegenerative diseases including Alzheimer’s [26], Parkinson’s [27,28], Amyloid lateral sclerosis (ALS) [29], prion diseases [30] among others [31]. p-PERK and p-eIF2α were detected in post mortem brain tissues of patients with different neurodegenerative diseases and this was associated with the accumulation of misfolded and aggregated protein [23,32]. BiP, phosphorylated forms of PERK, IRE1α and eIF2α were found in Alzheimer’s disease neurons and substantia nigra of Parkinson’s disease patients [30]. Furthermore, UPR markers were found to be overexpressed in spinal cord samples of ALS patients, in striatum, parietal cortex and caudate putamen of Prion disease patients [30].
Neuronal protein aggregates lose their function leading to the development of degenerative disorders. The accumulation of protein aggregate generates ER stress, followed by the activation of UPR. First, an adaptive response will start to alleviate the ER protein overload and preserve neuronal viability. Second, if ER stress persists, the ER stress-mediated cell death program will lead to neuronal loss. Intriguingly, each aggregated protein has a distinct mechanism of action, so different molecular mechanisms contribute to the imbalance of ER proteostasis and therefore neurodegeneration [21].
Finally, although misfolded proteins are implicated in the pathogenesis of neurodegenerative diseases, the exact relationship remains unclear. Additionally, whether ER stress plays a central role in the neurodegenerative diseases remains controversial. Some researchers have suggested that ER stress may play only a minor role in the mechanisms of neurodegeneration [33,34]. This review aims to discuss most recent findings relating PMDs, ER stress and UPR.
2.2. Interaction between ER Stress, Autophagy and Neurodegeneration
Autophagy is an important mechanism delivering damaged organelles, abnormal and misfolded proteins to the lysosome for degradation and leading to the recycling of the resulting macromolecules [35,36]. It plays an essential role in tissue remodeling, cell survival and regeneration [37]. Many studies in several model organisms have shown that the inactivation of autophagy genes leads to extensive cell death and to the presence of unnecessary components in the cytoplasm [37]. Neurons are vulnerable to impairments in autophagy; autophagic activity is important for maintenance of neuronal functioning since these cells are unable to dispose of aggregated proteins [38,39]. ER stress in chronic neurodegenerative disorders often stimulates autophagic activities, however, failure in clearing the aggregated proteins and the impairment of UPR or autophagy lead to the accumulation of misfolded proteins and the progression of neurodegeneration [40]. Impairment of autophagy via the knockdown of AuTophaGy(ATG)-related proteins ATG1, -2, -6, or -18 impeded synaptic development [41], impaired axonal integrity via altering microtubules stability and underlaid the onset and progression of various neurodegenerative disorders [42]. Autophagy-deficient mice showed neuronal defects and an impairment of autophagy was found to be associated with neurodegenerative diseases such as ALS, PD, AD and prion diseases [39,40].
ER stress may regulate autophagy via the PERK/eIF2α/ATF4 signaling pathway [43]. It was shown that ATG5 and ATG7 connect autophagy with ER stress through PERK signaling [44]. In response to ER stress, ATF4 was reported to guide the induction of autophagy gene transcription such as Beclin-1 (BECN1), microtubule associated protein 1 light chain 3 beta (MAP1LC3B), ATG5, ATG7 and ATG12 [45]. The eIF2α-kinases, GCN2 and PERK, ATF4 and CHOP were found to increase the transcription of a set of genes implicated in the formation, elongation and function of the autophagosome [45]. eIF2α dephosphorylation inhibition accelerated human cell death in an ER stress and autophagy-dependent manner [46]. These findings show that PERK pathway is pivotal for autophagy.
Moreover, IRE1α was studied in the context of autophagy and neurodegenerative diseases. IRE1/XBP1 pathway may influence the pathogenesis of neurodegenerative diseases. XBP1s acts as a transcription factor activating Beclin-1, the mammalian ortholog of the yeast autophagy-related gene 6 (Atg6) [47]. In addition, XBP1-FoxO1 (Forkhead box protein O1) interaction regulates ER stress-induced autophagy [48]. Blocking IRE1 or ATG7 expression was associated with dopaminergic neuronal loss, progressive locomotor impairment, shorter lifespan and the progression of PD, these findings show that IRE1 pathway couples ER stress to autophagy-dependent neuron death [49]. An interplay between IRE1/XBP1s and IRE1/TRAF2 (TNF receptor-associated factor 2)/ASK1/JNK (c-Jun N-terminal kinase) with autophagy was found. A recent study showed that in a Parkinson’s-like neurological disorder, IRE1 signaling pathway mediated the activation of JNK signaling via the formation of the ASK1-TRAF2 complex which can initiate the autophagy [50].
The ATF6 pathway was shown to be involved in interferon-gamma (IFN-γ)-induced activation of death-associated kinase 1 (DAPK1), an important regulator of cell death and autophagy [51]. Additionally, ATF6-mediated upregulation of CHOP, XBP1 and GRP78 contributes to ATF6-induced autophagy [52]. Less attention has been paid to ATF6 pathway involvement in autophagy; thus, further investigation is needed on this topic.
Finally, increasing evidence has indicated that autophagy and ER-membrane communication at membrane contact sites are closely related to neurodegenerative disorders, such as PD, AD, and ALS [53]. Vacuole membrane protein 1 (VMP1), an ER-localized metazoan-specific protein, interacts with Beclin-1 and plays important roles in the formation of autophagosomes and communication between the ER and other organelles [54].
3. ER Stress in Neurodegenerative Diseases
3.1. Alzheimer’s Disease (AD)
3.1.1. ER Stress in AD
AD is a devastating neurodegenerative disease, affecting more than 25 million individuals worldwide, and characterized by progressive decline of cognitive functions. At the neuronal level, it involves excitotoxicity, neuronal oxidative stress, deregulation of intracellular signaling pathways, synapse damage/loss [55] and neurodegeneration in different brain regions among others within frontal cortex, the hippocampus and the basal forebrain [56]. The mechanisms leading to AD are complex, related to changes in synaptic transmission, an altered calcium homeostasis, increased ER stress and a chronic state of neuroinflammation [57]. The neuropathological hallmarks of AD are the accumulation of hyperphosphorylated tau and extracellular plaques consisting of amyloid-β (Aβ) peptides [57,58]. While Tau is a stabilizer of neuronal microtubules, phosphorylated tau (p-tau) is not and tends to aggregate into intracellular neurofibrillary tangles [59]. The amyloid β-precursor protein (APP) is cleaved by the β-secretase (β-APP cleaving enzyme 1; BACE1) and γ-secretase complexes to generate Aβ peptides, insoluble plaques, fibrils or soluble and diffusible oligomers that are described as highly neurotoxic leading to neurodegeneration and apoptosis [60]. γ-secretase is a membrane-associated complex consisting of the following four different proteins: presenilin-1/2 (PS1/2), nicastrin, Aph1 and Pen2 [61]. The catalytically active site of γ-secretase resides within PS1/2. Mutations in APP and in the Presenilin’s, PS1 and PS2 are associated with increased aggregation of the Aβ peptides in the brain’s parenchyma through an increased overall production of all Aβ species (e.g., APP duplications or APP mutations located around the β cleavage site) or production of a more aggregated form of the Aβ peptide, leading to AD [62]. During AD, the continuous accumulation of Aβ or p-tau results in a drastic alteration of ER calcium homeostasis, abnormal protein folding and ER stress. Tau has been shown to block the ERAD pathway leading to the accumulation of misfolded proteins in the ER lumen [63]. Aβ oligomers interacts with neuronal N-methyl-D-aspartate receptors (NMDA-Rs) and induces the disruption of the cytosolic calcium balance, provoking ER stress-dependent cell death, synaptic depression and spine elimination [64]. Aβ peptides neurotoxicity is related to ER stress-mediated apoptosis associated with ASK1 and JNK activation [65].
Several studies have noted the occurrence of abnormal levels of ER stress in the human AD brain. Under mild ER stress, UPR has a proadaptive role. However, in long-termed ER stress, UPR-pro-apoptotic branch is activated, which is considered a possible cause of neurodegeneration. BiP, along with other chaperones such as Hsp72, and Hsp73, Grp94, PDI and calreticulin were found upregulated in cerebrospinal fluids and AD brains [26,66]. Phosphorylated forms of PERK and eIF2α are markedly increased in AD brains especially in the hippocampal pyramidal cells and the frontal cortex [32,67]. This phosphorylation can be activated by the accumulation of tau aggregates [26]. The activation of PERK was shown to be associated with increased expression of the transcription factor ATF4 and BACE1 [68], since their mRNAs contain upstream open reading frames allowing a higher translation rate after eIF2α phosphorylation [69]. PERK insufficiency was associated with a decrease in BACE1 expression, thus reduced levels of Aβ peptides and plaque burden in a mouse model of AD [69]. Treating post-mortem frontal cortex tissues with salubrinal, an inhibitor of p-eIF2α phosphatase PP1c, directly increased BACE1 and Aβ production in primary neurons [70]. The presence of Aβ oligomers in the hippocampus was associated with increased ATF4 expression in the axonal compartment; ATF4 is a key regulator of the neuronal plasticity and hippocampal-dependent long-term spatial memory [71].
Furthermore, a positive correlation between the progression of AD histopathology and the activation of IRE1 was detected in human brain tissue. Genetic ablation of the RNAse domain of IRE1 in the nervous system significantly reduced amyloid deposition, the content of Aβ oligomers, and the neuronal loss. It is worth noting that IRE1 deficiency restored the learning and memory capacity of AD mouse model [72]. Moreover, a polymorphism in the promoter of the transcription factor XBP1 was identified as a risk factor of Alzheimer’s disease [73]. Studies on AD transgenic animal models have shown that XBP1 is linked to ADAM10 transcriptional regulation; ADAM10 (ADAM Metallopeptidase Domain 10) is the main α-secretase that cleaves APP [74]. Furthermore, XBP1 can reduce the expression of BACE1, through HRD1, leading to the reduction in Aβ plaques [75] and can upregulate the expression of catalytic components of the ERAD machinery [76].
The role of ATF6 in AD has not been reported until recently where Du et al. found that in an AD mouse model, the expression of ATF6 was reduced. ATF6 was found to reduce the expression of APP to suppress the Aβ level, downregulate the promoter activity and expression of BACE1, and protect the retention of spatial memory in AD model mice [77].
On the other hand, as a consequence of prolonged ER stress, an elevation of pro-apoptotic UPR components has been found in AD brains, likely causing neurodegeneration. The main transcription factor linking ER stress to apoptosis, CHOP, was found upregulated in the brains of AD patients, along with downstream effectors such as caspase-12, and GADD34 [55]. ER stress-mediated CHOP activation plays a central role in the triggering of AD pathological hallmarks. Upregulation of CHOP generates ROS, oxidative stress, high level of Aβ oligomers, inflammation of the neurons and eventually neuronal cell death via apoptosis [78].
Finally, the involvement of UPR in AD is still considered debatable, since in the 5XFAD (familial Alzheimer’s disease) transgenic mice for example, having enhanced expression of APP and PS1, UPR was not activated. The expression of BiP, p-IRE1, p-eIF2α, ATF4 and CHOP was not significantly elevated as compared to the non-transgenic control mice, suggesting that ER stress might not be a specific hallmark of AD, or at least is not induced by overexpression of APP and PS1 [34]. Additional studies are needed to further confirm the involvement of UPR in AD.
3.1.2. ER Stress, Neuroinflammation and AD
A positive feedback loop exists between ER stress and inflammation, with clear implications for neurodegeneration and AD [55]. The three pathways of the UPR increase the production and the release of pro-inflammatory mediators such as the transcription factor NF-κB, known to activate the transcription of TNFα, IL-1, IL-6 and IL-8. Activated NF-κB regulates the expression of specific genes, including isoforms of the nuclear proto-oncogene SET, known to be elevated and mislocalized in the neuronal cytoplasm in brains of AD and directly implicated in the pathogenesis of AD [79]. In this context, p-IRE1 binds to the protein TRAF-2 and recruits the IKK (IκB kinase) complex, leading to the phosphorylation of IκB and resulting in its degradation and NF-κB activation [80]. In addition, the TRAF2-IRE1 complex may also bind ASK-1 to activate the JNK pathway, promoting inflammatory signaling through the transcription factor known as AP-1. JNK-AP1 signaling pathway is responsible for Aβ-induced neuroinflammation in an Alzheimer’s affected brain [81]. P-PERK, via P-eIF2α, inhibits the expression of IκB leading to an additional activation of NF-κB [55]. Furthermore, in the mouse brain, ER stress-induced activation of JAK1/STAT3 via P-PERK leads to neuroinflammation [82]. PERK may directly phosphorylate and activate GSK-3β (glycogen synthase kinase 3β), leading to the hyper-phosphorylation of tau, increased Aβ generation and deficits in learning and memory accompanied with neurodegeneration [83,84]. The role of ATF6 in proinflammatory action in brain pathology is under reported. It was shown that ATF6 siRNA abrogated the ER stress-mediated pro-inflammatory through protein kinase B(Akt)/NFκB activation [65] and the CREBH (cyclic adenosine monophosphate (cAMP)-responsive element-binding protein H) transcription factor [55].
3.2. Parkinson’s Disease (PD)
PD is a progressive neurodegenerative disease mostly diagnosed at a late stage of progression. PD comes after Alzheimer’s disease as the second most prevalent neurodegenerative disease [85]; it mainly affects people over 40 with a universal prevalence of 2% in men and 1.3% in women [86,87,88,89]. Tremor, impaired balance, posture and motion are the main symptoms of PD, and which progress with the severity of the disease. PD cases are mainly sporadic, only 10–15% of cases are caused by mutations in a variety of genes including SNCA/PARK1 (encoding α-synuclein (α-SYN)), Parkin (PRKN), Parkinson’s disease protein 9 (PARK9/ATP13A2), Parkinson’s disease protein 7 (PARK7/DJ1), leucine-rich repeat kinase 2 (LRRK2), PTEN-induced putative kinase 1 (PINK1) and among other genes [90].
PD is generally characterized by two major hallmarks: (1) selective loss of dopaminergic (DA) neurons in substantia nigra pars compacta (SNpc), the region of the brain implicated in movement and muscle contraction control, and (2) the accumulation of misfolded, aggregated alpha-synuclein (α-SYN) fibrils within neuronal somas called Lewy bodies (LBs) or dendrites and axons as Lewy neurites (LNs) [91]. α-SYN is highly expressed in the presynaptic terminals of the CNS neurons but can also be found in peripheral tissues and blood [92,93]. A53T, A30P and E46K mutations of the SNCA gene, duplication and triplications of the wild-type gene were identified as a hereditary cause of PD [94,95,96], they act as dominant variants inducing the disorder. α-SYN monomers tend to aggregate, β-sheet conformation elongates into insoluble protofibrils and fibrils [27]. Misfolded α-SYN spreads from cell-to-cell through interconnected circuits of the nervous system [97], induces neuronal damage and has been associated with many neurodegenerative diseases other than PD, referred as α-synucleinpathies.
Accumulating evidence supports the role of ER stress as a principal player in α-SYN toxicity and mediated dopaminergic neurons cell death. Numerous studies have shown that α-SYN aggregates interact with BiP, leading to the activation of the UPR signaling pathway when it binds to misfolded proteins [98,99]. In human brain tissues, ER stress markers, such as phosphorylated PERK and phosphorylated eIF2α, were detected in neuromelanin containing dopaminergic neurons of the substantia nigra of PD patients but not in control cases’ tissues [100]. Moreover, α-SYN was found to be more abundant in ER/microsomes fractions of human and mice PD brain tissues as compared to non-PD controls [101]. In an A53T transgenic mouse model, α-synucleinopathy coincides with the induction of ER chaperones and abnormal UPR in pathologic neurons. This is supported by the increase in ER stress associated polyubiquitin chains accumulation and caspase-12 activation. Interestingly, administration of an ER stress inhibitor, salubrinal, significantly ameliorated the onset of α-synucleinopathy in A53T transgenic mice and in an adeno-associated virus-transduced rat model of A53TαS-dependent dopaminergic neurodegeneration [101]. It was found to protect against cell death and reduce A53T cytotoxicity showing that ER stress is directly related to α-SYN mediated cell death [102,103]. In differentiated rat sympathetic-like neuron cells (PC12), overexpression of A53T α-SYN led to elevated intracellular ROS activity and decreased proteasome activity at a first stage followed by ER stress activation as a final protective stage before the induction of a caspase-dependent cell death. ER stress activation was detected by the elevation of eIF2 phosphorylation and the upregulation of ER stress related genes (GRP17 and GADD153).
In dopaminergic differentiated SH-SY5Y cells (SH-SY5Y+), glucose deprivation induced α-SYN/ER stress-dependent cell death. In this model, α-SYN acts as a stress sensor where glucose deprivation leads to its overexpression and aggregation resulting in its interaction with GRP78/BiP, and consequent activation of the PERK-dependent pathway and ATF4/CREB-2 transcription factors [98]. Overexpression of ATF4/CREB-2 was detected in the substantia nigra of SYN120 transgenic mice as compared to control mice [98]. The role of IRE1-XBP1 pathway in linking ER stress to PD is controversial. While in animals injected with 6-hydroxydopamine (PD-inducing neurotoxin), gene therapy using the active form of XBP1 was found to be neuroprotective [104]. Recently, in a PD model in the fruit fly Drosophila melanogaster, IRE1 activation was found to cause cell loss in photoreceptor neurons in an XBP1-independent manner, which can be prevented by the inhibition of autophagy (knockdown of ATG genes, ATG7 or ATG8B) [49].
On the other hand, using a genome-wide overexpression screen approach in yeast, Cooper AA. and colleagues showed that α-SYN accumulation causes ER stress by inhibiting the ER-Golgi trafficking [105,106]. It was reported that this trafficking impairment is caused by either a direct interaction with the ras-associated binding 1 (RAB1) GTPase [105,107] or by ATF6. In fact, co-expression of RAB1 with α-SYN rescued the loss of dopaminergic neurons in the animal models of Drosophila, C. elegans and in primary rat midbrain neurons [105]. Moreover, α-SYN inhibited ATF6 activation via coat protein complex II (COPII)-mediated ER–Golgi transit induced upon ER stress, leading to the attenuation of its cytoprotective effect, and apoptosis [108].
Another proposed model of ER stress induction by α-SYN aggregation is through ER Ca2+ homeostasis destabilization. In fact, α-SYN aggregates activate ER calcium pump SERCA (Sarco/endoplasmic reticulum Ca2+-ATPase) in neurons, leading to an alteration of the calcium metabolism, ROS production and apoptosis [109]. In mice, knockout of the CaBP-9k gene, a calcium binding protein expressed in the cytosol of dopaminergic neurons in the brain, increased α-SYN in dopaminergic neurons and induced ER stress-mediated apoptosis in neurons. Treatment of CaBP-9k KO mice with TUDCA (tauroursodeoxycholic acid), a pharmacological ER stress inhibitor, restored the expression of ER stress markers and cleaved caspase-12 to normal levels [110].
3.3. Amyotrophic Lateral Sclerosis (ALS)
ALS is an adult neurodegenerative disease marked by the degeneration of motor neurons in the cortex, spinal cord and brain stem resulting in muscle atrophy, paralysis, weakness and spasticity [111]. Most people with ALS have the sporadic form. Only a small proportion of people, estimated at 5 to 10 percent, have a familial ALS (fALS).
Many genes have been identified as ALS disease-causative, such as protein disulfide isomerase (PDI) and the superoxide dismutase1 (SOD1), TAR DNA-binding protein (TARDBP or TDP-43), fused in sarcoma (FUS/TLS), chromosome 9 open reading frame 72 (C9ORF72), vesicle-associated protein-associated protein B (VAPB) and Sigma-1 receptor (SIGR1) among others [24,111]. These mutations are linked to alterations in mRNA metabolism, proteostasis and aggregation of the affected protein, leading to neurotoxicity and neuroinflammation, thus ALS [111,112]. Many groups supported the involvement of ER stress in the pathophysiology of ALS, both in patients and animal models [113,114,115,116]. The upregulation of UPR markers was detected in ALS patients [113,114,115,116].
Approximately 20% of familial ALS, and 1–2% of all cases are caused by mutations in the gene encoding Sod1 [117]. SOD1-mutant female ALS patients had a better survival rate than males [118]. The involvement of this mutation in ALS is the most investigated in research. Transcriptional analysis of motor neurons derived from induced pluripotent stem cells (iPSCs) of patients carrying Sod1 mutation demonstrated that ER stress was high and UPR markers were upregulated [119]. Lumbal spinal cord sections’ examination of G93A SOD1 mice showed an upregulation of PERK, IRE1 α and ATF6 [119]. UPR markers BiP, calnexin and PDI were found co-localized with mutant SOD1 in post-mortem tissue [21], where it might sequester these vital chaperones [120]. Alterations in ER morphology have been found in ALS patients and SOD1 mouse model, including fragmentation of the rough ER, irregular distension of cisternae and detachment of ribosomes [121]. Mutant SOD1 interacts with Derlin-1, a component of ERAD machinery, triggers ER stress through dysfunction of ERAD, activates ASK1 and motor neuron death [122]. Initial studies indicated that Perk haploinsufficiency accelerates experimental ALS in Sod1 transgenic mice, these mice had a reduced capacity to turn down synthesis of misfolded SOD1, leading to an early overload of the UPR [123]. Furthermore, the genetic ablation of GADD34 [124] and the inhibition of eIF2α phosphatase [125] delayed the disease onset and prolonged motor neuron survival. However, more recent studies performed on SOD1 mice showed that neither PERK haploinsufficiency, nor genetic UPR enhancement via ablation of GADD34, is beneficial for mutant SOD1-induced motor neuron disease [126]. ATF4 ablation in mutant SOD1 G85R mice led to the protection against ALS, these mice appeared more resistant to ALS than mutant mice expressing normal levels of ATF4, possibly because of the reduced levels of apoptosis components, such as CHOP [127]. The role of IRE1 in ALS was shown by studying the homeodomain interacting protein kinase 2 (HIPK2), an essential component of the IRE1-ASK1 apoptotic cascade activating JNK under ER stress [128]. In SOD1 G93A mice, loss of HIPK2 was associated with delayed disease onset, reduced cell death in spinal motor neurons, and improved survival [128]. Furthermore, the ablation of XBP1 led to reduced motor neuron death and aggregation of mutant SOD1, mainly due to a homeostatic link between the UPR and the autophagy pathway [114]. The role of ATF6 in ALS is under reported. However, it was found that ATF6 levels were elevated in a SOD1 G93A mouse model, suggesting that they play an important role in the progression of ALS [129].
The ER chaperones PDI, have a broad protective role in ALS. In fact, an increased amount of PDI was detected in the cerebrospinal fluid and in the blood of ALS patients indicating that any dysfunction of this chaperone contributes to the ALS pathology and aids in diagnosis [130]. A total of nine PDIA1 missense variants and seven PDIA3 missense variants were identified in ALS patients. Single nucleotide polymorphisms (SNPs) in both genes were also found enriched in ALS cases [131]. PDI variants were found to induce motor defects associated with a disruption of motoneuron connectivity and impaired dendritic outgrowth in motoneuron cell culture models [132]. Additionally, the over-expression of ALS-linked mutant TDP-43 induced ER stress pathways in neuroblastoma cells and an interaction between PDI and TDP-43 was found in transfected cell lysates and the spinal cords of mutant A315T TDP-43 transgenic mice [133]. The phosphorylation of eIF2α was found to be abnormally upregulated by TDP-43 aggregates in Drosophila and therapeutic modulation of eIF2α-phosphorylation attenuated TDP-43 toxicity in ALS [134]. In ALS models, FUS, which is normally located in the nucleus, translocates to the cytoplasm and forms inclusions; this has been linked to ER stress. Mutant FUS also colocalized with PDI in human ALS lumbar spinal cords and mutant FUS-linked familial ALS tissues [135]. In addition, C9ORF72 mutation, a common cause of familial ALS, was found to be associated with an activation of the UPR; this mutation is associated with a vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity (AMPAR) known to be dysregulated in lower motor neurons in all ALS cases [136,137]. Mutations in genes encoding for ER proteins have been also linked to familial and sporadic cases of ALS, such as the mutation E102Q in the ER chaperone gene SIGR1; this mutation was found to lead to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins [138]. Furthermore, the disruption of the vesicle-associated protein-associated protein B (VAPB) was associated to ALS. The mutant protein VAPB P56S affects intracellular Ca2+ storage, Ca2+ signaling capacities and inhibits ATF6 and XBP1 [139]. Patients with ALS-associated VAPB mutations present chronic ER stress, synaptic loss and cell death [140]. Finally, alteration of DNA methylation and histone modification has been reported in ALS [141]. However, the picture of the impact of epigenetic modifications on the clinical aspects of ALS is still not clear [142].
3.4. Prion Diseases
Prion diseases or transmissible spongiform encephalopathies (TSEs) are a family of rare progressive neurodegenerative disorders that occur in both humans and animals. They include Creutzfeldt–Jakob disease (CJD), Gertmann–Straussler–Sheinker syndrome and fatal familial insomnia in humans, scrapie in sheep and goat, bovine spongiform encephalopathy (BSE, more commonly known as “mad cow disease”) in cattle, and chronic wasting disease (CWD) in cervids. Although there are three different etiologies of prion diseases, infectious, sporadic and hereditary, however, they are all characterized by the formation of an abnormal folding of the cellular prion protein (PrP), leading to the development of an abnormal protease resistant form [143]. In this context, the main molecular event in the pathogenesis of prion diseases is the conversion of the normal cellular α-helical prion protein (termed PrPC) into the pathological, β-sheet-rich, misfolded form known as PrPSc (for scrapie associated PrP) [144]. PrPSc is a marker for prion infection [145]; its deposition and accumulation are not intrinsically toxic [146]. It was proposed that the accumulation of PrPSc might stabilize ER misfolded subtypes inducing toxicity through different signaling cascades such as caspase activation, ER stress, autophagy, and calcium dysregulation—features found in prion diseases [146,147]. ER stress plays a significant role in the pathogenesis of prion diseases, not only by leading to the accumulation of PrPSc levels but also by increasing the misfolded form of PrPC susceptible to prion conversion [148,149,150,151].
The involvement of ER stress in prion diseases was shown by the upregulation of the ER chaperone Grp58/ERp57 (ER protein) in parallel with PrPSc in scarpie-infected mice [152], Grp78/BiP, Grp94, PDIA1 and Grp58/ERp57 in the cortex of patients affected with variant CJD and sporadic CJD [153], and the heat shock protein 70 (HSP70) in CJD patients and scarpie-infected rodents [154,155]. Prion infection of mice lacking HSP70 showed accelerated disease progression suggesting that HSP70 may play an important role in suppressing or delaying prion disease progression [154].
Mainly PERK but also IRE1α and ATF6 pathways were shown to be involved in the prion disease. The modulation of UPR signaling prevented neuronal loss and increased survival in prion-diseased mice [54]. Recently, astrocytes from prion-infected mice presented increased p-PERK levels and neuronal degeneration; however, the authors were not able to determine if PERK signaling was activated because of the accumulation of misfolded prion protein or not [156]. The PERK pathway was reported to be activated in the hippocampus of prion-infected mice and those overexpressing PrPc [157]. PrP accumulation induced an increase in p-eIF2α, leading to a reduction in the expression of relevant synaptic proteins, synaptic failure and neuronal loss in prion-diseased mice [23,157] and CJD patients [143]. The modulation of astrocytic PERK-eIF2α signaling in mice with prion disease was profoundly neuroprotective [54]. Interestingly, targeting p-PERK signaling in astrocytes during prion disease was alone sufficient to prevent neuronal loss and prolong survival thus UPR over-activation is neuroprotective [54]. Furthermore, in prion infected animals, gene therapy to deliver eIF2α phosphatase [157], oral treatment with an inhibitor of PERK [158] and treatment with ISRIB [159], a compound that blocks the consequences of p-eIF2α, provided neuroprotection, attenuated disease progression and protected against prion disease. Additionally, prion infection in mice induced the splicing of XBP1 mRNA and the activation of the stress kinases JNK and ERK (extracellular signal-regulated kinase) through IRE1α [160,161]. In a cellular model of prion disease, XBP1s overexpression prevented PrP misfolding [149,162], whereas a dominant negative form of XBP1 and IRE1α significantly increased PrP aggregation [149,162]. The neuroprotective activity of XBP1 in prion diseases was tested in mice lacking XBP1 in the brain; these animals showed normal prion replication and neuropathology, suggesting that other UPR pathways may compensate the absence of XBP1 [161]. Furthermore, in a cellular model of prion disease, the overexpression of ATF4 or an active mutant form of ATF6 prevented PrP aggregation [115]. Overall data suggest that prion diseases are associated with ER stress response, where the PERK pathway is mostly involved. Future studies are needed to investigate in depth the role of IRE1 and ATF6 in the pathogenesis of these diseases.
4. Therapeutic Approaches: Chemical Compounds Targeting the UPR Pathways
Targeting the UPR pathway may be a valuable therapeutic strategy to control ER stress response associated with disorders such as neurodegenerative diseases. Recently, many chemical compounds and small molecules capable of targeting the UPR through different molecular mechanisms have been identified and tested. A selection of some of the most widely used drugs as promising therapeutic candidates will be presented in this review.
4.1. Chemical Chaperones
The use of chemical chaperones is a promising approach to alleviate ER stress in a range of diseases. The antidiabetic compound azoramide and BIP/GRP78 inducer X (BiX) were found to improve ER protein-folding capability in multiple systems [163,164]. In addition, 4-Phenylbutyrate (4-PBA) and tauroursodeoxycholic acid (TUDCA) have shown therapeutic benefits for a wide variety of diseases, such as AD, ALS and diabetes [165,166]. Furthermore, 2-phenylimidazo[2,1-b]benzothiazole derivatives (IBTs) were recently identified as chemical chaperones in a cell-based high-throughput screen. It was shown that IBT21 binds to unfolded or misfolded proteins, inhibits protein aggregation and prevents cell death caused by induced ER stress [165].
4.2. GlaxoSmithKline(GSK) 2606414
GSK2606414 is the first-generation PERK inhibitor. In fact, this inhibitor is a promising treatment strategy against neurodegeneration [167]. The reduction in PERK expression by GSK2606414 improved neuronal excitability and cognitive function in young normal mice; the hippocampal memory in middle aged mice was restored to the normal performance levels observed in young individuals [67]. In an in vivo experimental model of Marinesco–Sjögren syndrome, the inhibition of PERK pathway via GSK2606414 delayed Purkinje cell degeneration, prolonged the asymptomatic phase and improved the motor performance during the symptomatic phase [168]. As mentioned earlier, PERK signaling is activated in PD patients and rodent models of the disease. The oral administration of GSK2606414 inhibited this pathway in the SNpc after experimental ER stress stimulation, improved motor performance and increased dopamine levels and the expression of synaptic proteins [169]. The treatment of a mouse model of prion disease with this inhibitor induced a repression of protein synthesis associated with decreased levels of p-PERK, p-eIF2α, ATF4 and CHOP, showing the neuroprotective effect of GSK2606414 [158,167]. Moreover, in the frontotemporal dementia disease, the high levels of p-PERK, p-eIF2α and ATF4 were reduced after orally treating transgenic mouse model with GSK2606414 [170]. Recently, it was found that this compound is a potential therapy of cancer since it inhibits KIT, a type III receptor tyrosine kinase (RTK), known to activate many signal transduction pathways including RAS/ERK (Extracellular Signal-Regulated Kinase), PI3K/AKT or PKB (Phosphoinositide 3-kinase/ Protein kinase B), phospholipase C, JAK/STAT (Janus kinase/signal transducers and activators of transcription), and Src kinase pathways [171]. However, it has been reported that GSK2606414 may cause side effects related to pancreatic toxicity [169] maybe due to the extent of UPR inhibition, weight loss and hyperglycemia [158].
4.3. ISRIB
ISRIB, a small molecule which reverses the phosphorylation of eIF2α [172], inhibits the downstream targets of all eIF2α kinases including ATF4, CHOP, and GADD34, but it does not induce significant translational changes [167,173]. ISRIB has a promising therapeutic potential in vivo, it was found to enhance cognitive memory processes in brain-injured mice without inducing side effects [174]. In prion-diseased mice, the inhibition of the PERK pathway using GSK2606414 was profoundly neuroprotective but toxic to the secretory tissues. However, the treatment with ISRIB was neuroprotective without inducing any adverse effect on the pancreas, but it only partially restored global protein translation, as compared with GSK2606414 [159]. Recently, in an ALS rodent model, the inhibition of PERK signaling with ISRIB, but not with GSK2606414, significantly enhanced the neuronal survival via a partial inhibition of the translation imposed by PERK and a reduction in IRE1-dependent signaling [175]. In addition to neurodegenerative diseases, ISRIB was recently found to inhibit stress response signaling in the aging lung epithelium leading to decreased apoptosis and reduced recruitment of pathogenic monocyte-derived alveolar macrophages [176]. Moreover, the inhibition of PERK pathway by ISRIB reduced ATF4 and CHOP and two other eIF2α kinases: GCN2 (general control nonderepressible 2) and HRI (heme-regulated inhibitor) [177]. Finally, despite its neuroprotective effect, ISRIB cannot be used against human neurodegenerative diseases because of its insolubility [178].
4.4. Guanabenz
Guanabenz is an alpha-2-adrenergic receptor agonist safely used to treat hypertension. It is a small molecule that inhibits GADD34-mediated dephosphorylation of p-eIF2α, leading to increased p-eIF2α levels, modulates the synthesis of proteins and increases chaperone synthesis resulting in an adaptive response under ER stress conditions in human cells [179]. Guanabenz rescued motoneurons from misfolding protein stress both in in vitro and in vivo ALS models [167]. In an ALS transgenic mouse model, Guanabenz treatment prolonged the early phase of disease and survival as compared to untreated transgenic mice [180].
4.5. Sephin1
In mouse models, Sephin1, a derivative of guanabenz, was shown to inhibit eIF2α dephosphorylation via the inhibition of GADD34, as well as inhibit the global protein translation recovery, prevent stress-induced damage in cells and delay PMD development, such as Charcot-Marie-Tooth 1B, in ALS mice models [181]. However, Crespillo-Casado et al. found that in mammalian integrated stress response (ISR) deficient cells, Sephin1’s ability to suppress neurodegeneration was not due to the inhibition of eIF2α dephosphorylation but maybe due to its ability to attenuate IRE1 branch [182]. Finally, it was recently shown that intraperitoneal treatment with Sephin1 reduced PrPSc levels and ER stress-induced PrP aggregates, and prolonged survival of prion-infected mice [147].
4.6. Trazodone Hydrochloride and Dibenzoylmethane
Trazodone hydrochloride and dibenzoylmethane were identified by the National Institute of Neurological Disorders and Stroke (NINDS) small-molecule library screening performed on 1040 drugs. Both drugs reversed p-eIF2α mediated translational attenuation and were neuroprotective in two mouse models of neurodegeneration [178]. In prion-diseased mice, both drugs restored memory deficits, abrogated development of neurological signs, prevented neurodegeneration and significantly prolonged survival. Furthermore, in an animal model of tauopathy-FTD (frontotemporal dementia), these compounds rescued memory deficits and hippocampal atrophy [178]. Finally, both compounds were not toxic to the pancreas [178].
4.7. Salubrinal
Salubrinal acts as an activator of UPR branches, raises BiP levels, selectively inhibits eIF2α dephosphorylation, resulting in increased levels of p-eIF2α [183], interferes with death/survival-related signaling pathways such as ATF4 or ASK1 [184,185] and attenuates neuronal apoptosis [183]. Salubrinal may promote amyloidogenesis thus plays a key role in the mechanisms leading to AD development and progression [70]. Moreover, it was found to be effective in several models of PD. In A53T mutant mice, salubrinal reduced the accumulation of α-SYN in the ER, extended the life span, and delayed the onset of motor dysfunction [101]. In an A53T rat model, it was found to prevent neuronal loss and alleviate the symptoms [101]. In prion mice models, administration of salubrinal exacerbated neurodegeneration and reduced survival time [157].
4.8. Kinase Inhibiting RNase Attenuators (KIRA)
KIRA are aldehyde derivatives which bind to the active site of IRE1 by interacting with the lysine 309, target the RNase activity leading to IRE1 inhibition and the blockage of XBP1 splicing [186]. Recently, it was found that KIRA inhibitors interfere with IRE1 face-to-face dimer formation thus disabling the activation of the RNase domain [187]. KIRA may be a promising compound to treat diseases involving the activation of IRE1 pathway such as neurodegenerative diseases.
4.9. N-[2-hydroxy-5-methylphenyl)-3-phenylpropanamide
N-[2-hydroxy-5-methylphenyl)-3-phenylpropanamide is a small molecule known to activate the ATF6 arm of the UPR [188]. The activation of ATF6 was found to ameliorate disease-associated imbalances in ER proteostasis and function [189]. N-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide was found to reduce the aggregation of amyloid disease-associated proteins in relevant cell models [188]. This treatment activated endogenous ATF6 through an increased processing by S1P and S2P [188], and through the modifications of a subset of ER proteins, including multiple PDIs [189].
5. Conclusions
ER stress plays an important role in many neurodegenerative disorders characterized by the accumulation of misfolded proteins and aggregates, such as AD, PD, ALS and prion diseases, among others. However, the involvement of the UPR and the mechanisms by which ER stress contributes to the pathogenesis remain not fully clear and may have contrasting and even opposing effects. This may be caused by the crosstalk between ER stress, UPR, energy control, global proteostasis and neuroinflammation. As discussed earlier, evidence from in vivo and in vitro neurodegenerative disease models showed that the accumulation of particular misfolded proteins associated with the disease leads to neuronal and synaptic dysfunction. Targeting UPR pathways to treat a range of neurodegenerative disorders is promising. Full understanding of the signaling pathways and physiological functions of UPR-related molecules will help to develop novel therapeutics. Identifying new drugs that can interfere with UPR signaling and target ER stress associated genes in different animal and cell models is crucial and will provide solid knowledge regarding the involvement of the UPR in PMD progression.
Author Contributions
R.G. and M.K. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 4-PBA | 4-Phenylbutyrate |
| AD | Alzheimer’s disease |
| ADAM10 | A disintegrin and metalloprotease domain 10 |
| ALS | Amyloid lateral sclerosis |
| AMPAR | AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor |
| APP | Amyloid β-precursor protein |
| ASK1 | Apoptotic-signaling kinase-1 |
| ATF4 | Activating transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| ATG | AuTophaGy(ATG)-related proteins |
| Aβ | Amyloid-β |
| BACE1 | β-APP cleaving enzyme 1 |
| BECN1 | Beclin-1 |
| BiX | BIP/GRP78 inducer X |
| BSE | Bovine spongiform encephalopathy |
| C9orf72 | Chromosome 9 open reading frame 72 |
| CaBP-9k | Calbindin-D9K |
| Ca2+ | Calcium |
| CHOP | C/EBPα-homologous protein |
| CJD | Creutzfeldt–Jakob disease |
| CNS | Central nervous system |
| COPII | Coat protein complex II |
| CRAC | Ca2+-release-activated Ca2+ |
| CREBH | Cyclic adenosine monophosphate (cAMP)-responsive element-binding protein H |
| CWD | Chronic wasting disease |
| DAPK1 | Death-associated kinase 1 |
| eIF2α | Eukaryotic translation initiation factor α |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation |
| ERK | Extracellular signal-regulated kinase |
| ERMES | ER-mitochondria encounter structures |
| ERp | ER protein |
| FoxO1 | Forkhead box protein O1 |
| FTD | Frontotemporal dementia |
| FUS | Fused in sarcoma |
| GADD34 | Growth arrest and DNA-damage-inducible 34 |
| GCN2 | General control nonderepressible 2 |
| GRP17 | Glycine rich protein 17 |
| GRP78 | 78 KDa glucose-regulated protein |
| GSK-3β | Glycogen Synthase Kinase 3β |
| HD | Huntington disease |
| Herp | Homocysteine-inducible ER stress protein |
| HIPK2 | Homeodomain interacting protein kinase 2 |
| HRD1 | HMG-CoA reductase degradation protein 1 |
| HRI | Heme-regulated inhibitor |
| Hsp | Heat shock proteins |
| IBTs | 2-phenylimidazo[2,1-b]benzothiazole |
| IFN-γ | Interferon-gamma |
| IKK | IκB kinase |
| IL | Interleukin |
| IP3R | IP3 receptor |
| iPSCs | Induced pluripotent stem cells |
| IRE | Inositol-requiring transmembrane kinase/endoribonuclease |
| ISRIB | Integrated stress response inhibitor |
| JAK/STAT JNK | Janus kinase/signal transducers and activators of transcription Jun-N-terminal kinase |
| KIRA | Kinase inhibiting RNase attenuators |
| LBs | Lewy bodies |
| LNs | Lewy neurites |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAM | Mitochondria-associated ER membrane |
| MAP1LC3B | Microtubule associated protein 1 light chain 3 beta |
| Mfn2 | Mitochondrial fusion protein mitofusin 2 |
| NE | Nuclear envelope |
| NF-κB | Nuclear factor-κB |
| NINDS | National Institute of Neurological Disorders and Stroke |
| NMDA-Rs | N-methyl-d-aspartate receptors |
| Osh | Oxysterol-binding homology |
| p38 MAPK | p38 mitogen-activated protein kinase |
| PARK | Parkinson’s disease protein |
| PC12 | Pheochromocytoma of the rat adrenal medulla |
| PD | Parkinson’s disease |
| PDI | Protein disulfide isomerase |
| PE | Phosphatidylethanolamine |
| PERK | PKR-like ER kinase |
| PI3K/AKT PI4P | Phosphoinositide 3-kinase/ Protein kinase B Phosphatidylinositol 4-phosphate |
| PINK1 | PTEN-induced putative kinase 1 |
| PM | Plasma membrane |
| PMDs | Protein misfolding disorders |
| Pp1 | Protein phosphatase 1 |
| PRKN | Parkin |
| PrP | Prion protein |
| PS | Phosphatidylserine |
| PS1/2 | Presenilin-1/2 (PS1/2) |
| PTSD | Post-traumatic stress disorder |
| RAB1 | Ras-associated binding 1 |
| RIDD | Regulated IRE1-dependent decay |
| ROS | Reactive oxygen species |
| RTK | Receptor tyrosine kinase |
| S1P | Site 1 protease |
| S2P | Site 2 protease |
| SERCA | Sarco/endoplasmic reticulum Ca2+-ATPase |
| Sigr1 | Sigma-1 receptor |
| SNCA | Synuclein Alpha gene |
| SNpc | Substantia nigra pars compacta |
| SNPs | Single nucleotide polymorphisms |
| SOCE | Store operated Ca2+ entry |
| SOD1 | Superoxide dismutase1 |
| STIM | Stromal-interacting molecule |
| TARDBP or TDP-43 | TAR DNA-binding protein |
| TNF | Tumor necrosis factor |
| TRAF2 | TNF receptor-associated factor 2 |
| TSE | Transmissible spongiform encephalopathies |
| TUDCA | Tauroursodeoxycholic acid |
| uORF | Upstream open reading frame |
| UPR | Unfolded protein response |
| VAP VAPB | Vesicle-associated membrane protein-associated protein Vesicle-associated membrane protein-associated protein B |
| VDAC1 | Voltage-dependent anion selective channel protein 1 |
| VMP1 | Vacuole membrane protein 1 |
| WT | Wild type |
| XBP1 | X-box binding protein 1 |
| α-SYN | α-synuclein |
References
- English, A.R.; Voeltz, G.K. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 2013, 5, a013227. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, T.; Patel, S.; Eden, E.R. Calcium signaling at ER membrane contact sites. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1853, 2012–2017. [Google Scholar] [CrossRef] [PubMed]
- Stefan, C.J.; Manford, A.G.; Baird, D.; Yamada-Hanff, J.; Mao, Y.; Emr, S.D. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 2011, 144, 389–401. [Google Scholar] [CrossRef]
- Martinvalet, D. The role of the mitochondria and the endoplasmic reticulum contact sites in the development of the immune responses. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Xia, M.F.; Zhang, Y.Z.; Jin, K.; Lu, Z.T.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 1–19. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Area-Gomez, E.; De Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Bravo-San Pedro, J.M.; González-Polo, R.A.; Fuentes, J.M.; Niso-Santano, M. ER-mitochondria signaling in Parkinson’s disease review-article. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 1–18. [Google Scholar] [CrossRef]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M.U. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 2015, 4, e03522. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Xiang, C.; Wang, Y.; Zhang, H.; Han, F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 2017, 22, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Hubbard, S.R. How IRE1 Reacts to ER Stress. Cell 2008, 132, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Hillary, R.F.; Fitzgerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 1–10. [Google Scholar] [CrossRef]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.Y.; Kaufman, R.J. ATF6α Optimizes Long-Term Endoplasmic Reticulum Function to Protect Cells from Chronic Stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Ashraf, G.; Greig, N.; Khan, T.; Hassan, I.; Tabrez, S.; Shakil, S.; Sheikh, I.; Zaidi, S.; Akram, M.; Jabir, N.; et al. Protein Misfolding and Aggregation in Alzheimer’s Disease and Type 2 Diabetes Mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1280–1293. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Remondelli, P.; Renna, M. The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front. Mol. Neurosci. 2017, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 1–13. [Google Scholar] [CrossRef]
- Hughes, D.; Mallucci, G.R. The unfolded protein response in neurodegenerative disorders—Therapeutic modulation of the PERK pathway. FEBS J. 2019, 286, 342–355. [Google Scholar] [CrossRef]
- Cabral-Miranda, F.; Hetz, C. ER stress in neurodegenerative disease: From disease mechanisms to therapeutic interventions. Endoplasmic Reticulum Stress Dis. 2017, 4, 11–26. [Google Scholar] [CrossRef]
- García-González, P.; Cabral-Miranda, F.; Hetz, C.; Osorio, F. Function in the development of neurodegenerative diseases. Front. Immunol. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed]
- Colla, E. Linking the endoplasmic reticulum to Parkinson’s disease and alpha-synucleinopathy. Front. Neurosci. 2019, 13, 1–10. [Google Scholar] [CrossRef]
- Prell, T.; Stubendorff, B.; Le, T.T.; Gaur, N.; Tadić, V.; Rödiger, A.; Witte, O.W.; Grosskreutz, J. Reaction to endoplasmic reticulum stress via ATF6 in amyotrophic lateral sclerosis deteriorates with aging. Front. Aging Neurosci. 2019, 11, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent insights into the role of unfolded protein response in ER stress in health and disease. Front. Cell Dev. Biol. 2017, 5, 1–16. [Google Scholar] [CrossRef]
- Marotta, D.; Tinelli, E.; Mole, S.E. NCLs and ER: A stressful relationship. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Stutzbach, L.D.; Xie, S.X.; Naj, A.C.; Albin, R.; Gilman, S.; Lee, V.M.Y.; Trojanowski, J.Q.; Devlin, B.; Schellenberg, G.D. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 31. [Google Scholar] [CrossRef]
- Zhao, L.; Ackerman, S.L. Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 2006, 18, 444–452. [Google Scholar] [CrossRef]
- Sadleir, K.R.; Popovic, J.; Vassar, R. ER stress is not elevated in the 5XFAD mouse model of Alzheimer’s disease. J. Biol. Chem. 2018, 293, 18434–18443. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef]
- Tettamanti, G.; Carata, E.; Montali, A.; Dini, L.; Fimia, G.M. Autophagy in development and regeneration: Role in tissue remodelling and cell survival. Eur. Zool. J. 2019, 86, 113–131. [Google Scholar] [CrossRef]
- Son, J.H.; Shim, J.H.; Kim, K.H.; Ha, J.Y.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89–98. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association between autophagy and neurodegenerative diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef]
- Birdsall, V.; Waites, C.L. Autophagy at the synapse. Neurosci. Lett. 2019, 697, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Negrete-Hurtado, A.; Overhoff, M.; Bera, S.; De Bruyckere, E.; Schätzmüller, K.; Kye, M.J.; Qin, C.; Lammers, M.; Kondylis, V.; Neundorf, I.; et al. Autophagy lipidation machinery regulates axonal microtubule dynamics but is dispensable for survival of mammalian neurons. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yan, D.Y.; Wang, C.; Ma, Z.; Deng, Y.; Liu, W.; Xu, B. Manganese activates autophagy to alleviate endoplasmic reticulum stress–induced apoptosis via PERK pathway. J. Cell. Mol. Med. 2020, 24, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Xie, W.; Yin, D.; Luo, R.; Liu, M.; Guo, F. ATG5 and ATG7 induced autophagy interplays with UPR via PERK signaling. Cell Commun. Signal. 2019, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- Yu, C.L.; Yang, S.F.; Hung, T.W.; Lin, C.L.; Hsieh, Y.H.; Chiou, H.L. Inhibition of eIF2α dephosphorylation accelerates pterostilbene-induced cell death in human hepatocellular carcinoma cells in an ER stress and autophagy-dependent manner. Cell Death Dis. 2019, 10, 418. [Google Scholar] [CrossRef]
- Hosoi, T.; Nomura, J.; Tanaka, K.; Ozawa, K.; Nishi, A.; Nomura, Y. Link between endoplasmic reticulum stress and autophagy in neurodegenerative diseases. Endoplasmic Reticulum Stress Dis. 2017, 4, 37–45. [Google Scholar] [CrossRef]
- Kishino, A.; Hayashi, K.; Hidai, C.; Masuda, T.; Nomura, Y.; Oshima, T. XBP1-FoxO1 interaction regulates ER stress-induced autophagy in auditory cells. Sci. Rep. 2017, 7, 4442. [Google Scholar] [CrossRef]
- Yan, C.; Liu, J.; Gao, J.; Sun, Y.; Zhang, L.; Song, H.; Xue, L.; Zhan, L.; Gao, G.; Ke, Z.; et al. IRE1 promotes neurodegeneration through autophagy-dependent neuron death in the Drosophila model of Parkinson’s disease. Cell Death Dis. 2019, 10, 800–815. [Google Scholar] [CrossRef]
- Liu, C.; Yan, D.Y.; Wang, C.; Ma, Z.; Deng, Y.; Liu, W.; Xu, B. IRE1 signaling pathway mediates protective autophagic response against manganese-induced neuronal apoptosis in vivo and in vitro. Sci. Total Environ. 2020, 712, 136480. [Google Scholar] [CrossRef]
- Gade, P.; Ramachandran, G.; Maachani, U.B.; Rizzo, M.A.; Okada, T.; Prywes, R.; Cross, A.S.; Mori, K.; Kalvakolanu, D.V. An IFN-γ-stimulated ATF6-C/EBP-β-signaling pathway critical for the expression of death associated protein kinase 1 and induction of autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, 10316–10321. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.M. Endoplasmic Reticulum Stress and Related Pathological Processes. J. Diagn. Tech. Biomed. Anal. 2013, 1, 1000107. [Google Scholar] [CrossRef]
- Wang, P.; Kou, D.; Le, W. Roles of VMP1 in Autophagy and ER–Membrane Contact: Potential Implications in Neurodegenerative Disorders. Front. Mol. Neurosci. 2020, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schätzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.E.; Ferreira, S.T. Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 2018, 136, 350–360. [Google Scholar] [CrossRef]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Aβ oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Gendron, T.F. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 1–19. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, Q.; Zhang, Y.; Xu, H. Proteolytic processing of APP. J. Neurochem. 2012, 120 (Suppl. 1), 9–21. [Google Scholar] [CrossRef]
- Haass, C. Take five-BACE and the γ-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Wallon, D.; Rovelet-lecrux, A.; Richard, A.; Pasquier, F.; Lacour, M.; Rollin-sillaire, A.; Martinaud, O.; Quillard-muraine, M.; De, V.; Boutoleau-bretonniere, C.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 1, 1–16. [Google Scholar]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C.; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The role of NMDA receptors in Alzheimer’s disease. Front. Neurosci. 2019, 13, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Park, K.A.; Lee, W.T.; Lee, J.E. Apoptosis signal regulating kinase 1 (ASK1): Potential as a therapeutic target for Alzheimer’s disease. Int. J. Mol. Sci. 2014, 15, 2119–2129. [Google Scholar] [CrossRef]
- Lin, Q.; Cao, Y.; Gao, J. Serum calreticulin is a negative biomarker in patients with Alzheimer’s disease. Int. J. Mol. Sci. 2014, 15, 21740–21753. [Google Scholar] [CrossRef]
- Sharma, V.; Ounallah-Saad, H.; Chakraborty, D.; Hleihil, M.; Sood, R.; Barrera, I.; Edry, E.; Chandran, S.K.; de Leon, S.B.T.; Kaphzan, H.; et al. Local inhibition of PERK enhances memory and reverses age-related deterioration of cognitive and neuronal properties. J. Neurosci. 2018, 38, 648–658. [Google Scholar] [CrossRef]
- Hugon, J.; Mouton-Liger, F.; Dumurgier, J.; Paquet, C. PKR involvement in Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 83. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2272–2281. [Google Scholar] [CrossRef]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the Translation Initiation Factor eIF2α Increases BACE1 Levels and Promotes Amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef]
- Pasini, S.; Corona, C.; Liu, J.; Greene, L.A.; Shelanski, M.L. Specific downregulation of hippocampal ATF4 reveals a necessary role in synaptic plasticity and memory. Cell Rep. 2015, 11, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Duran-Aniotz, C.; Cornejo, V.H.; Espinoza, S.; Ardiles, Á.O.; Medinas, D.B.; Salazar, C.; Foley, A.; Gajardo, I.; Thielen, P.; Iwawaki, T.; et al. IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 2017, 134, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.; Vidal, R.L.; Mardones, P.; Serrano, F.G.; Ardiles, A.O.; Wirth, C.; Valdés, P.; Thielen, P.; Schneider, B.L.; Kerr, B.; et al. Regulation of Memory Formation by the Transcription Factor XBP1. Cell Rep. 2016, 14, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Peron, R.; Vatanabe, I.P.; Manzine, P.R.; Camins, A.; Cominetti, M.R. Alpha-secretase ADAM10 regulation: Insights into Alzheimer’s disease treatment. Pharmaceuticals 2018, 11, 12. [Google Scholar] [CrossRef]
- Cissé, M.; Duplan, E.; Checler, F. The transcription factor XBP1 in memory and cognition: Implications in Alzheimer’s disease. Mol. Med. 2017, 22, 905–917. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Du, Y.; Liu, X.; Zhu, X.; Liu, Y.; Wang, X.; Wu, X. Activating transcription factor 6 reduces Aβ1–42 and restores memory in Alzheimer’s disease model mice. Int. J. Neurosci. 2020, 1–9. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress Structure and Properties of C/EBP Homologous Protein Roles of CHOP in ER Stress-Mediated Apoptosis. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef]
- Feng, Y.; Li, X.; Zhou, W.; Lou, D.; Huang, D.; Li, Y.; Kang, Y.; Xiang, Y.; Li, T.; Zhou, W.; et al. Regulation of SET Gene Expression by NFkB. Mol. Neurobiol. 2017, 54, 4477–4485. [Google Scholar] [CrossRef]
- Mohammed-Ali, Z.; Cruz, G.L.; Dickhout, J.G. Crosstalk between the unfolded protein response and NF-κ B-mediated inflammation in the progression of chronic kidney disease. J. Immunol. Res. 2015, 2015. [Google Scholar] [CrossRef]
- Vukic, V.; Callaghan, D.; Walker, D.; Lue, L.F.; Liu, Q.Y.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Stanimirovic, D.B.; Zhang, W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 2009, 34, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-Dependent Activation of JAK1 and STAT3 Contributes to Endoplasmic Reticulum Stress-Induced Inflammation. Mol. Cell. Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Ávila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [PubMed]
- Hirtz, D.; Thurman, D.J.; Gwinn-Hardy, K.; Mohamed, M.; Chaudhuri, A.R.; Zalutsky, R. How common are the “common” neurologic disorders? Neurology 2007, 68, 326–337. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Roman, G.C.; Hong, Z.; Wu, C.B.; Qu, Q.M.; Huang, J.B.; Zhou, B.; Geng, Z.P.; Wu, J.X.; Wen, H.B.; et al. Parkinson’s disease in China: Prevalence in Beijing, Xian, and Shanghai. Lancet 2005, 365, 595–597. [Google Scholar] [CrossRef]
- Tan, L.C.S.; Venketasubramanian, N.; Jamora, R.D.G.; Heng, D. Incidence of Parkinson’s disease in Singapore. Park. Relat. Disord. 2007, 13, 40–43. [Google Scholar] [CrossRef]
- Tian, Y.Y.; Tang, C.J.; Wu, J.; Zhou, J.S. Parkinson’s disease in China. Neurol. Sci. 2011, 32, 23–30. [Google Scholar] [CrossRef]
- Elbaz, A.; Carcaillon, L.; Kab, S.; Moisan, F. Epidemiology of Parkinson’s disease. Rev. Neurol. 2016, 172, 14–26. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.N. Molecular chaperones, α-synuclein, and neurodegeneration. Mol. Neurobiol. 2013, 47, 552–560. [Google Scholar] [CrossRef]
- Malek, N.; Swallow, D.; Grosset, K.A.; Anichtchik, O.; Spillantini, M.; Grosset, D.G. Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson’s disease—A systematic review. Acta Neurol. Scand. 2014, 130, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Golbe, L.I.; Di Iorio, G.; Bonavita, V.; Miller, D.C.; Duvoisin, R.C. A large kindred with autosomal dominant Parkinson’s disease. Ann. Neurol. 1990, 27, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The New Mutation, E46K, of α-Synuclein Causes Parkinson and Lewy Body Dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Soto, C. Transmissible proteins: Expanding the prion heresy. Cell 2012, 149, 968–977. [Google Scholar] [CrossRef]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef]
- Bellani, S.; Mescola, A.; Ronzitti, G.; Tsushima, H.; Tilve, S.; Canale, C.; Valtorta, F.; Chieregatti, E. GRP78 clustering at the cell surface of neurons transduces the action of exogenous alpha-synuclein. Cell Death Differ. 2014, 21, 1971–1983. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.I.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of elF2α dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef]
- Credle, J.J.; Forcelli, P.A.; Delannoy, M.; Oaks, A.W.; Permaul, E.; Berry, D.L.; Duka, V.; Wills, J.; Sidhu, A. α-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol. Dis. 2015, 76, 112–125. [Google Scholar] [CrossRef]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Calì, T.; Gai, W.; Chen, T.; et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19, e44617. [Google Scholar] [CrossRef]
- Jung, E.M.; Yoo, Y.M.; Park, S.Y.; Ahn, C.; Jeon, B.H.; Hong, E.J.; Kim, W.Y.; Jeung, E.B. Calbindin-D9k is a novel risk gene for neurodegenerative disease. Cell. Physiol. Biochem. 2020, 54, 438–456. [Google Scholar]
- Matus, S.; Valenzuela, V.; Medinas, D.B.; Hetz, C. ER dysfunction and protein folding stress in ALS. Int. J. Cell Biol. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Yamada, M.; Tanaka, H.; Aida, K.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Inuzuka, T.; Takahashi, H.; Hara, H. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 2009, 36, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S. Endoplasmic Reticulum Stress in Motor Neurons of the Spinal Cord in Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 346–355. [Google Scholar] [CrossRef]
- Dion, P.A.; Daoud, H.; Rouleau, G.A. Genetics of motor neuron disorders: New insights into pathogenic mechanisms. Nat. Rev. Genet. 2009, 10, 769–782. [Google Scholar] [CrossRef]
- Tang, L.; Ma, Y.; Liu, X.L.; Chen, L.; Fan, D.S. Better survival in female SOD1-mutant patients with ALS: A study of SOD1-related natural history. Transl. Neurodegener. 2019, 8, 1–10. [Google Scholar]
- Chen, D.; Wang, Y.; Chin, E.R. Activation of the endoplasmic reticulum stress response in skeletal muscle of G93a*SOD1 amyotrophic lateral sclerosis mice. Front. Cell. Neurosci. 2015, 9, 170. [Google Scholar] [CrossRef]
- Kikuchi, H.; Almer, G.; Yamashita, S.; Guégan, C.; Nagai, M.; Xu, Z.; Sosunov, A.A.; McKhann, G.M.; Przedborski, S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. USA 2006, 103, 6025–6030. [Google Scholar] [CrossRef]
- Lautenschlaeger, J.; Prell, T.; Grosskreutz, J. Endoplasmic reticulum stress and the ER mitochondrial calcium cycle in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 166–177. [Google Scholar] [CrossRef]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Popko, B.; Roos, R.P. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2011, 20, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Popko, B.; Roos, R.P. An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum. Mol. Genet. 2014, 23, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Q.; Ren, M.; Jiang, H.Z.; Wang, J.; Zhang, J.; Yin, X.; Wang, S.Y.; Qi, Y.; Wang, X.D.; Feng, H.L. Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 2014, 277, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Dzhashiashvili, Y.; Monckton, C.P.; Shah, H.S.; Kunjamma, R.B.; Popko, B. The UPR-PERK pathway is not a promising therapeutic target for mutant SOD1-induced ALS. Neurobiol. Dis. 2019, 127, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Matus, S.; Lopez, E.; Valenzuela, V.; Nassif, M.; Hetz, C. Functional Contribution of the Transcription Factor ATF4 to the Pathogenesis of Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e66672. [Google Scholar] [CrossRef]
- Lee, S.; Shang, Y.; Redmond, S.A.; Urisman, A.; Tang, A.A.; Li, K.H.; Burlingame, A.L.; Pak, R.A.; Jovičić, A.; Gitler, A.D.; et al. Activation of HIPK2 Promotes ER Stress-Mediated Neurodegeneration in Amyotrophic Lateral Sclerosis. Neuron 2016, 91, 41–55. [Google Scholar] [CrossRef]
- Clark, E.M.; Nonarath, H.J.T.; Bostrom, J.R.; Link, B.A. Establishment and validation of an endoplasmic reticulum stress reporter to monitor zebrafish ATF6 activity in development and disease. DMM Dis. Model. Mech. 2020, 13, dmm041426. [Google Scholar] [CrossRef]
- Walker, A.K.; Atkin, J.D. Mechanisms of neuroprotection by protein disulphide isomerase in amyotrophic lateral sclerosis. Neurol. Res. Int. 2011, 2011. [Google Scholar] [CrossRef]
- Gonzalez-Perez, P.; Woehlbier, U.; Chian, R.J.; Sapp, P.; Rouleau, G.A.; Leblond, C.S.; Daoud, H.; Dion, P.A.; Landers, J.E.; Hetz, C.; et al. Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene 2015, 566, 158–165. [Google Scholar] [CrossRef]
- Woehlbier, U.; Colombo, A.; Saaranen, M.J.; Pérez, V.; Ojeda, J.; Bustos, F.J.; Andreu, C.I.; Torres, M.; Valenzuela, V.; Medinas, D.B.; et al. ALS -linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 2016, 35, 845–865. [Google Scholar] [CrossRef]
- Walker, A.K.; Soo, K.Y.; Sundaramoorthy, V.; Parakh, S.; Ma, Y.; Farg, M.A.; Wallace, R.H.; Crouch, P.J.; Turner, B.J.; Horne, M.K.; et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 2013, 8, e81170. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Raphael, A.R.; Ladow, E.S.; Mcgurk, L.; Weber, R.A.; Trojanowski, J.Q.; Lee, V.M.Y.; Finkbeiner, S.; Gitler, A.D.; Bonini, N.M. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 2014, 46, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Soo, K.Y.; Walker, A.K.; Pham, H.; Orian, J.; Horne, M.K.; Warraich, S.T.; Williams, K.L.; Blair, I.P.; Atkin, J.D. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 2012, 33, 2855–2868. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.M.; Livesey, M.R.; McDade, K.; Selvaraj, B.T.; Barton, S.K.; Chandran, S.; Smith, C. Dysregulation of AMPA receptor subunit expression in sporadic ALS post-mortem brain. J. Pathol. 2020, 250, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Dreser, A.; Vollrath, J.T.; Sechi, A.; Johann, S.; Roos, A.; Yamoah, A.; Katona, I.; Bohlega, S.; Wiemuth, D.; Tian, Y.; et al. The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 2017, 24, 1655–1671. [Google Scholar] [CrossRef]
- Gkogkas, C.; Middleton, S.; Kremer, A.M.; Wardrope, C.; Hannah, M.; Gillingwater, T.H.; Skehel, P. VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 2008, 17, 1517–1526. [Google Scholar] [CrossRef]
- Hall, C.E.; Yao, Z.; Choi, M.; Tyzack, G.E.; Serio, A.; Luisier, R.; Harley, J.; Preza, E.; Arber, C.; Crisp, S.J.; et al. Progressive Motor Neuron Pathology and the Role of Astrocytes in a Human Stem Cell Model of VCP-Related ALS. Cell Rep. 2017, 19, 1739–1749. [Google Scholar] [CrossRef]
- Belzil, V.V.; Katzman, R.B.; Petrucelli, L. ALS and FTD: An epigenetic perspective. Acta Neuropathol. 2016, 132, 487–502. [Google Scholar] [CrossRef]
- Liscic, R.M.; Alberici, A.; Cairns, N.J.; Romano, M.; Buratti, E. From basic research to the clinic: Innovative therapies for ALS and FTD in the pipeline. Mol. Neurodegener. 2020, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Satterfield, T.; Pritchett, J.; Cruz, S.; Kemp, K. Prion disease and endoplasmic reticulum stress pathway correlations and treatment pursuits. Endoplasmic Reticulum Stress Dis. 2017, 4, 27–36. [Google Scholar] [CrossRef]
- Mays, C.E.; Soto, C. The stress of prion disease. Brain Res. 2016, 1648, 553–560. [Google Scholar] [CrossRef]
- Hughes, D.; Halliday, M. What is our current understanding of prpsc-associated neurotoxicity and its molecular underpinnings? Pathogens 2017, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, O.; Ashok, A.; Hegde, R.S. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem. Sci. 2009, 34, 287–295. [Google Scholar] [CrossRef]
- Thapa, S.; Abdelaziz, D.H.; Abdulrahman, B.A.; Schatzl, H.M. Sephin1 Reduces Prion Infection in Prion-Infected Cells and Animal Model. Mol. Neurobiol. 2020, 57, 1–14. [Google Scholar] [CrossRef]
- Orsi, A.; Fioriti, L.; Chiesa, R.; Sitia, R. Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J. Biol. Chem. 2006, 281, 30431–30438. [Google Scholar] [CrossRef]
- Hetz, C.; Castilla, J.; Soto, C. Perturbation of endoplasmic reticulum homeostasis facilitates prion replication. J. Biol. Chem. 2007, 282, 12725–12733. [Google Scholar] [CrossRef]
- Rane, N.S.; Kang, S.W.; Chakrabarti, O.; Feigenbaum, L.; Hegde, R.S. Reduced Translocation of Nascent Prion Protein During ER Stress Contributes to Neurodegeneration. Dev. Cell 2008, 15, 359–370. [Google Scholar] [CrossRef]
- Nunziante, M.; Ackermann, K.; Dietrich, K.; Wolf, H.; Gädtke, L.; Gilch, S.; Vorberg, I.; Groschup, M.; Schätzl, H.M. Proteasomal dysfunction and endoplasmic reticulum stress enhance trafficking of prion protein aggregates through the secretory pathway and increase accumulation of pathologic prion protein. J. Biol. Chem. 2011, 286, 33942–33953. [Google Scholar] [CrossRef]
- Hetz, C.; Russelakis-Carneiro, M.; Wälchli, S.; Carboni, S.; Vial-Knecht, E.; Maundrell, K.; Castilla, J.; Soto, C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J. Neurosci. 2005, 25, 2793–2802. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Russelakis-Carneiro, M.; Maundrell, K.; Castilla, J.; Soto, C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003, 22, 5435–5445. [Google Scholar] [CrossRef]
- Mays, C.E.; Armijo, E.; Morales, R.; Kramm, C.; Flores, A.; Tiwari, A.; Bian, J.; Telling, G.C.; Pandita, T.K.; Hunt, C.R.; et al. Prion disease is accelerated in mice lacking stress-induced heat shock protein 70 (HSP70). J. Biol. Chem. 2019, 294, 13619–13628. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Eun Kim, G.; Morales, R.; Moda, F.; Moreno-Gonzalez, I.; Concha-Marambio, L.; Lee, A.S.; Hetz, C.; Soto, C. The Endoplasmic Reticulum Chaperone GRP78/BiP Modulates Prion Propagation in vitro and in vivo. Sci. Rep. 2017, 7, 44723. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, G. Unfolding the mystery of UPR in astrocytes. Sci. Transl. Med. 2020, 12, 14. [Google Scholar] [CrossRef]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; Le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.Z.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Hetz, C.; Lee, A.H.; Gonzalez-Romero, D.; Thielen, P.; Castilla, J.; Soto, C.; Glimcher, L.H. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 757–762. [Google Scholar] [CrossRef]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-β neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yalcin, A.; Lee, G.Y.; Li, P.; Fan, J.; Arruda, A.P.; Pers, B.M.; Yilmaz, M.; Eguchi, K.; Hotamisligil, G.S. Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci. Transl. Med. 2015, 7, 292ra98. [Google Scholar] [CrossRef]
- Kudo, T.; Kanemoto, S.; Hara, H.; Morimoto, N.; Morihara, T.; Kimura, R.; Tabira, T.; Imaizumi, K.; Takeda, M. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008, 15, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Kitakaze, K.; Taniuchi, S.; Kawano, E.; Hamada, Y.; Miyake, M.; Oyadomari, M.; Kojima, H.; Kosako, H.; Kuribara, T.; Yoshida, S.; et al. Cell-based hts identifies a chemical chaperone for preventing er protein aggregation and proteotoxicity. Elife 2019, 8, e43302. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.S.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M.P. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454. [Google Scholar] [CrossRef]
- Rozpędek-Kamińska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-dependent molecular mechanisms as a novel therapeutic target for neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 2108. [Google Scholar] [CrossRef]
- Grande, V.; Ornaghi, F.; Comerio, L.; Restelli, E.; Masone, A.; Corbelli, A.; Tolomeo, D.; Capone, V.; Axten, J.M.; Laping, N.J.; et al. PERK inhibition delays neurodegeneration and improves motor function in a mouse model of Marinesco-Sjögren syndrome. Hum. Mol. Genet. 2018, 27, 2477–2489. [Google Scholar] [CrossRef]
- Mercado, G.; Castillo, V.; Soto, P.; López, N.; Axten, J.M.; Sardi, S.P.; Hoozemans, J.J.M.; Hetz, C. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis. 2018, 112, 136–148. [Google Scholar] [CrossRef]
- Radford, H.; Moreno, J.A.; Verity, N.; Halliday, M.; Mallucci, G.R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015, 130, 633–642. [Google Scholar] [CrossRef]
- Mahameed, M.; Wilhelm, T.; Darawshi, O.; Obiedat, A.; Tommy, W.S.; Chintha, C.; Schubert, T.; Samali, A.; Chevet, E.; Eriksson, L.A.; et al. The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 2019, 10, 300. [Google Scholar] [CrossRef]
- Li, A.; Song, N.J.; Riesenberg, B.P.; Li, Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front. Immunol. 2020, 10, 3154. [Google Scholar] [CrossRef] [PubMed]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife 2015, 4, e05033. [Google Scholar] [CrossRef] [PubMed]
- Rabouw, H.H.; Langereis, M.A.; Anand, A.A.; Visser, L.J.; De Groot, R.J.; Walter, P.; Van Kuppeveld, F.J.M. Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2097–2102. [Google Scholar] [CrossRef] [PubMed]
- Bugallo, R.; Marlin, E.; Baltanás, A.; Toledo, E.; Ferrero, R.; Vinueza-Gavilanes, R.; Larrea, L.; Arrasate, M.; Aragón, T. Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Markov, N.S.; Lu, Z.; Aillon, R.P.; Soberanes, S.; Runyan, C.E.; Ren, Z.; Grant, R.A.; Maciel, M.; Abdala-Valencia, H.; et al. Resetting proteostasis with ISRIB prevents pulmonary fibrosis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sidrauski, C.; Acosta-Alvear, D.; Khoutorsky, A.; Vedantham, P.; Hearn, B.R.; Li, H.; Gamache, K.; Gallagher, C.M.; Ang, K.K.H.; Wilson, C.; et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2013, 2, e00498. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Wang, L.; Popko, B.; Tixier, E.; Roos, R.P. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis. 2014, 71, 317–324. [Google Scholar] [CrossRef]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef]
- Chen, Y.; Podojil, J.R.; Kunjamma, R.B.; Jones, J.; Weiner, M.; Lin, W.; Miller, S.D.; Popko, B. Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 2019, 142, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Crespillo-Casado, A.; Chambers, J.E.; Fischer, P.M.; Marciniak, S.J.; Ron, D. PPP1R15A-mediated dephosphorylation of eIF2a is unaffected by sephin1 or guanabenz. Elife 2017, 6, e26109. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, K.S.; Lee, H.J.; Kim, D.H.; Noh, Y.H.; Yu, K.; Jung, H.Y.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; et al. Activation of PERK signaling attenuates Aβ-mediated ER stress. PLoS ONE 2010, 5, e10489. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Luo, N.; Zhao, H.R.; Gao, Q.; Lu, J.; Pan, Y.; Shi, J.P.; Tian, Y.Y.; Zhang, Y.D. Salubrinal protects against rotenone-induced SH-SY5Y cell death via ATF4-parkin pathway. Brain Res. 2014, 1549, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Niso-Santano, M.; Pedro, J.M.B.S.; Gómez-Sánchez, R.; Climent, V.; Soler, G.; Fuentes, J.M.; González-Polo, R.A. ASK1 overexpression accelerates paraquat-induced autophagy via endoplasmic reticulum stress. Toxicol. Sci. 2011, 119, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef]
- Carlesso, A.; Chintha, C.; Gorman, A.M.; Samali, A.; Eriksson, L.A. Effect of kinase inhibiting rnase attenuator (Kira) compounds on the formation of face-to-face dimers of inositol-requiring enzyme 1: Insights from computational modeling. Int. J. Mol. Sci. 2019, 20, 5538. [Google Scholar] [CrossRef]
- Plate, L.; Cooley, C.B.; Chen, J.J.; Paxman, R.J.; Gallagher, C.M.; Madoux, F.; Genereux, J.C.; Dobbs, W.; Garza, D.; Spicer, T.P.; et al. Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. Elife 2016, 5, 75. [Google Scholar] [CrossRef]
- Paxman, R.; Plate, L.; Blackwood, E.A.; Glembotski, C.; Powers, E.T.; Wiseman, R.L.; Kelly, J.W. Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. Elife 2018, 7, e37168. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).