Current Methods of Magnetic Resonance for Noninvasive Assessment of Molecular Aspects of Pathoetiology in Multiple Sclerosis

 ,
, 
Abstract
1. Introduction
2. General MRI Techniques
- -
- Magnetization transfer MRI is based on the interactions that occur between free-water protons and protons bound to macromolecules and that are typically evaluated as their quantitative ratio/magnetization transfer ratio [29,32]. This technique reveals subtle brain tissue integrity loss during MS progression better than conventional MRI [28]. The magnetization transfer ratio has been demonstrated continuously to decrease in MS lesions and normal-appearing WM (NAWM) and has been related to the percentage of residual axons and the degree of myelin content [28,31].
- -
- Diffusion-weighted MRI is a suitable method for the measurement of the water molecule motion in tissue, especially in the WM, in which water diffusion is preferably oriented along the axons and thus follows the WM tracts [29]. Any neuronal tract failure or axonal membrane permeability disruption should lead to an increase in the mean diffusivity (the averaged molecular motion) and to a decrease in the fractional anisotropy (the directional preponderance), as reported in chronic MS lesions and NAWM [28,31,33].
- -
- Susceptibility-weighted MRI enhances the inhomogeneity of the magnetic field caused by the paramagnetic properties of venous deoxygenated hemoglobin and other nonheme iron, all of which affect the local magnetic susceptibility in the MS brain [29]. The highest potential of this technique is in the long-term monitoring of the disturbed microenvironment of MS lesions and veins, showing an increasing trend in the accumulation of iron deposits in these areas [28].
- -
- Cerebral perfusion MRI provides information on capillary microcirculation in the tissue based on generating a blood flow contrast between multiple images by using a wide variety of blood-labeling methods such as exogenous tracers (Gd-contrast agent) or water protons from the arterial blood (arterial spin labeling) [32]. Perfusion studies reveal increased perfusion in acute inflammatory MS lesions, and thus vasodilation and decreased cerebral blood flow in non-enhancing persisting MS lesions [28,32]. Perfusion is also altered in NAWM and in cortical and subcortical GM, reflecting not only microvascular abnormalities, but also tissue degradation and blood–brain barrier permeability failure [28].
- -
- Functional MRI enables the assessment of brain activation manifesting as differences in the deoxyhemoglobin concentration in the blood in activated brain areas, in response to the applied motor or visual stimuli [30,32]. Observed abnormalities in MS patients usually occur in the visual, cognitive, and motor systems with early manifestation, and tend to vary throughout the disease [28].
Limitations of General MRI Techniques
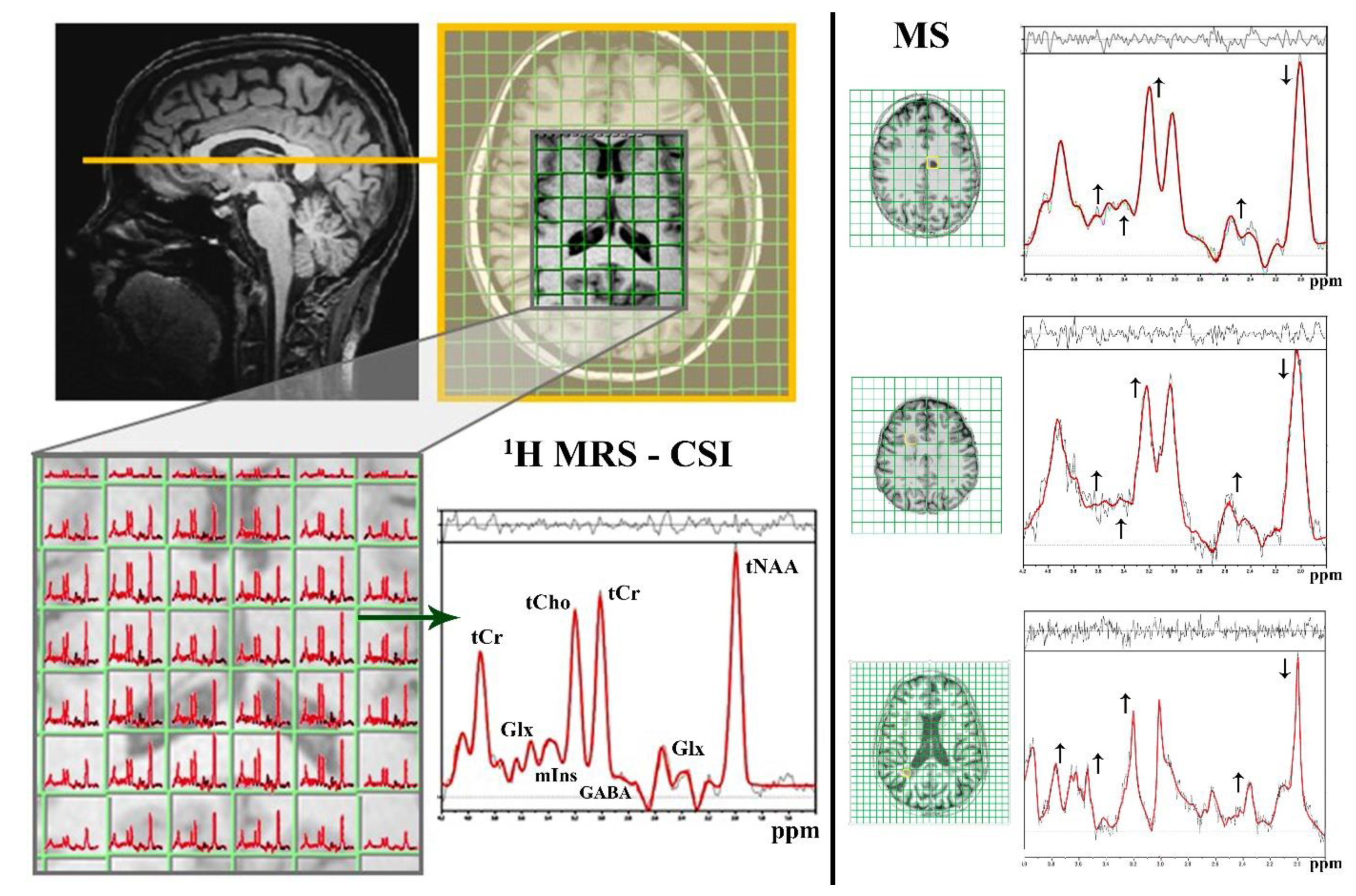
3. Magnetic Resonance Spectroscopy
3.1. 1H MRS
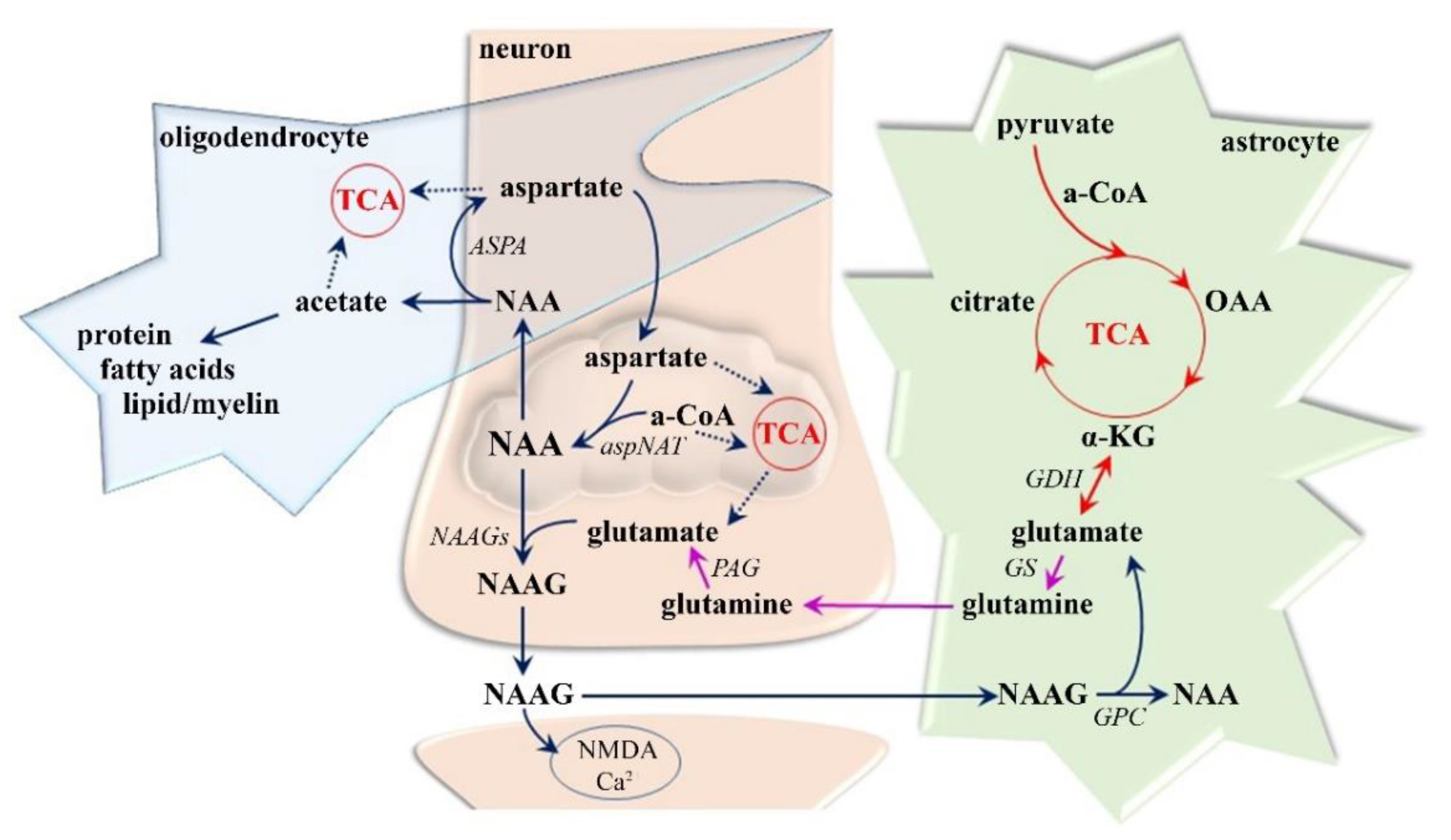
3.1.1. Neuro-Axonal Degradation
3.1.2. Demyelization and Reactive Gliosis
tCho—Cell Membrane Predictor
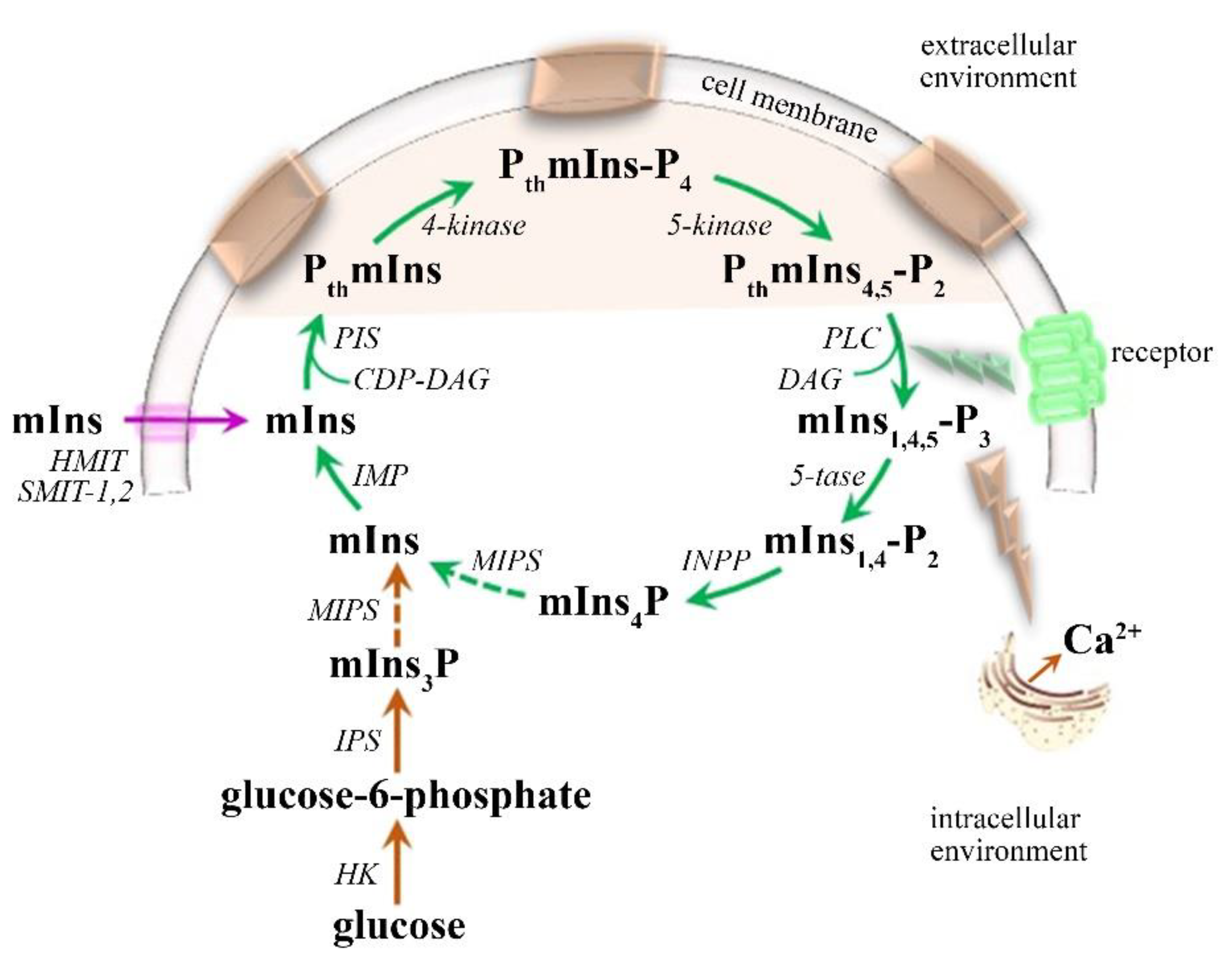
mIns—Glial Marker
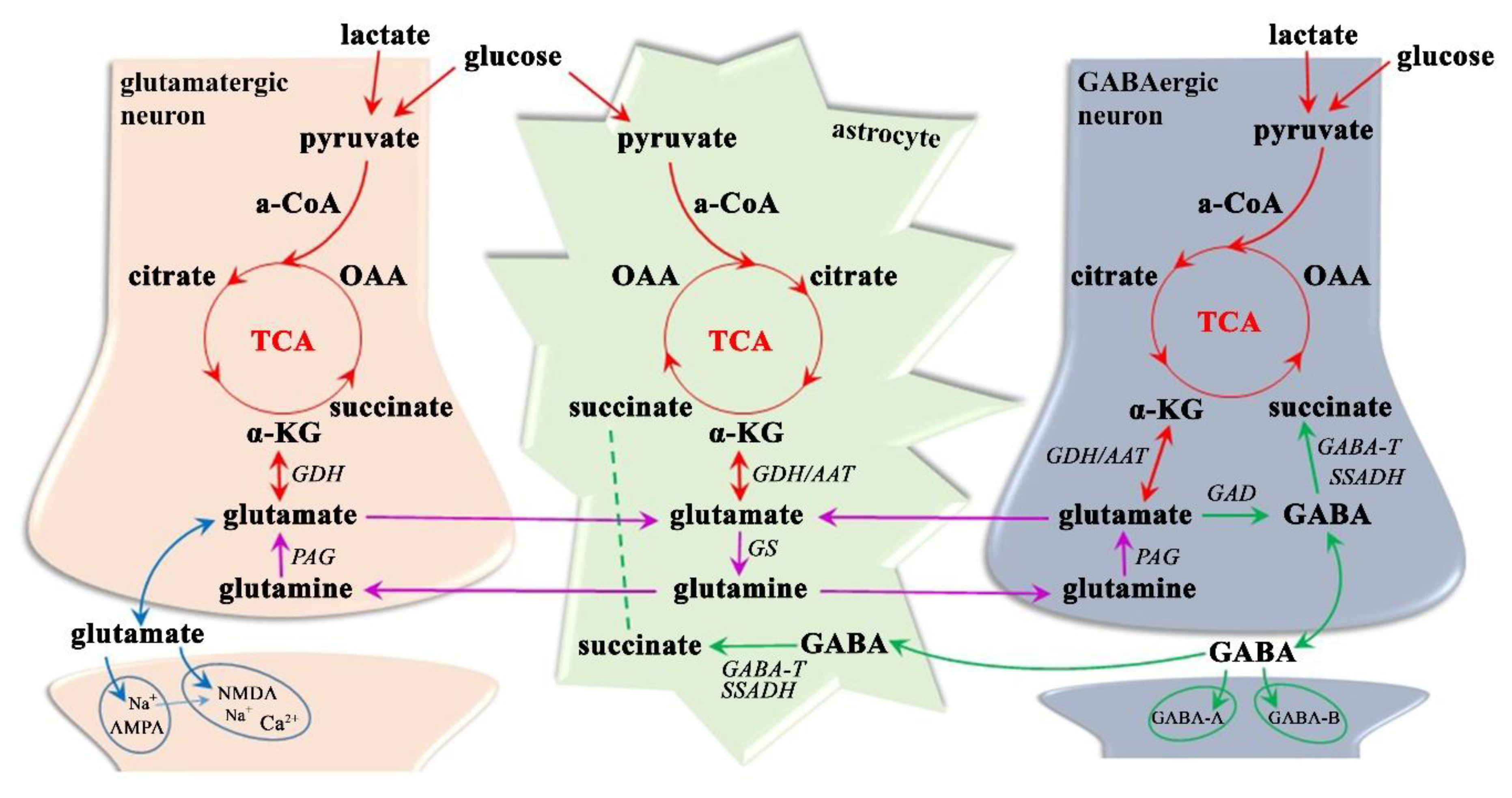
3.1.3. Neurotransmitter Dysregulation–Glx Excitotoxicity
3.1.4. Neurotransmitter Dysregulation–GABA Inhibition
3.1.5. Limitations of 1H MRS
3.2. 31P MRS
3.2.1. Phospholipids Metabolism
3.2.2. Cellular Energy Metabolism
3.2.3. Intracellular pH
3.2.4. Intracellular Mg2+
3.2.5. Limitations of 31P MRS
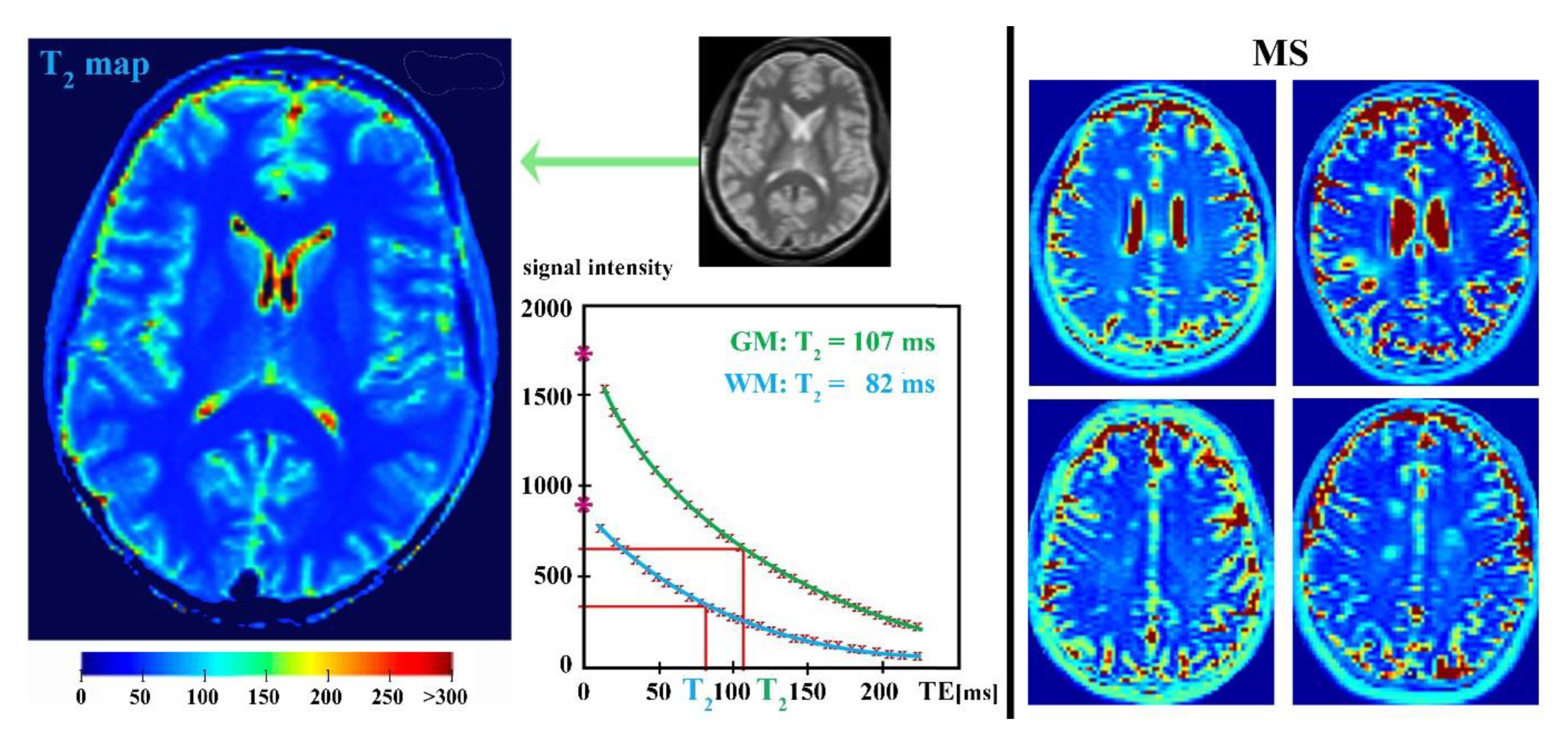
4. Parametric T2-Relaxation Mapping
4.1. Myelin Water Mapping
4.2. Iron Mapping Quantification
4.3. Limitations of Parametric MR-Relaxation Mapping
Funding
Acknowledgments
Conflicts of Interest
References
- Haussleiter, I.S.; Brune, M.; Juckel, G. Psychopathology in multiple sclerosis: Diagnosis, prevalence and treatment. Ther. Adv. Neurol. Disord. 2009, 2, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Jafari, N. Risk Factors in Cause and Course of Multiple Sclerosis. Ph.D. Thesis, Erasmus University Rotterdam, Rotterdam, The Netherlands, 2012. [Google Scholar]
- Swanberg, K.M.; Landheer, K.; Pitt, D.; Juchem, C. Quantifying the Metabolic Signature of Multiple Sclerosis by in vivo Proton Magnetic Resonance Spectroscopy: Current Challenges and Future Outlook in the Translation From Proton Signal to Diagnostic Biomarker. Front. Neurol. 2019, 10, 1173. [Google Scholar] [CrossRef]
- Kantarci, O.H. Phases and Phenotypes of Multiple Sclerosis. Continuum 2019, 25, 636–654. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Oksenberg, J.R. The neurobiology of multiple sclerosis: Genes, inflammation, and neurodegeneration. Neuron 2006, 52, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Stys, P.K.; Tsutsui, S. Recent advances in understanding multiple sclerosis. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Popescu, B.F.; Bunyan, R.F.; Parisi, J.E.; Ransohoff, R.M.; Lucchinetti, C.F. A case of multiple sclerosis presenting with inflammatory cortical demyelination. Neurology 2011, 76, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Haider, L.; Simeonidou, C.; Steinberger, G.; Hametner, S.; Grigoriadis, N.; Deretzi, G.; Kovacs, G.G.; Kutzelnigg, A.; Lassmann, H.; Frischer, J.M. Multiple sclerosis deep grey matter: The relation between demyelination, neurodegeneration, inflammation and iron. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1386–1395. [Google Scholar] [CrossRef]
- van Rensburg, S.J.; Kotze, M.J.; van Toorn, R. The conundrum of iron in multiple sclerosis—Time for an individualised approach. Metab. Brain Dis. 2012, 27, 239–253. [Google Scholar] [CrossRef]
- Zhang, J.M. Human Brain Glutamate, Glutamine, γ-Aminobutyric Acid Proton Magnetic Resonance Spectral Quantification with the Fast Pade Transform. Ph.D. Thesis, The University of California, Los Angeles, CA, USA, 2013. [Google Scholar]
- Puts, N.A.; Edden, R.A. In vivo magnetic resonance spectroscopy of GABA: A methodological review. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 60, 29–41. [Google Scholar] [CrossRef]
- Agarwal, N.; Renshaw, P.F. Proton MR spectroscopy-detectable major neurotransmitters of the brain: Biology and possible clinical applications. AJNR Am. J. Neuroradiol. 2012, 33, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Patti, F. Gray matters in multiple sclerosis: Cognitive impairment and structural MRI. Mult. Scler. Int. 2014, 2014, 609694. [Google Scholar] [CrossRef] [PubMed]
- Hohlfeld, R. ‘Gimme five’: Future challenges in multiple sclerosis. ECTRIMS Lecture 2009. Mult. Scler. 2010, 16, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Steen, C.; Wilczak, N.; Hoogduin, J.M.; Koch, M.; De Keyser, J. Reduced creatine kinase B activity in multiple sclerosis normal appearing white matter. PLoS ONE 2010, 5, e10811. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stankiewicz, J.; Panter, S.S.; Neema, M.; Arora, A.; Batt, C.E.; Bakshi, R. Iron in chronic brain disorders: Imaging and neurotherapeutic implications. Neurother. J. Am. Soc. Exp. Neurother. 2007, 4, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Enzinger, C.; Langkammer, C.; Tscherner, M.; Wallner-Blazek, M.; Jehna, M.; Ropele, S.; Fuchs, S.; Fazekas, F. Quantitative assessment of brain iron by R2* relaxometry in patients with clinically isolated syndrome and relapsing-remitting multiple sclerosis. Mult. Scler. 2009, 15, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.L.; Nydes, M.; Shanley, K.L.; Morales Pantoja, I.E.; Howard, T.A.; Bizzozero, O.A. Reduced expression of the ferroptosis inhibitor glutathione peroxidase-4 in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurochem. 2019, 148, 426–439. [Google Scholar] [CrossRef]
- Stojanovic, I.R.; Kostic, M.; Ljubisavljevic, S. The role of glutamate and its receptors in multiple sclerosis. J. Neural Transm. 2014, 121, 945–955. [Google Scholar] [CrossRef]
- Azevedo, C.J.; Kornak, J.; Chu, P.; Sampat, M.; Okuda, D.T.; Cree, B.A.; Nelson, S.J.; Hauser, S.L.; Pelletier, D. In vivo evidence of glutamate toxicity in multiple sclerosis. Ann. Neurol. 2014, 76, 269–278. [Google Scholar] [CrossRef]
- Zivadinov, R.; Pirko, I. Advances in understanding gray matter pathology in multiple sclerosis: Are we ready to redefine disease pathogenesis? BMC Neurol. 2012, 12, 9. [Google Scholar] [CrossRef]
- Cawley, N.; Solanky, B.S.; Muhlert, N.; Tur, C.; Edden, R.A.; Wheeler-Kingshott, C.A.; Miller, D.H.; Thompson, A.J.; Ciccarelli, O. Reduced gamma-aminobutyric acid concentration is associated with physical disability in progressive multiple sclerosis. Brain J. Neurol. 2015, 138, 2584–2595. [Google Scholar] [CrossRef] [PubMed]
- Hnilicova, P.; Kantorova, E.; Polacek, H.; Grendar, M.; Bittsansky, M.; Cierny, D.; Sivak, S.; Zelenak, K.; Lehotsky, J.; Dobrota, D.; et al. Altered hypothalamic metabolism in early multiple sclerosis—MR spectroscopy study. J. Neurol. Sci. 2019, 407, 116458. [Google Scholar] [CrossRef] [PubMed]
- Sajja, B.R.; Wolinsky, J.S.; Narayana, P.A. Proton magnetic resonance spectroscopy in multiple sclerosis. Neuroimaging Clin. N. Am. 2009, 19, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Barker, P.B. Fundamentals of MR spectroscopy. In Clinical MR Neuroimaging: Physiological and Functional Techniques; Gillard, J.H., Waldman, A.D., Barker, P.B., Eds.; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Baranovicova, E.; Mlynarik, V.; Kantorova, E.; Hnilicova, P.; Dobrota, D. Quantitative evaluation of cerebral white matter in patients with multiple sclerosis using multicomponent T2 mapping. Neurol. Res. 2016, 38, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Rocca, M.A.; De Stefano, N.; Enzinger, C.; Fisher, E.; Horsfield, M.A.; Inglese, M.; Pelletier, D.; Comi, G. Magnetic resonance techniques in multiple sclerosis: The present and the future. Arch. Neurol. 2011, 68, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Hemond, C.C.; Bakshi, R. Magnetic Resonance Imaging in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028969. [Google Scholar] [CrossRef]
- Napoli, S.Q.; Bakshi, R. Magnetic resonance imaging in multiple sclerosis. Rev. Neurol. Dis. 2005, 2, 109–116. [Google Scholar]
- MacKay, A.; Laule, C.; Li, D.K.; Meyers, S.M.; Russell-Schulz, B.; Vavasour, I.M. Magnetic Resonance Techniques for Investigation of Multiple Sclerosis. In Proceedings of the XIII Mexican Symposium on Medical Physics; American Institute of Physics Publishing LLC: New York, NY, USA, 2014; pp. 22–35. [Google Scholar] [CrossRef]
- Bakshi, R.; Thompson, A.J.; Rocca, M.A.; Pelletier, D.; Dousset, V.; Barkhof, F.; Inglese, M.; Guttmann, C.R.; Horsfield, M.A.; Filippi, M. MRI in multiple sclerosis: Current status and future prospects. Lancet Neurol. 2008, 7, 615–625. [Google Scholar] [CrossRef]
- Trip, S.A.; Miller, D.H. Imaging in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2005, 76, 11–18. [Google Scholar] [CrossRef]
- McFarland, H.F. Examination of the role of magnetic resonance imaging in multiple sclerosis: A problem-orientated approach. Ann. Indian Acad. Neurol. 2009, 12, 254–263. [Google Scholar] [CrossRef]
- Tognarelli, J.M.; Dawood, M.; Shariff, M.I.; Grover, V.P.; Crossey, M.M.; Cox, I.J.; Taylor-Robinson, S.D.; McPhail, M.J. Magnetic Resonance Spectroscopy: Principles and Techniques: Lessons for Clinicians. J. Clin. Exp. Hepatol. 2015, 5, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, N.R. Role of Magnetic Resonance Imaging and in vivo MR Spectroscopy in Clinical, Experimental and Biological Research. Proc. Indian Natl. Sci. Acad. 2003, B69, 423–446. [Google Scholar]
- Zhu, H.; Barker, P.B. MR spectroscopy and spectroscopic imaging of the brain. Methods Mol. Biol. 2011, 711, 203–226. [Google Scholar] [CrossRef] [PubMed]
- Bogner, W.; Gagoski, B.; Hess, A.T.; Bhat, H.; Tisdall, M.D.; van der Kouwe, A.J.W.; Strasser, B.; Marjanska, M.; Trattnig, S.; Grant, E.; et al. 3D GABA imaging with real-time motion correction, shim update and reacquisition of adiabatic spiral MRSI. Neuroimage 2014, 103, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Hnilicova, P.; Povazan, M.; Strasser, B.; Andronesi, O.C.; Gajdosik, M.; Dydak, U.; Ukropec, J.; Dobrota, D.; Trattnig, S.; Bogner, W. Spatial variability and reproducibility of GABA-edited MEGA-LASER 3D-MRSI in the brain at 3 T. NMR Biomed. 2016, 29, 1656–1665. [Google Scholar] [CrossRef]
- Opstad, K.S.; Bell, B.A.; Griffiths, J.R.; Howe, F.A. An investigation of human brain tumour lipids by high-resolution magic angle spinning 1H MRS and histological analysis. NMR Biomed. 2008, 21, 677–685. [Google Scholar] [CrossRef]
- Soares, D.P.; Law, M. Magnetic resonance spectroscopy of the brain: Review of metabolites and clinical applications. Clin. Radiol. 2009, 64, 12–21. [Google Scholar] [CrossRef]
- Moffett, J.R.; Ross, B.; Arun, P.; Madhavarao, C.N.; Namboodiri, A.M. N-Acetylaspartate in the CNS: From neurodiagnostics to neurobiology. Prog. Neurobiol. 2007, 81, 89–131. [Google Scholar] [CrossRef]
- Clark, J.F.; Doepke, A.; Filosa, J.A.; Wardle, R.L.; Lu, A.; Meeker, T.J.; Pyne-Geithman, G.J. N-acetylaspartate as a reservoir for glutamate. Med. Hypotheses 2006, 67, 506–512. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wang, J.; Wu, G.; Lu, Z.H.; Chakraborty, G.; Meyenhofer, M.; Tyring, S.K.; Matalon, R. Physiological role of N-acetylaspartate: Contribution to myelinogenesis. Adv. Exp. Med. Biol. 2006, 576, 131–143. [Google Scholar] [CrossRef]
- Madhavarao, C.N.; Moffett, J.R.; Moore, R.A.; Viola, R.E.; Namboodiri, M.A.; Jacobowitz, D.M. Immunohistochemical localization of aspartoacylase in the rat central nervous system. J. Comp. Neurol. 2004, 472, 318–329. [Google Scholar] [CrossRef]
- Baslow, M.H. Functions of N-acetyl-L-aspartate and N-acetyl-L-aspartylglutamate in the vertebrate brain: Role in glial cell-specific signaling. J. Neurochem. 2000, 75, 453–459. [Google Scholar] [CrossRef]
- Nguyen, T.; Kirsch, B.J.; Asaka, R.; Nabi, K.; Quinones, A.; Tan, J.; Antonio, M.J.; Camelo, F.; Li, T.; Nguyen, S.; et al. Uncovering the Role of N-Acetyl-Aspartyl-Glutamate as a Glutamate Reservoir in Cancer. Cell Rep. 2019, 27, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, K.; Hamilton, N.B.; Wade, A.; Karadottir, R.; Attwell, D. The effect of N-acetyl-aspartyl-glutamate and N-acetyl-aspartate on white matter oligodendrocytes. Brain J. Neurol. 2009, 132, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, R.; Coyle, J.T.; Tsai, G.; Greene, R.W. NAAG reduces NMDA receptor current in CA1 hippocampal pyramidal neurons of acute slices and dissociated neurons. Neuropsychopharmacology 2005, 30, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Pena, J.L.; Pinero, P.; Sellers, G.; Argente, J.; Casado, A.; Foronda, J.; Ucles, A.; Izquierdo, G. Magnetic resonance spectroscopy of normal appearing white matter in early relapsing-remitting multiple sclerosis: Correlations between disability and spectroscopy. BMC Neurol. 2004, 4, 8. [Google Scholar] [CrossRef]
- Tisell, A.; Leinhard, O.D.; Warntjes, J.B.; Lundberg, P. Procedure for quantitative (1) H magnetic resonance spectroscopy and tissue characterization of human brain tissue based on the use of quantitative magnetic resonance imaging. Magn. Reson. Med. 2013, 70, 905–915. [Google Scholar] [CrossRef]
- Kantorova, E.; Polacek, H.; Bittsansky, M.; Baranovicova, E.; Hnilicova, P.; Cierny, D.; Sivak, S.; Nosal, V.; Zelenak, K.; Kurca, E. Hypothalamic damage in multiple sclerosis correlates with disease activity, disability, depression, and fatigue. Neurol. Res. 2017, 39, 323–330. [Google Scholar] [CrossRef]
- Quarantelli, M. MRI/MRS in neuroinflammation: Methodology and applications. Clin. Transl. Imaging 2015, 3, 475–489. [Google Scholar] [CrossRef]
- De Stefano, N.; Bartolozzi, M.L.; Guidi, L.; Stromillo, M.L.; Federico, A. Magnetic resonance spectroscopy as a measure of brain damage in multiple sclerosis. J. Neurol. Sci. 2005, 233, 203–208. [Google Scholar] [CrossRef]
- Inglese, M.; Liu, S.; Babb, J.S.; Mannon, L.J.; Grossman, R.I.; Gonen, O. Three-dimensional proton spectroscopy of deep gray matter nuclei in relapsing-remitting MS. Neurology 2004, 63, 170–172. [Google Scholar] [CrossRef]
- Wylezinska, M.; Cifelli, A.; Jezzard, P.; Palace, J.; Alecci, M.; Matthews, P.M. Thalamic neurodegeneration in relapsing-remitting multiple sclerosis. Neurology 2003, 60, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Geurts, J.J.; Reuling, I.E.; Vrenken, H.; Uitdehaag, B.M.; Polman, C.H.; Castelijns, J.A.; Barkhof, F.; Pouwels, P.J. MR spectroscopic evidence for thalamic and hippocampal, but not cortical, damage in multiple sclerosis. Magn. Reson. Med. 2006, 55, 478–483. [Google Scholar] [CrossRef]
- Polacek, H.; Kantorova, E.; Hnilicova, P.; Grendar, M.; Zelenak, K.; Kurca, E. Increased glutamate and deep brain atrophy can predict the severity of multiple sclerosis. Biomed. Pap. 2019, 163, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Mattisi, I.; Rinaldi, F.; Favaretto, A.; Atzori, M.; Bernardi, V.; Barachino, L.; Romualdi, C.; Rinaldi, L.; Perini, P.; et al. Magnetic resonance evidence of cerebellar cortical pathology in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Geurts, J.J.; Stys, P.K.; Minagar, A.; Amor, S.; Zivadinov, R. Gray matter pathology in (chronic) MS: Modern views on an early observation. J. Neurol. Sci. 2009, 282, 12–20. [Google Scholar] [CrossRef]
- Minagar, A.; Barnett, M.H.; Benedict, R.H.; Pelletier, D.; Pirko, I.; Sahraian, M.A.; Frohman, E.; Zivadinov, R. The thalamus and multiple sclerosis: Modern views on pathologic, imaging, and clinical aspects. Neurology 2013, 80, 210–219. [Google Scholar] [CrossRef]
- Calabrese, M.; Rinaldi, F.; Mattisi, I.; Grossi, P.; Favaretto, A.; Atzori, M.; Bernardi, V.; Barachino, L.; Romualdi, C.; Rinaldi, L.; et al. Widespread cortical thinning characterizes patients with MS with mild cognitive impairment. Neurology 2010, 74, 321–328. [Google Scholar] [CrossRef]
- Houtchens, M.K.; Benedict, R.H.; Killiany, R.; Sharma, J.; Jaisani, Z.; Singh, B.; Weinstock-Guttman, B.; Guttmann, C.R.; Bakshi, R. Thalamic atrophy and cognition in multiple sclerosis. Neurology 2007, 69, 1213–1223. [Google Scholar] [CrossRef]
- Pirko, I.; Lucchinetti, C.F.; Sriram, S.; Bakshi, R. Gray matter involvement in multiple sclerosis. Neurology 2007, 68, 634–642. [Google Scholar] [CrossRef]
- Fisher, E.; Lee, J.C.; Nakamura, K.; Rudick, R.A. Gray matter atrophy in multiple sclerosis: A longitudinal study. Ann. Neurol. 2008, 64, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Rudick, R.A.; Lee, J.C.; Nakamura, K.; Fisher, E. Gray matter atrophy correlates with MS disability progression measured with MSFC but not EDSS. J. Neurol. Sci. 2009, 282, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, G.; Lavorgna, L.; Russo, P.; Prinster, A.; Dinacci, D.; Savettieri, G.; Quattrone, A.; Livrea, P.; Messina, C.; Reggio, A.; et al. Brain atrophy and lesion load in a large population of patients with multiple sclerosis. Neurology 2005, 65, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Elahy, M.; Jackaman, C.; Mamo, J.C.; Lam, V.; Dhaliwal, S.S.; Giles, C.; Nelson, D.; Takechi, R. Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun. Ageing 2015, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Staffen, W.; Zauner, H.; Mair, A.; Kutzelnigg, A.; Kapeller, P.; Stangl, H.; Raffer, E.; Niederhofer, H.; Ladurner, G. Magnetic resonance spectroscopy of memory and frontal brain region in early multiple sclerosis. J. Neuropsychiatry Clin. Neurosci. 2005, 17, 357–363. [Google Scholar] [CrossRef]
- Zaini, W.H.; Giuliani, F.; Beaulieu, C.; Kalra, S.; Hanstock, C. Fatigue in Multiple Sclerosis: Assessing Pontine Involvement Using Proton MR Spectroscopic Imaging. PLoS ONE 2016, 11, e0149622. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; De Stefano, N.; Francis, G.S.; Arnaoutelis, R.; Caramanos, Z.; Collins, D.L.; Pelletier, D.; Arnason, B.G.W.; Antel, J.P.; Arnold, D.L. Axonal metabolic recovery in multiple sclerosis patients treated with interferon beta-1b. J. Neurol. 2001, 248, 979–986. [Google Scholar] [CrossRef]
- Khan, O.; Shen, Y.; Caon, C.; Bao, F.; Ching, W.; Reznar, M.; Buccheister, A.; Hu, J.; Latif, Z.; Tselis, A.; et al. Axonal metabolic recovery and potential neuroprotective effect of glatiramer acetate in relapsing-remitting multiple sclerosis. Mult. Scler. 2005, 11, 646–651. [Google Scholar] [CrossRef]
- Cambron, M.; Reynders, T.; Debruyne, J.; Reyngoudt, H.; Ribbens, A.; Achten, E.; Laureys, G. Targeting phosphocreatine metabolism in relapsing-remitting multiple sclerosis: Evaluation with brain MRI, (1)H and (31)P MRS, and clinical and cognitive testing. J. Neurol. 2018, 265, 2614–2624. [Google Scholar] [CrossRef]
- Mostert, J.P.; Sijens, P.E.; Oudkerk, M.; De Keyser, J. Fluoxetine increases cerebral white matter NAA/Cr ratio in patients with multiple sclerosis. Neurosci. Lett. 2006, 402, 22–24. [Google Scholar] [CrossRef]
- Llufriu, S.; Kornak, J.; Ratiney, H.; Oh, J.; Brenneman, D.; Cree, B.A.; Sampat, M.; Hauser, S.L.; Nelson, S.J.; Pelletier, D. Magnetic resonance spectroscopy markers of disease progression in multiple sclerosis. JAMA Neurol. 2014, 71, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Bagory, M.; Durand-Dubief, F.; Ibarrola, D.; Comte, J.C.; Cotton, F.; Confavreux, C.; Sappey-Marinier, D. Implementation of an absolute brain 1H-MRS quantification method to assess different tissue alterations in multiple sclerosis. IEEE Trans. Biol. Med. Eng. 2012, 59, 2687–2694. [Google Scholar] [CrossRef]
- Kirov, I.I.; Tal, A.; Babb, J.S.; Herbert, J.; Gonen, O. Serial proton MR spectroscopy of gray and white matter in relapsing-remitting MS. Neurology 2013, 80, 39–46. [Google Scholar] [CrossRef]
- Chang, L.; Munsaka, S.M.; Kraft-Terry, S.; Ernst, T. Magnetic resonance spectroscopy to assess neuroinflammation and neuropathic pain. J. Neuroimmune Pharmacol. 2013, 8, 576–593. [Google Scholar] [CrossRef] [PubMed]
- Narayana, P.A. Magnetic resonance spectroscopy in the monitoring of multiple sclerosis. J. Neuroimaging 2005, 15, 46S–57S. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Inglese, M.; Li, B.S.; Babb, J.S.; Grossman, R.I.; Gonen, O. Relapsing-remitting multiple sclerosis: Metabolic abnormality in nonenhancing lesions and normal-appearing white matter at MR imaging: Initial experience. Radiology 2005, 234, 211–217. [Google Scholar] [CrossRef]
- Neema, M.; Arora, A.; Healy, B.C.; Guss, Z.D.; Brass, S.D.; Duan, Y.; Buckle, G.J.; Glanz, B.I.; Stazzone, L.; Khoury, S.J.; et al. Deep gray matter involvement on brain MRI scans is associated with clinical progression in multiple sclerosis. J. Neuroimaging 2009, 19, 3–8. [Google Scholar] [CrossRef]
- Hulst, H.E.; Geurts, J.J. Gray matter imaging in multiple sclerosis: What have we learned? BMC Neurol. 2011, 11, 153. [Google Scholar] [CrossRef]
- Chhetri, D.R. Myo-Inositol and Its Derivatives: Their Emerging Role in the Treatment of Human Diseases. Front. Pharmacol. 2019, 10, 1172. [Google Scholar] [CrossRef]
- Kim, H.; McGrath, B.M.; Silverstone, P.H. A review of the possible relevance of inositol and the phosphatidylinositol second messenger system (PI-cycle) to psychiatric disorders--focus on magnetic resonance spectroscopy (MRS) studies. Hum. Psychopharmacol. 2005, 20, 309–326. [Google Scholar] [CrossRef]
- Rango, M.; Cogiamanian, F.; Marceglia, S.; Barberis, B.; Arighi, A.; Biondetti, P.; Priori, A. Myoinositol content in the human brain is modified by transcranial direct current stimulation in a matter of minutes: A 1H-MRS study. Magn. Reson. Med. 2008, 60, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Croze, M.L.; Soulage, C.O. Potential role and therapeutic interests of myo-inositol in metabolic diseases. Biochimie 2013, 95, 1811–1827. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.S.; Mitoma, H.; Manto, M. GABA and Glutamate: Their Transmitter Role in the CNS and Pancreatic Islets. In GABA and Glutamate: New Developments in Neurotransmission Research; IntechOpen: London, UK, 2017; pp. 65–90. [Google Scholar] [CrossRef]
- Gamper, N.; Shapiro, M.S. Regulation of ion transport proteins by membrane phosphoinositides. Nat. Rev. Neurosci. 2007, 8, 921–934. [Google Scholar] [CrossRef]
- Banerjee, S.; Hasan, G. The InsP3 receptor: Its role in neuronal physiology and neurodegeneration. Bioessays 2005, 27, 1035–1047. [Google Scholar] [CrossRef]
- Srinivasan, R.; Sailasuta, N.; Hurd, R.; Nelson, S.; Pelletier, D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain J. Neurol. 2005, 128, 1016–1025. [Google Scholar] [CrossRef]
- Kirov, I.I.; Patil, V.; Babb, J.S.; Rusinek, H.; Herbert, J.; Gonen, O. MR spectroscopy indicates diffuse multiple sclerosis activity during remission. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1330–1336. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fernando, K.T.; McLean, M.A.; Chard, D.T.; MacManus, D.G.; Dalton, C.M.; Miszkiel, K.A.; Gordon, R.M.; Plant, G.T.; Thompson, A.J.; Miller, D.H. Elevated white matter myo-inositol in clinically isolated syndromes suggestive of multiple sclerosis. Brain J. Neurol. 2004, 127, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Moheet, A.; Emir, U.E.; Terpstra, M.; Kumar, A.; Eberly, L.E.; Seaquist, E.R.; Oz, G. Initial experience with seven tesla magnetic resonance spectroscopy of hypothalamic GABA during hyperinsulinemic euglycemia and hypoglycemia in healthy humans. Magn. Reson. Med. 2014, 71, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef]
- Kolisek, M.; Sponder, G.; Pilchova, I.; Cibulka, M.; Tatarkova, Z.; Werner, T.; Racay, P. Magnesium Extravaganza: A Critical Compendium of Current Research into Cellular Mg(2+) Transporters Other than TRPM6/7. Rev. Physiol. Biochem. Pharmacol. 2019, 176, 65–105. [Google Scholar] [CrossRef]
- Horakova, D.; Kalincik, T.; Dusankova, J.B.; Dolezal, O. Clinical correlates of grey matter pathology in multiple sclerosis. BMC Neurol. 2012, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Klaver, R.; De Vries, H.E.; Schenk, G.J.; Geurts, J.J. Grey matter damage in multiple sclerosis: A pathology perspective. Prion 2013, 7, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Muhlert, N.; Atzori, M.; De Vita, E.; Thomas, D.L.; Samson, R.S.; Wheeler-Kingshott, C.A.; Geurts, J.J.; Miller, D.H.; Thompson, A.J.; Ciccarelli, O. Memory in multiple sclerosis is linked to glutamate concentration in grey matter regions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Shirayama, Y.; Takahashi, M.; Osone, F.; Hara, A.; Okubo, T. Myo-inositol, Glutamate, and Glutamine in the Prefrontal Cortex, Hippocampus, and Amygdala in Major Depression. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2017, 2, 196–204. [Google Scholar] [CrossRef]
- Urrila, A.S.; Hakkarainen, A.; Castaneda, A.; Paunio, T.; Marttunen, M.; Lundbom, N. Frontal Cortex Myo-Inositol Is Associated with Sleep and Depression in Adolescents: A Proton Magnetic Resonance Spectroscopy Study. Neuropsychobiology 2017, 75, 21–31. [Google Scholar] [CrossRef]
- Daikhin, Y.; Yudkoff, M. Compartmentation of brain glutamate metabolism in neurons and glia. J. Nutr. 2000, 130, 1026S–1031S. [Google Scholar] [CrossRef]
- Hassel, B.; Dingledine, R. Glutamate and Glutamate Receptors. In Basic Neurochemistry; Academic Press, Elsevier Inc.: Oxford, UK, 2012; pp. 342–366. [Google Scholar] [CrossRef]
- Shen, J. Glutamate. In Magnetic Resonance Spectroscopy; Academic Press, Elsevier Inc.: Oxford, UK, 2014; pp. 111–121. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef]
- Hertz, L. The Glutamate-Glutamine (GABA) Cycle: Importance of Late Postnatal Development and Potential Reciprocal Interactions between Biosynthesis and Degradation. Front. Endocrinol. 2013, 4, 59. [Google Scholar] [CrossRef]
- Macrez, R.; Stys, P.K.; Vivien, D.; Lipton, S.A.; Docagne, F. Mechanisms of glutamate toxicity in multiple sclerosis: Biomarker and therapeutic opportunities. Lancet Neurol. 2016, 15, 1089–1102. [Google Scholar] [CrossRef]
- Deckx, N.; Lee, W.P.; Berneman, Z.N.; Cools, N. Neuroendocrine immunoregulation in multiple sclerosis. Clin. Dev. Immunol. 2013, 2013, 705232. [Google Scholar] [CrossRef] [PubMed]
- Trapani, V.; Mastrototaro, L.; Wolf, F.I. Magnesium and the Yin-Yang interplay in apoptosis. In Magnesium in the Central Nervous System; Vink, R., Nechifor, M., Eds.; University of Adelaide Press: Adelaide, Australia, 2011. [Google Scholar]
- Stagg, C.J. Magnetic Resonance Spectroscopy as a tool to study the role of GABA in motor-cortical plasticity. Neuroimage 2014, 86, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; Seraji-Bozorgzad, N.; Bao, F.; Razmjou, S.; Caon, C.; Santiago, C.; Latif, Z.; Aronov, R.; Zak, I.; Ashtamker, N.; et al. The Relationship between Brain MR Spectroscopy and Disability in Multiple Sclerosis: 20-Year Data from the U.S. Glatiramer Acetate Extension Study. J. Neuroimaging 2017, 27, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sun, W.; Shi, Y.; Shi, R.; Cheng, J.X. Glutamate excitotoxicity inflicts paranodal myelin splitting and retraction. PLoS ONE 2009, 4, e6705. [Google Scholar] [CrossRef][Green Version]
- Matute, C.; Domercq, M.; Sanchez-Gomez, M.V. Glutamate-mediated glial injury: Mechanisms and clinical importance. Glia 2006, 53, 212–224. [Google Scholar] [CrossRef]
- Buzsaki, G.; Kaila, K.; Raichle, M. Inhibition and brain work. Neuron 2007, 56, 771–783. [Google Scholar] [CrossRef]
- Novotny, E.J., Jr.; Fulbright, R.K.; Pearl, P.L.; Gibson, K.M.; Rothman, D.L. Magnetic resonance spectroscopy of neurotransmitters in human brain. Ann. Neurol. 2003, 54, S25–S31. [Google Scholar] [CrossRef]
- Cambron, M.; D’Haeseleer, M.; Laureys, G.; Clinckers, R.; Debruyne, J.; De Keyser, J. White-matter astrocytes, axonal energy metabolism, and axonal degeneration in multiple sclerosis. J. Cereb. Blood Flow Metab. 2012, 32, 413–424. [Google Scholar] [CrossRef]
- Gilani, A.A.; Dash, R.P.; Jivrajani, M.N.; Thakur, S.K.; Nivsarkar, M. Evaluation of GABAergic Transmission Modulation as a Novel Functional Target for Management of Multiple Sclerosis: Exploring Inhibitory Effect of GABA on Glutamate-Mediated Excitotoxicity. Adv. Pharmacol. Sci. 2014, 2014, 632376. [Google Scholar] [CrossRef]
- Davies, C.H.; Starkey, S.J.; Pozza, M.F.; Collingridge, G.L. GABA autoreceptors regulate the induction of LTP. Nature 1991, 349, 609–611. [Google Scholar] [CrossRef]
- Bhattacharyya, P.K.; Phillips, M.D.; Stone, L.A.; Bermel, R.A.; Lowe, M.J. Sensorimotor cortex gamma-aminobutyric acid concentration correlates with impaired performance in patients with MS. AJNR Am. J. Neuroradiol. 2013, 34, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Kiljan, S.; Prins, M.; Baselmans, B.M.; Bol, J.; Schenk, G.J.; van Dam, A.M. Enhanced GABAergic Immunoreactivity in Hippocampal Neurons and Astroglia of Multiple Sclerosis Patients. J. Neuropathol. Exp. Neurol. 2019, 78, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Quadrelli, S.; Ribbons, K.; Arm, J.; Al-Iedani, O.; Lechner-Scott, J.; Lea, R.; Ramadan, S. 2D in-vivo L-COSY spectroscopy identifies neurometabolite alterations in treated multiple sclerosis. Ther. Adv. Neurol. Disord. 2019, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yin, X.; Edden, R.A.E.; Evans, A.C.; Xu, J.; Cao, G.; Li, H.; Li, M.; Zhao, B.; Wang, J.; et al. Altered hippocampal GABA and glutamate levels and uncoupling from functional connectivity in multiple sclerosis. Hippocampus 2018, 28, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Edden, R.A.E.; Gao, F.; Li, H.; Gong, T.; Chen, W.; Liu, X.; Wang, G.; Zhao, B. Reduced GABA levels correlate with cognitive impairment in patients with relapsing-remitting multiple sclerosis. Eur. Radiol. 2018, 28, 1140–1148. [Google Scholar] [CrossRef]
- Bertholdo, D.; Watcharakorn, A.; Castillo, M. Brain proton magnetic resonance spectroscopy: Introduction and overview. Neuroimaging Clin. N. Am. 2013, 23, 359–380. [Google Scholar] [CrossRef]
- Buonocore, M.H.; Maddock, R.J. Magnetic resonance spectroscopy of the brain: A review of physical principles and technical methods. Rev. Neurosci. 2015, 26, 609–632. [Google Scholar] [CrossRef]
- Choi, I.Y.; Lee, S.P.; Merkle, H.; Shen, J. In vivo detection of gray and white matter differences in GABA concentration in the human brain. Neuroimage 2006, 33, 85–93. [Google Scholar] [CrossRef]
- Snyder, J.; Thompson, R.B.; Wilman, A.H. Difference spectroscopy using PRESS asymmetry: Application to glutamate, glutamine, and myo-inositol. NMR Biomed. 2010, 23, 41–47. [Google Scholar] [CrossRef]
- Björkman-Burtscher, I.M.; Sundgren, P.C. Metabolic Imaging: MR Spectroscopy. In Neurological Imaging; Radiology Key, WordPress: San Francisco, CA, USA, 2016. [Google Scholar] [CrossRef]
- Al-Iedani, O.; Lechner-Scott, J.; Ribbons, K.; Ramadan, S. Fast magnetic resonance spectroscopic imaging techniques in human brain- applications in multiple sclerosis. J. Biomed. Sci. 2017, 24, 17. [Google Scholar] [CrossRef]
- Kurhanewicz, J.; Vigneron, D.B.; Nelson, S.J. Three-dimensional magnetic resonance spectroscopic imaging of brain and prostate cancer. Neoplasia 2000, 2, 166–189. [Google Scholar] [CrossRef]
- Hingerl, L.; Strasser, B.; Moser, P.; Hangel, G.; Motyka, S.; Heckova, E.; Gruber, S.; Trattnig, S.; Bogner, W. Clinical High-Resolution 3D-MR Spectroscopic Imaging of the Human Brain at 7 T. Investig. Radiol. 2020, 55, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C.S.; Otaduy, C.G.; Park, E.J.; Leite, C.C. Phosphorus-31 MR spectroscopy of the human brain: Technical aspects and biomedical applications. Int. J. Curr. Res. Rev. 2014, 6, 41–57. [Google Scholar]
- Faghihi, R.; Zeinali-Rafsanjani, B.; Mosleh-Shirazi, M.A.; Saeedi-Moghadam, M.; Lotfi, M.; Jalli, R.; Iravani, V. Magnetic Resonance Spectroscopy and its Clinical Applications: A Review. J. Med. Imaging Radiat. Sci. 2017, 48, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, J.P. Multi-Nuclear Magnetic Resonance Spectroscopy of Human Brain Tumours. Ph.D. Thesis, Radboud University Nijmegen, Nijmegen, The Netherlands, 2010. [Google Scholar]
- Marie, S.K.; Shinjo, S.M. Metabolism and brain cancer. Clinics 2011, 66, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H. High-Field 31P Magnetic Resonance Spectroscopy (MRS) in Human Brain. Ph.D. Thesis, University of Minnesota, Minneapolis, MN, USA, 2010. [Google Scholar]
- Kauv, P.; Ayache, S.S.; Creange, A.; Chalah, M.A.; Lefaucheur, J.P.; Hodel, J.; Brugieres, P. Adenosine Triphosphate Metabolism Measured by Phosphorus Magnetic Resonance Spectroscopy: A Potential Biomarker for Multiple Sclerosis Severity. Eur. Neurol. 2017, 77, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Solivera, J.; Cerdan, S.; Pascual, J.M.; Barrios, L.; Roda, J.M. Assessment of 31P-NMR analysis of phospholipid profiles for potential differential diagnosis of human cerebral tumors. NMR Biomed. 2009, 22, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Komoroski, R.A.; Pearce, J.M.; Mrak, R.E. 31P NMR spectroscopy of phospholipid metabolites in postmortem schizophrenic brain. Magn. Reson. Med. 2008, 59, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.K.; Treasaden, I.H. A human in vivo study of the extent to which 31-phosphorus neurospectroscopy phosphomonoesters index cerebral cell membrane phospholipid anabolism. Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.E.; Drost, D.J.; Menon, R.S.; Williamson, P.C. In vivo brain (31)P-MRS: Measuring the phospholipid resonances at 4 Tesla from small voxels. NMR Biomed. 2002, 15, 338–347. [Google Scholar] [CrossRef]
- Forester, B.P.; Berlow, Y.A.; Harper, D.G.; Jensen, J.E.; Lange, N.; Froimowitz, M.P.; Ravichandran, C.; Iosifescu, D.V.; Lukas, S.E.; Renshaw, P.F.; et al. Age-related changes in brain energetics and phospholipid metabolism. NMR Biomed. 2010, 23, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Kadota, T.; Horinouchi, T.; Kuroda, C. Development and aging of the cerebrum: Assessment with proton MR spectroscopy. AJNR Am. J. Neuroradiol. 2001, 22, 128–135. [Google Scholar]
- Moreno-Torres, A.; Pujol, J.; Soriano-Mas, C.; Deus, J.; Iranzo, A.; Santamaria, J. Age-related metabolic changes in the upper brainstem tegmentum by MR spectroscopy. Neurobiol. Aging 2005, 26, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Cadoux-Hudson, T.A.; Kermode, A.; Rajagopalan, B.; Taylor, D.; Thompson, A.J.; Ormerod, I.E.; McDonald, W.I.; Radda, G.K. Biochemical changes within a multiple sclerosis plaque in vivo. J. Neurol. Neurosurg. Psychiatry 1991, 54, 1004–1006. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, C.; Agius, P.; Naudin, M.; Herpe, G.; Ragot, S.; Maubeuge, N.; Philippe Neau, J.; Guillevin, R. (1) H-(31) P magnetic resonance spectroscopy: Effect of biotin in multiple sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 1332–1337. [Google Scholar] [CrossRef]
- Husted, C.A.; Matson, G.B.; Adams, D.A.; Goodin, D.S.; Weiner, M.W. In vivo detection of myelin phospholipids in multiple sclerosis with phosphorus magnetic resonance spectroscopic imaging. Ann. Neurol. 1994, 36, 239–241. [Google Scholar] [CrossRef]
- Minderhoud, J.M.; Mooyaart, E.L.; Kamman, R.L.; Teelken, A.W.; Hoogstraten, M.C.; Vencken, L.M.; Gravenmade, E.J.; van den Burg, W. In vivo phosphorus magnetic resonance spectroscopy in multiple sclerosis. Arch. Neurol. 1992, 49, 161–165. [Google Scholar] [CrossRef]
- Du, F.; Zhu, X.H.; Zhang, Y.; Friedman, M.; Zhang, N.; Ugurbil, K.; Chen, W. Tightly coupled brain activity and cerebral ATP metabolic rate. Proc. Natl. Acad. Sci. USA 2008, 105, 6409–6414. [Google Scholar] [CrossRef]
- Zhu, X.H.; Qiao, H.; Du, F.; Xiong, Q.; Liu, X.; Zhang, X.; Ugurbil, K.; Chen, W. Quantitative imaging of energy expenditure in human brain. Neuroimage 2012, 60, 2107–2117. [Google Scholar] [CrossRef]
- Kuiper, J.W.; Oerlemans, F.T.; Fransen, J.A.; Wieringa, B. Creatine kinase B deficient neurons exhibit an increased fraction of motile mitochondria. BMC Neurosci. 2008, 9, 73. [Google Scholar] [CrossRef]
- de Souza, A.C.; Justo, G.Z.; de Araujo, D.R.; Cavagis, A.D. Defining the molecular basis of tumor metabolism: A continuing challenge since Warburg’s discovery. Cell. Physiol. Biochem. 2011, 28, 771–792. [Google Scholar] [CrossRef]
- Beloueche-Babari, M.; Chung, Y.L.; Al-Saffar, N.M.; Falck-Miniotis, M.; Leach, M.O. Metabolic assessment of the action of targeted cancer therapeutics using magnetic resonance spectroscopy. Br. J. Cancer 2010, 102, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Sherry, A.D.; Malloy, C.R. (31)P-MRS of healthy human brain: ATP synthesis, metabolite concentrations, pH, and T1 relaxation times. NMR Biomed. 2015, 28, 1455–1462. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Steen, C.; D’Haeseleer, M.; Hoogduin, J.M.; Fierens, Y.; Cambron, M.; Mostert, J.P.; Heersema, D.J.; Koch, M.W.; De Keyser, J. Cerebral white matter blood flow and energy metabolism in multiple sclerosis. Mult. Scler. 2013, 19, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain J. Neurol. 2008, 131, 1722–1735. [Google Scholar] [CrossRef]
- Stys, P.K. General mechanisms of axonal damage and its prevention. J. Neurol. Sci. 2005, 233, 3–13. [Google Scholar] [CrossRef]
- Chesler, M. Regulation and modulation of pH in the brain. Physiol. Rev. 2003, 83, 1183–1221. [Google Scholar] [CrossRef]
- Cichocka, M. Possibilities of phosphorous magnetic resonance spectroscopy (31P MRS) in brain diagnostics. Acta Bio-Opt. Inform. Med. 2017, 23, 246–252. [Google Scholar]
- Ha, D.H.; Choi, S.; Oh, J.Y.; Yoon, S.K.; Kang, M.J.; Kim, K.U. Application of 31P MR spectroscopy to the brain tumors. Korean J. Radiol. 2013, 14, 477–486. [Google Scholar] [CrossRef]
- Dixon, R.M.; Styles, P. In vivo NMR, Applications, 31P. In Encyclopedia of Spectroscopy and Spectrometry; Lindon, J.C., Tranter, G.E., Koppenaal, D., Eds.; University of Oxford: Oxford, UK, 2017. [Google Scholar]
- Maintz, D.; Heindel, W.; Kugel, H.; Jaeger, R.; Lackner, K.J. Phosphorus-31 MR spectroscopy of normal adult human brain and brain tumours. NMR Biomed. 2002, 15, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Iotti, S.; Malucelli, E. Free magnesium concentration in the human brain. In Magnesium in the Central Nervous System; Vink, R., Nechifor, M., Eds.; University of Adelaide Press: Adelaide, Australia, 2011. [Google Scholar]
- Billard, J.M. Brain free magnesium homeostasis as a target for reducing cognitive aging. In Magnesium in the Central Nervous System; Vink, R., Nechifor, M., Eds.; University of Adelaide Press: Adelaide, Australia, 2011. [Google Scholar]
- Karpinska, E.; Socha, K.; Soroczynska, J.; Kochanowicz, J.; Jakoniuk, M.; Mariak, Z.; Borawska, M.H. Concentration of magnesium in the serum and the ability status of patients with relapsing-remitting multiple sclerosis. J. Elem. 2017, 22, 671–679. [Google Scholar] [CrossRef]
- Ramsaransing, G.S.; Mellema, S.A.; De Keyser, J. Dietary patterns in clinical subtypes of multiple sclerosis: An exploratory study. Nutr. J. 2009, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Romani, A.M.P. Intracellular magnesium homeostasis. In Magnesium in the Central Nervous System; Vink, R., Nechifor, M., Eds.; University of Adelaide Press: Adelaide, Australia, 2011. [Google Scholar]
- Rubin, H. Intracellular free Mg(2+) and MgATP(2-) in coordinate control of protein synthesis and cell proliferation. In Magnesium in the Central Nervous System; Vink, R., Nechifor, M., Eds.; University of Adelaide Press: Adelaide, Australia, 2011. [Google Scholar]
- Ghabriel, M.N.; Vink, R. Magnesium transport across the blood-brain barriers. In Magnesium in the Central Nervous System; Vink, R., Nechifor, M., Eds.; University of Adelaide Press: Adelaide, Australia, 2011. [Google Scholar]
- Fagan, T.E.; Cefaratti, C.; Romani, A. Streptozotocin-induced diabetes impairs Mg2+ homeostasis and uptake in rat liver cells. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E184–E193. [Google Scholar] [CrossRef] [PubMed]
- Vink, R.; Cook, N.L.; van den Heuvel, C. Magnesium in acute and chronic brain injury: An update. Magnes. Res. 2009, 22, 158S–162S. [Google Scholar] [CrossRef] [PubMed]
- Eby, G.A.; Eby, K.L. Rapid recovery from major depression using magnesium treatment. Med. Hypotheses 2006, 67, 362–370. [Google Scholar] [CrossRef]
- Kara, H.; Sahin, N.; Ulusan, V.; Aydogdu, T. Magnesium infusion reduces perioperative pain. Eur. J. Anaesthesiol. 2002, 19, 52–56. [Google Scholar] [CrossRef]
- Takahashi, H.; Imai, K.; Katanuma, A.; Sugaya, T.; Hisano, K.; Motoya, S.; Aoki, S.; Sugiyama, T.; Yachi, A. A case of chronic fatigue syndrome who showed a beneficial effect by intravenous administration of magnesium sulphate. Arerugi Allergy 1992, 41, 1605–1610. [Google Scholar]
- Soave, P.M.; Conti, G.; Costa, R.; Arcangeli, A. Magnesium and anaesthesia. Curr. Drug Targets 2009, 10, 734–743. [Google Scholar] [CrossRef]
- Guerrera, M.P.; Volpe, S.L.; Mao, J.J. Therapeutic uses of magnesium. Am. Fam. Physician 2009, 80, 157–162. [Google Scholar]
- Cernak, I.; Vink, R.; Zapple, D.N.; Cruz, M.I.; Ahmed, F.; Chang, T.; Fricke, S.T.; Faden, A.I. The pathobiology of moderate diffuse traumatic brain injury as identified using a new experimental model of injury in rats. Neurobiol. Dis. 2004, 17, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Dasilva, K.W.; O’Connor, C.; van den Heuvel, C.; Vink, R. Magnesium gluconate offers no more protection than magnesium sulphate following diffuse traumatic brain injury in rats. J. Am. Coll. Nutr. 2004, 23, 541S–544S. [Google Scholar] [CrossRef] [PubMed]
- Imer, M.; Omay, B.; Uzunkol, A.; Erdem, T.; Sabanci, P.A.; Karasu, A.; Albayrak, S.B.; Sencer, A.; Hepgul, K.; Kaya, M. Effect of magnesium, MK-801 and combination of magnesium and MK-801 on blood-brain barrier permeability and brain edema after experimental traumatic diffuse brain injury. Neurol. Res. 2009, 31, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Davies, M.; Blaker, P.A.; Hall, S.M.; Smith, K.J. Blockers of sodium and calcium entry protect axons from nitric oxide-mediated degeneration. Ann. Neurol. 2003, 53, 174–180. [Google Scholar] [CrossRef]
- Euser, A.G.; Cipolla, M.J. Magnesium sulfate for the treatment of eclampsia: A brief review. Stroke 2009, 40, 1169–1175. [Google Scholar] [CrossRef]
- Johnson, A.C.; Tremble, S.M.; Chan, S.L.; Moseley, J.; LaMarca, B.; Nagle, K.J.; Cipolla, M.J. Magnesium sulfate treatment reverses seizure susceptibility and decreases neuroinflammation in a rat model of severe preeclampsia. PLoS ONE 2014, 9, e113670. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, Z.H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402. [Google Scholar] [CrossRef]
- Nelander, M.; Weis, J.; Bergman, L.; Larsson, A.; Wikstrom, A.K.; Wikstrom, J. Cerebral Magnesium Levels in Preeclampsia; A Phosphorus Magnetic Resonance Spectroscopy Study. Am. J. Hypertens. 2017, 30, 667–672. [Google Scholar] [CrossRef]
- Yasui, M.; Yase, Y.; Ando, K.; Adachi, K.; Mukoyama, M.; Ohsugi, K. Magnesium concentration in brains from multiple sclerosis patients. Acta Neurol. Scand. 1990, 81, 197–200. [Google Scholar] [CrossRef]
- Fanea, L.; Sfrangeu, S.A. Relaxation times mapping using magnetic resonance imaging. Rom. Rep. Phys. 2011, 63, 456–464. [Google Scholar]
- Deoni, S.C. Magnetic resonance relaxation and quantitative measurement in the brain. Methods Mol. Biol. 2011, 711, 65–108. [Google Scholar] [CrossRef] [PubMed]
- Hnilicova, P.; Bittsansky, M.; Dobrota, D. Optimization of Brain T2 Mapping using Standard CPMG Sequence in a Clinical Scanner. Meas. Sci. Rev. 2014, 14, 117–125. [Google Scholar] [CrossRef]
- Carneiro, A.A.O.; Vilela, G.R.; de Araujo, D.B.; Baffa, O. MRI Relaxometry: Methods and Applications. Braz. J. Phys. 2006, 36, 9–15. [Google Scholar] [CrossRef]
- Cudalbu, C.; Mlynarik, V.; Gruetter, R. Handling macromolecule signals in the quantification of the neurochemical profile. J. Alzheimers Dis. JAD 2012, 31, S101–S115. [Google Scholar] [CrossRef] [PubMed]
- Stanisz, G.J.; Odrobina, E.E.; Pun, J.; Escaravage, M.; Graham, S.J.; Bronskill, M.J.; Henkelman, R.M. T1, T2 relaxation and magnetization transfer in tissue at 3T. Magn. Reson. Med. 2005, 54, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Hasan, K.M.; Walimuni, I.S.; Kramer, L.A.; Frye, R.E. Human brain atlas-based volumetry and relaxometry: Application to healthy development and natural aging. Magn. Reson. Med. 2010, 64, 1382–1389. [Google Scholar] [CrossRef]
- Kumar, R.; Delshad, S.; Woo, M.A.; Macey, P.M.; Harper, R.M. Age-related regional brain T2-relaxation changes in healthy adults. J. Magn. Reson. Imaging JMRI 2012, 35, 300–308. [Google Scholar] [CrossRef]
- Lu, H.; Nagae-Poetscher, L.M.; Golay, X.; Lin, D.; Pomper, M.; van Zijl, P.C. Routine clinical brain MRI sequences for use at 3.0 Tesla. J. Magn. Reson. Imaging JMRI 2005, 22, 13–22. [Google Scholar] [CrossRef]
- Pell, G.S.; Briellmann, R.S.; Waites, A.B.; Abbott, D.F.; Jackson, G.D. Voxel-based relaxometry: A new approach for analysis of T2 relaxometry changes in epilepsy. Neuroimage 2004, 21, 707–713. [Google Scholar] [CrossRef]
- Freedman, M.S. Multiple Sclerosis and Demyelinating Diseases; Lippincott Williams & Wilkins: Ottawa, ON, Canada, 2006. [Google Scholar]
- West, J.; Aalto, A.; Tisell, A.; Leinhard, O.D.; Landtblom, A.M.; Smedby, O.; Lundberg, P. Normal appearing and diffusely abnormal white matter in patients with multiple sclerosis assessed with quantitative MR. PLoS ONE 2014, 9, e095161. [Google Scholar] [CrossRef]
- Stevenson, V.L.; Parker, G.J.; Barker, G.J.; Birnie, K.; Tofts, P.S.; Miller, D.H.; Thompson, A.J. Variations in T1 and T2 relaxation times of normal appearing white matter and lesions in multiple sclerosis. J. Neurol. Sci. 2000, 178, 81–87. [Google Scholar] [CrossRef]
- Whittall, K.P.; MacKay, A.L.; Li, D.K.; Vavasour, I.M.; Jones, C.K.; Paty, D.W. Normal-appearing white matter in multiple sclerosis has heterogeneous, diffusely prolonged T(2). Magn. Reson. Med. 2002, 47, 403–408. [Google Scholar] [CrossRef]
- Bot, J.C.; Barkhof, F. Spinal-cord MRI in multiple sclerosis: Conventional and nonconventional MR techniques. Neuroimaging Clin. N. Am. 2009, 19, 81–99. [Google Scholar] [CrossRef] [PubMed]
- Hocq, A.; Luhmer, M.; Saussez, S.; Louryan, S.; Gillis, P.; Gossuin, Y. Effect of magnetic field and iron content on NMR proton relaxation of liver, spleen and brain tissues. Contrast Media Mol. Imaging 2015, 10, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Birkl, C.; Birkl-Toeglhofer, A.M.; Kames, C.; Goessler, W.; Haybaeck, J.; Fazekas, F.; Ropele, S.; Rauscher, A. The influence of iron oxidation state on quantitative MRI parameters in post mortem human brain. Neuroimage 2020, 220, 117080. [Google Scholar] [CrossRef]
- Hare, D.; Ayton, S.; Bush, A.; Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013, 5, 34. [Google Scholar] [CrossRef]
- Williams, R.; Buchheit, C.L.; Berman, N.E.; LeVine, S.M. Pathogenic implications of iron accumulation in multiple sclerosis. J. Neurochem. 2012, 120, 7–25. [Google Scholar] [CrossRef]
- Bakshi, R.; Benedict, R.H.; Bermel, R.A.; Caruthers, S.D.; Puli, S.R.; Tjoa, C.W.; Fabiano, A.J.; Jacobs, L. T2 hypointensity in the deep gray matter of patients with multiple sclerosis: A quantitative magnetic resonance imaging study. Arch. Neurol. 2002, 59, 62–68. [Google Scholar] [CrossRef]
- Brem, F.; Hirt, A.M.; Winklhofer, M.; Frei, K.; Yonekawa, Y.; Wieser, H.G.; Dobson, J. Magnetic iron compounds in the human brain: A comparison of tumour and hippocampal tissue. J. R. Soc. Interface 2006, 3, 833–841. [Google Scholar] [CrossRef]
- Hagemeier, J.; Heininen-Brown, M.; Poloni, G.U.; Bergsland, N.; Magnano, C.R.; Durfee, J.; Kennedy, C.; Carl, E.; Weinstock-Guttman, B.; Dwyer, M.G.; et al. Iron deposition in multiple sclerosis lesions measured by susceptibility-weighted imaging filtered phase: A case control study. J. Magn. Reson. Imaging JMRI 2012, 36, 73–83. [Google Scholar] [CrossRef]
- Hametner, S.; Wimmer, I.; Haider, L.; Pfeifenbring, S.; Bruck, W.; Lassmann, H. Iron and neurodegeneration in the multiple sclerosis brain. Ann. Neurol. 2013, 74, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Weigel, K.J.; Lynch, S.G.; LeVine, S.M. Iron chelation and multiple sclerosis. ASN Neuro 2014, 6, e00136. [Google Scholar] [CrossRef] [PubMed]
- Worthington, V.; Killestein, J.; Eikelenboom, M.J.; Teunissen, C.E.; Barkhof, F.; Polman, C.H.; Uitdehaag, B.M.; Petzold, A. Normal CSF ferritin levels in MS suggest against etiologic role of chronic venous insufficiency. Neurology 2010, 75, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebron, J.A.; Bjorkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef]
- Ganz, T. Hepcidin and iron regulation, 10 years later. Blood 2011, 117, 4425–4433. [Google Scholar] [CrossRef]
- Zecca, L.; Zucca, F.A.; Albertini, A.; Rizzio, E.; Fariello, R.G. A proposed dual role of neuromelanin in the pathogenesis of Parkinson’s disease. Neurology 2006, 67, S8–S11. [Google Scholar] [CrossRef]
- Dusek, P.; Dezortova, M.; Wuerfel, J. Imaging of iron. Int. Rev. Neurobiol. 2013, 110, 195–239. [Google Scholar] [CrossRef]
- Bakshi, R.; Shaikh, Z.A.; Janardhan, V. MRI T2 shortening (‘black T2′) in multiple sclerosis: Frequency, location, and clinical correlation. Neuroreport 2000, 11, 15–21. [Google Scholar] [CrossRef]
- Ceccarelli, A.; Rocca, M.A.; Neema, M.; Martinelli, V.; Arora, A.; Tauhid, S.; Ghezzi, A.; Comi, G.; Bakshi, R.; Filippi, M. Deep gray matter T2 hypointensity is present in patients with clinically isolated syndromes suggestive of multiple sclerosis. Mult. Scler. 2010, 16, 39–44. [Google Scholar] [CrossRef]
- Hagemeier, J.; Weinstock-Guttman, B.; Bergsland, N.; Heininen-Brown, M.; Carl, E.; Kennedy, C.; Magnano, C.; Hojnacki, D.; Dwyer, M.G.; Zivadinov, R. Iron deposition on SWI-filtered phase in the subcortical deep gray matter of patients with clinically isolated syndrome may precede structure-specific atrophy. AJNR Am. J. Neuroradiol. 2012, 33, 1596–1601. [Google Scholar] [CrossRef]
- Burgetova, A.; Seidl, Z.; Krasensky, J.; Horakova, D.; Vaneckova, M. Multiple sclerosis and the accumulation of iron in the Basal Ganglia: Quantitative assessment of brain iron using MRI t(2) relaxometry. Eur. Neurol. 2010, 63, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Brass, S.D.; Benedict, R.H.; Weinstock-Guttman, B.; Munschauer, F.; Bakshi, R. Cognitive impairment is associated with subcortical magnetic resonance imaging grey matter T2 hypointensity in multiple sclerosis. Mult. Scler. 2006, 12, 437–444. [Google Scholar] [CrossRef] [PubMed]
- van Rensburg, S.J.; Kotze, M.J.; Hon, D.; Haug, P.; Kuyler, J.; Hendricks, M.; Botha, J.; Potocnik, F.C.; Matsha, T.; Erasmus, R.T. Iron and the folate-vitamin B12-methylation pathway in multiple sclerosis. Metab. Brain Dis. 2006, 21, 121–137. [Google Scholar] [CrossRef]
- Alcina, A.; Ramagopalan, S.V.; Fernandez, O.; Catala-Rabasa, A.; Fedetz, M.; Ndagire, D.; Leyva, L.; Arnal, C.; Delgado, C.; Lucas, M.; et al. Hexose-6-phosphate dehydrogenase: A new risk gene for multiple sclerosis. Eur. J. Hum. Genet. EJHG 2010, 18, 618–620. [Google Scholar] [CrossRef] [PubMed]
- Neema, M.; Goldberg-Zimring, D.; Guss, Z.D.; Healy, B.C.; Guttmann, C.R.; Houtchens, M.K.; Weiner, H.L.; Horsfield, M.A.; Hackney, D.B.; Alsop, D.C.; et al. 3 T MRI relaxometry detects T2 prolongation in the cerebral normal-appearing white matter in multiple sclerosis. Neuroimage 2009, 46, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, A.J.; Borthakur, A.; Corbo, M.T.; Moonis, G.; Melhem, E.; Reddy, R. T2rho-weighted contrast in MR images of the human brain. Magn. Reson. Med. 2004, 52, 1223–1227. [Google Scholar] [CrossRef]
- Dingwall, N.; Chalk, A.; Martin, T.I.; Scott, C.J.; Semedo, C.; Le, Q.; Orasanu, E.; Cardoso, J.M.; Melbourne, A.; Marlow, N.; et al. T2 relaxometry in the extremely-preterm brain at adolescence. Magn. Reson. Imaging 2016, 34, 508–514. [Google Scholar] [CrossRef]
- Jensen, J.H.; Chandra, R.; Ramani, A.; Lu, H.; Johnson, G.; Lee, S.P.; Kaczynski, K.; Helpern, J.A. Magnetic field correlation imaging. Magn. Reson. Med. 2006, 55, 1350–1361. [Google Scholar] [CrossRef]
- Michaeli, S.; Oz, G.; Sorce, D.J.; Garwood, M.; Ugurbil, K.; Majestic, S.; Tuite, P. Assessment of brain iron and neuronal integrity in patients with Parkinson’s disease using novel MRI contrasts. Mov. Disord. Off. J. Mov. Disord. Soc. 2007, 22, 334–340. [Google Scholar] [CrossRef]
- Hikita, T.; Abe, K.; Sakoda, S.; Tanaka, H.; Murase, K.; Fujita, N. Determination of transverse relaxation rate for estimating iron deposits in central nervous system. Neurosci. Res. 2005, 51, 67–71. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MRI | MS Assessment |
|---|---|
| T1-weighted | brain and spinal cord atrophy hypointensities (dark areas) ~ definitive distortion of the axonal structure, demyelination, neuronal loss, and edema |
| Gd-enhanced T1-weighted | hyperintensities (bright areas) ~ active inflammation; blood–brain barrier breakdown; active demyelination |
| T2-weighted | hyperintensities (bright areas) ~ edema, gliosis, demyelination; disease burden or lesion load |
| FLAIR | hyperintensities ~ demyelinated lesions; MS activity by reducing interference from the spinal fluid |
| Magnetization transfer | magnetization transfer ratio ~ degree of demyelination |
| Diffusion | hyperintensities, high diffusibility, low fractional anisotropy ~ demyelinated axons, and damaged nerve tracks |
| Susceptibility-weighted | hyperintensities ~ iron deposits |
| Perfusion | hyperintensities ~ acute inflammatory lesions hypointensities ~ tissue degradation |
| Functional | manifestation of disrupted brain activity |
| MS Manifestation | MRS Techniques | Other MR Techniques | |
|---|---|---|---|
| demyelination | 1H MRS: ↑tCho 31P MRS: ↑PDE, ↓[Mg2+] | ↑tCho in DGM PDE > PME (mainly in WM) ↓[Mg2+] (in MS lesions) | MRI-T2 lesion load MR-relaxometry: ↑T2 magnetization transfer |
| remyelination | 1H MRS: ↑tCho 31P MRS: ↑PME | PME > PDE (mainly in WM) | |
| neuro-axonal loss | 1H MRS: ↓tNAA, ↑Glx, ↓GABA 31P MRS: ↑PCr, ↓ATP, ↓[Mg2+] | ↓tNAA (in MS lesions, NAWM, cortical GM)↑Glx excitotoxicity (mainly in WM) ↓GABA (mainly in the hippocampus and sensorimotor cortex) ↑PCr and ↑PCr/β-ATP ↓[Mg2+] (mainly in WM) | diffusion: ↑ACD, ↓FA MRI: T1 hypointensities MRI: T2 hyperintensities MR-relaxometry: ↑T2 |
| gliosis, inflammation | 1H MRS: ↑tCho and ↑mIns | ↑tCho (in MS lesions) ↑mIns (in MS lesions, NAWM) | perfusion: hyperintensities Gd-T1 hyperintensities |
| cell debris accumulation | 1H MRS: ↑tCho 31P MRS: ↑PDE | ↑tCho (in MS lesions) | magnetization transfer |
| cell energy failure | 31P MRS: ↑PCr, ↓ATP | ↑PCr and ↑PCr/β-ATP (in acute MS lesions) | functional MRI |
| BBB permeability | unknown | unknown | Gd-T1 hyperintensities |
| iron deposits | unknown | unknown | MR-relaxometry: ↓T2 SWI: hyperintensities |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hnilicová, P.; Štrbák, O.; Kolisek, M.; Kurča, E.; Zeleňák, K.; Sivák, Š.; Kantorová, E. Current Methods of Magnetic Resonance for Noninvasive Assessment of Molecular Aspects of Pathoetiology in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 6117. https://doi.org/10.3390/ijms21176117
Hnilicová P, Štrbák O, Kolisek M, Kurča E, Zeleňák K, Sivák Š, Kantorová E. Current Methods of Magnetic Resonance for Noninvasive Assessment of Molecular Aspects of Pathoetiology in Multiple Sclerosis. International Journal of Molecular Sciences. 2020; 21(17):6117. https://doi.org/10.3390/ijms21176117
Chicago/Turabian StyleHnilicová, Petra, Oliver Štrbák, Martin Kolisek, Egon Kurča, Kamil Zeleňák, Štefan Sivák, and Ema Kantorová. 2020. "Current Methods of Magnetic Resonance for Noninvasive Assessment of Molecular Aspects of Pathoetiology in Multiple Sclerosis" International Journal of Molecular Sciences 21, no. 17: 6117. https://doi.org/10.3390/ijms21176117
APA StyleHnilicová, P., Štrbák, O., Kolisek, M., Kurča, E., Zeleňák, K., Sivák, Š., & Kantorová, E. (2020). Current Methods of Magnetic Resonance for Noninvasive Assessment of Molecular Aspects of Pathoetiology in Multiple Sclerosis. International Journal of Molecular Sciences, 21(17), 6117. https://doi.org/10.3390/ijms21176117