Histone Demethylase LSD1 Regulates Kidney Cancer Progression by Modulating Androgen Receptor Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
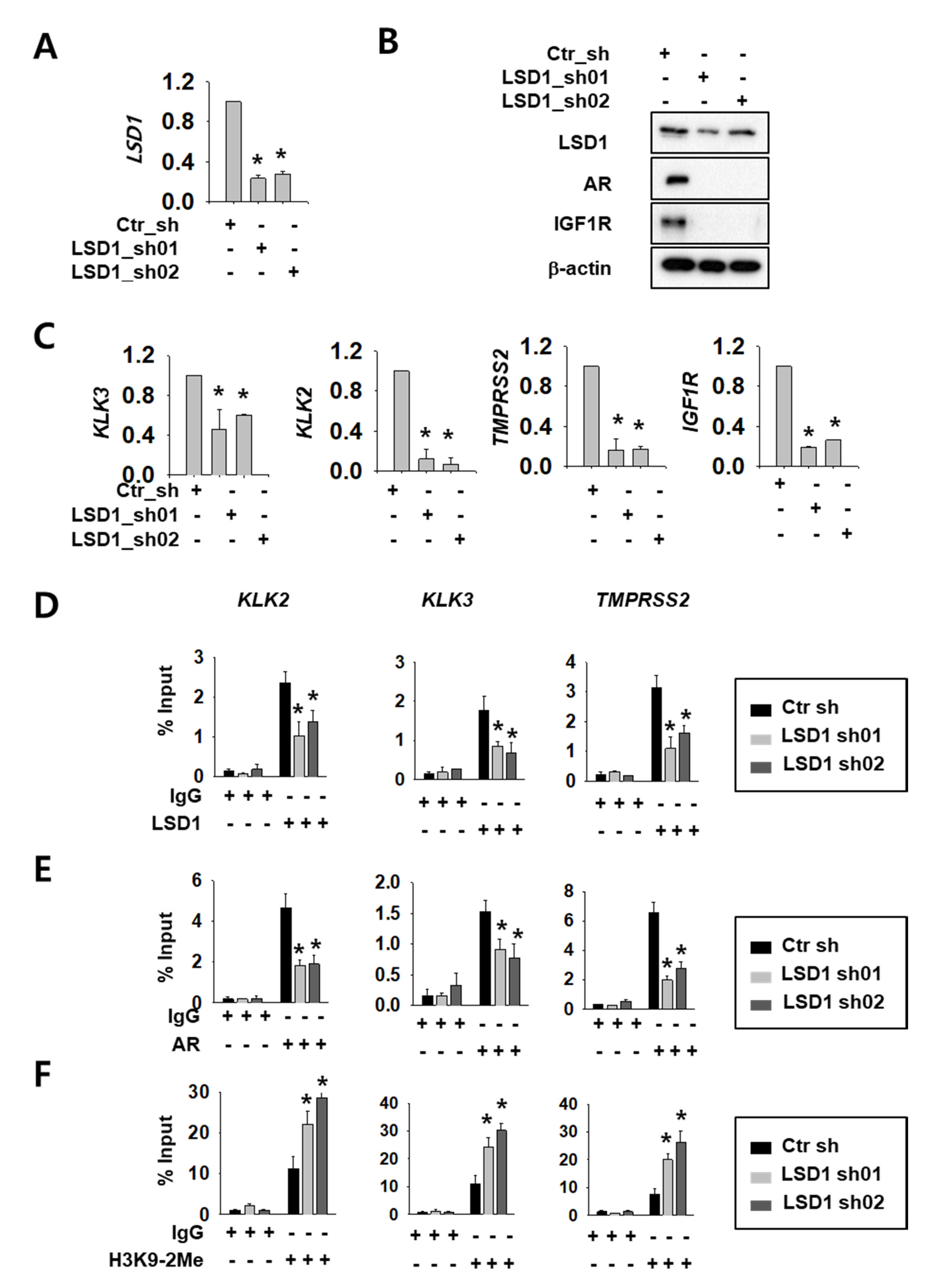
2.1. LSD1 Regulate AR Activity and Cell Proliferation in RCC
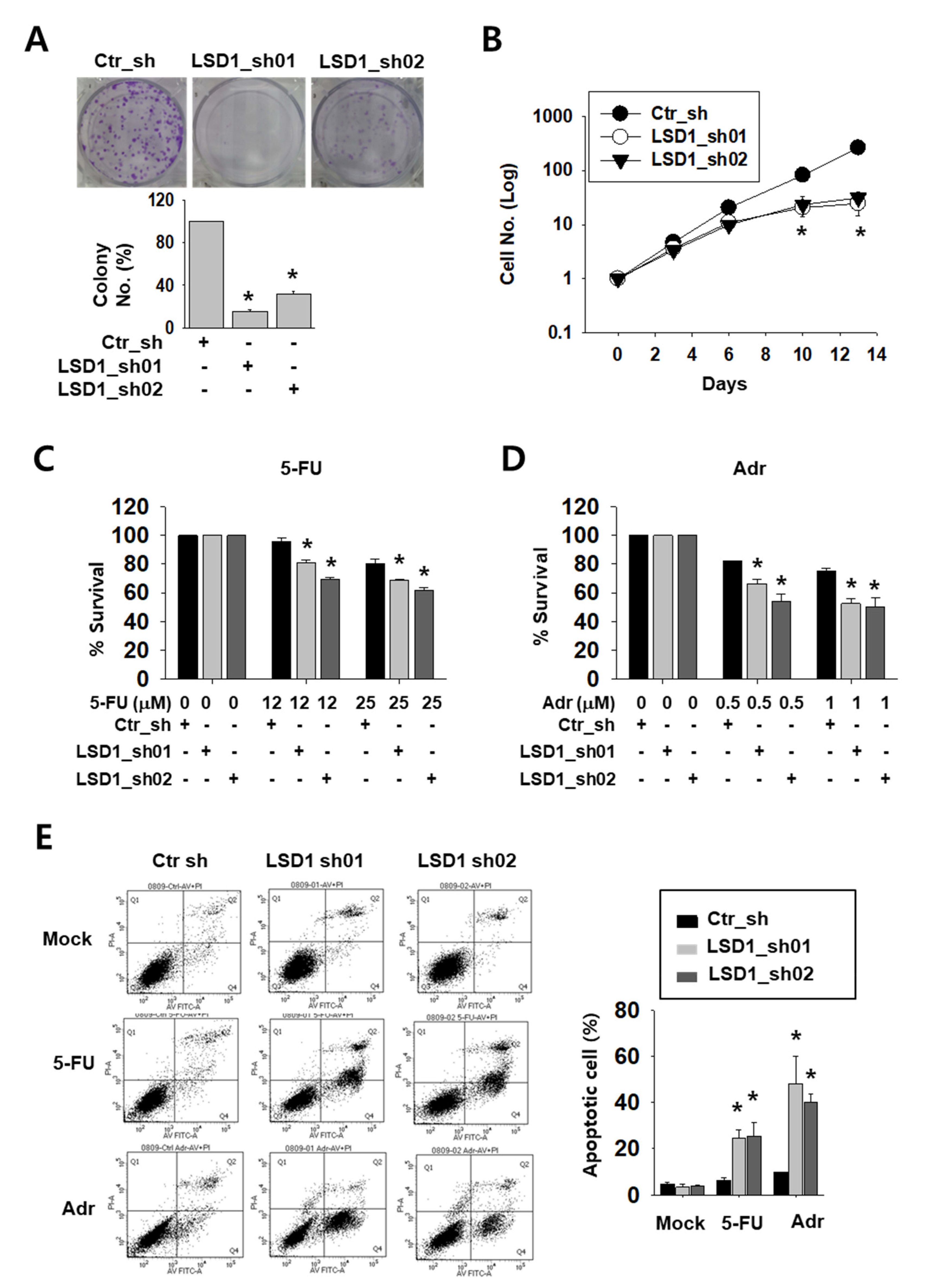
2.2. LSD1 Down-Regulation Reduces Kidney Cancer Cell Proliferation and Increases Anti-Cancer Drug Sensitivity
2.3. LSD1 Reduction Slowed Kidney Cancer Cell Migration and Decreased Epithelial–Mesenchymal Transition (EMT) Marker Expression
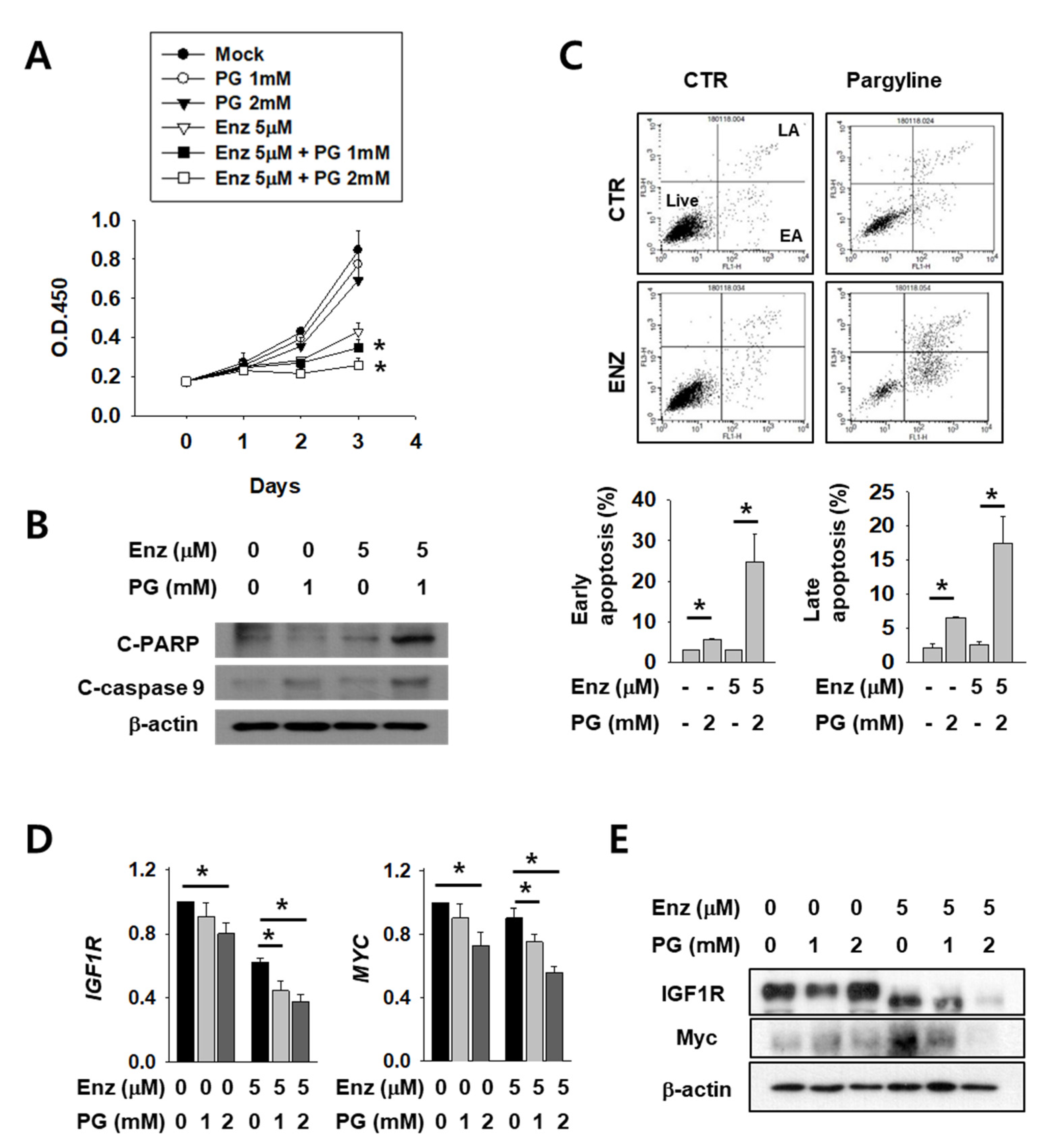
2.4. Growth Attenuation and Apoptosis Induction After Enzalutamide Treatment are Additively Increased by LSD1 Inhibitor in Kidney Cancer Cells
2.5. The Effect of Enzalutamide on EMT Marker Reduction is Enhanced by PG Co-Treatment in Kidney Cancer Cells
2.6. LSD1 is Essential for Kidney Cancer Growth in Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines, Cloning, and Virus Production
4.3. RNA Isolation and RT-qPCR
4.4. Western Blotting of Cultured Cells
4.5. Flow Cytometry for Analysis of Apoptosis and Cell Viability Assay
4.6. Colony Formation Assay and Cell Invasion Assay
4.7. Chromatin Immunoprecipitation (ChIP) Assays
4.8. Animal Study
4.9. Immunohistochemical Staining and Analysis
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AR | androgen receptor |
| ChIP | chromatin immunoprecipitation |
| DHT | dihydrotestosterone |
| E-CAD | E-cadherin |
| EMT | epithelial-mesenchymal transition |
| EZH2 | Enhancer of Zeste Homolog 2 |
| IGF1R | insulin-like growth factor 1 receptor |
| KDM | lysine demethylase |
| KLK | kallikrein related peptidase |
| LSD1 | lysine-specific histone demethylase 1 |
| N-CAD | N-cadherin |
| PCAF | P300/CBP-associated factor |
| VEGF | vascular endothelial growth factor |
| VIM | vimentin |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Roshan, M.H.; Tambo, A.; Pace, N.P. The role of testosterone in colorectal carcinoma: Pathomechanisms and open questions. EPMA J. 2016, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Ricciardelli, C.; Bianco-Miotto, T.; Jindal, S.; Butler, L.M.; Leung, S.; McNeil, C.M.; O’Toole, S.A.; Ebrahimie, E.; Millar, E.K.; Sakko, A.J.; et al. The Magnitude of Androgen Receptor Positivity in Breast Cancer Is Critical for Reliable Prediction of Disease Outcome. Clin. Cancer Res. 2018, 24, 2328–2341. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, L.; Zhang, N.; Liu, J.; Zhang, L.; Gao, H.; Wang, G.; Li, Y.; Zhang, Y.; Li, X.; et al. Androgen and AR contribute to breast cancer development and metastasis: An insight of mechanisms. Oncogene 2016, 36, 2775–2790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.-G.; Du, T.; Zang, M.-D.; Chang, Q.; Fan, Z.-Y.; Li, J.-F.; Yu, B.-Q.; Su, L.-P.; Li, C.; Yan, C.; et al. Androgen receptor promotes gastric cancer cell migration and invasion via AKT-phosphorylation dependent upregulation of matrix metalloproteinase 9. Oncotarget 2014, 5, 10584–10595. [Google Scholar] [CrossRef]
- Li, Y.; Izumi, K.; Miyamoto, H. The role of the androgen receptor in the development and progression of bladder cancer. Jpn. J. Clin. Oncol. 2012, 42, 569–577. [Google Scholar] [CrossRef]
- Zhuang, Y.H.; Blauer, M.; Tammela, T.; Tuohimaa, P. Immunodetection of androgen receptor in human urinary bladder cancer. Histopathology 1997, 30, 556–562. [Google Scholar] [CrossRef]
- Boorjian, S.; Ugras, S.; Mongan, N.P.; Gudas, L.J.; You, X.; Tickoo, S.K.; Scherr, D.S. Androgen receptor expression is inversely correlated with pathologic tumor stage in bladder cancer. Urology 2004, 64, 383–388. [Google Scholar] [CrossRef]
- Boorjian, S.A.; Heemers, H.V.; Frank, I.; Farmer, S.A.; Schmidt, L.J.; Sebo, T.J.; Tindall, D.J. Expression and significance of androgen receptor coactivators in urothelial carcinoma of the bladder. Endocr.-Relat. Cancer 2009, 16, 123–137. [Google Scholar] [CrossRef]
- Foersch, S.; Schindeldecker, M.; Keith, M.; Tagscherer, K.E.; Fernandez, A.; Stenzel, P.J.; Pahernik, S.; Hohenfellner, M.; Schirmacher, P.; Roth, W.; et al. Prognostic relevance of androgen receptor expression in renal cell carcinomas. Oncotarget 2017, 8, 78545–78555. [Google Scholar] [CrossRef]
- Zhao, H.; Leppert, J.T.; Peehl, D.M. A Protective Role for Androgen Receptor in Clear Cell Renal Cell Carcinoma Based on Mining TCGA Data. PLoS ONE 2016, 11, e0146505. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Li, L.; Zhu, G.; Liang, L.; Guan, Z.; Chang, L.; Chen, Y.; Yeh, S.; Chang, C. ASC-J9 suppresses renal cell carcinoma progression by targeting an androgen receptor-dependent HIF2alpha/VEGF signaling pathway. Cancer Res. 2014, 74, 4420–4430. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, Y.; Rao, Q.; Xu, H.; Li, L.; Chang, C. Androgen receptor (AR) suppresses miRNA-145 to promote renal cell carcinoma (RCC) progression independent of VHL status. Oncotarget 2015, 6, 31203–31215. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Sun, Y.; Guo, C.; Liu, T.; Fei, X.; Chang, C. Androgen receptor regulates ASS1P3/miR-34a-5p/ASS1 signaling to promote renal cell carcinoma cell growth. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pak, S.; Kim, W.; Kim, Y.; Song, C.; Ahn, H. Dihydrotestosterone promotes kidney cancer cell proliferation by activating the STAT5 pathway via androgen and glucocorticoid receptors. J. Cancer Res. Clin. Oncol. 2019, 145, 2293–2301. [Google Scholar] [CrossRef]
- Sun, Z.; Pan, J.; Hope, W.X.; Cohen, S.N.; Balk, S.P. Tumor susceptibility gene 101 protein represses androgen receptor transactivation and interacts with p300. Cancer 1999, 86, 689–696. [Google Scholar] [CrossRef]
- Brady, M.E.; Ozanne, D.M.; Gaughan, L.; Waite, I.; Cook, S.; Neal, D.E.; Robson, C.N. Tip60 Is a Nuclear Hormone Receptor Coactivator. J. Boil. Chem. 1999, 274, 17599–17604. [Google Scholar] [CrossRef]
- Veldscholte, J.; Berrevoets, C.A.; Brinkmann, A.O.; Grootegoed, J.A.; Mulder, E. Anti-androgens and the mutated androgen receptor of LNCaP cells: Differential effects on binding affinity, heat-shock protein interaction, and transcription activation. Biochemistry 1992, 31, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Marshall, T.W.; Link, K.A.; Petre-Draviam, C.E.; Knudsen, K.E. Differential requirement of SWI/SNF for androgen receptor activity. J. Boil. Chem. 2003, 278, 30605–30613. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 2010, 29, 3691–3702. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bassets, I.; Kwon, Y.-S.; Telese, F.; Prefontaine, G.G.; Hutt, K.R.; Cheng, C.S.; Ju, B.-G.; Ohgi, K.A.; Wang, J.; Escoubet-Lozach, L.; et al. Histone Methylation-Dependent Mechanisms Impose Ligand Dependency for Gene Activation by Nuclear Receptors. Cell 2007, 128, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, M.; Yin, N.; Müller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Günther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef]
- Oxenkrug, G.F. Antidepressive and antihypertensive effects of MAO-A inhibition: Role of N-acetylserotonin. A review. Neurobiology 1999, 7, 213–224. [Google Scholar]
- Wang, M.; Liu, X.; Guo, J.; Weng, X.; Jiang, G.; Wang, Z.; He, L. Inhibition of LSD1 by Pargyline inhibited process of EMT and delayed progression of prostate cancer in vivo. Biochem. Biophys. Res. Commun. 2015, 467, 310–315. [Google Scholar] [CrossRef]
- Konovalov, S.; Garcia-Bassets, I. Analysis of the levels of lysine-specific demethylase 1 (LSD1) mRNA in human ovarian tumors and the effects of chemical LSD1 inhibitors in ovarian cancer cell lines. J. Ovarian Res. 2013, 6, 75. [Google Scholar] [CrossRef]
- Colditz, J.; Rupf, B.; Maiwald, C.; Baniahmad, A. Androgens induce a distinct response of epithelial-mesenchymal transition factors in human prostate cancer cells. Mol. Cell. Biochem. 2016, 421, 139–147. [Google Scholar] [CrossRef]
- Zhu, G.; Liang, L.; Li, L.; Dang, Q.; Song, W.; Yeh, S.; He, D.; Chang, C. The Expression and Evaluation of Androgen Receptor in Human Renal Cell Carcinoma. Urology 2014, 83, 510.e19–510.e24. [Google Scholar] [CrossRef]
- Huang, Q.; Sun, Y.; Ma, X.; Gao, Y.; Li, X.; Niu, Y.; Zhang, X.; Chang, C. Androgen receptor increases hematogenous metastasis yet decreases lymphatic metastasis of renal cell carcinoma. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Adelaiye-Ogala, R.; Damayanti, N.P.; Orillion, A.R.; Arisa, S.; Chintala, S.; Titus, M.A.; Kao, C.; Pili, R. Therapeutic Targeting of Sunitinib-Induced AR Phosphorylation in Renal Cell Carcinoma. Cancer Res. 2018, 78, 2886–2896. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schule, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef]
- Huang, Y.; Vasilatos, S.N.; Boric, L.; Shaw, P.G.; Davidson, N.E. Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res. Treat. 2012, 131, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.M.; McCafferty, D.G. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Choi, M.R.; Doh, M.S.; Jung, K.H.; Chai, Y.G. Effects of the monoamine oxidase inhibitors pargyline and tranylcypromine on cellular proliferation in human prostate cancer cells. Oncol. Rep. 2013, 30, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Schalken, J.; Fitzpatrick, J.M. Enzalutamide: Targeting the androgen signalling pathway in metastatic castration-resistant prostate cancer. BJU Int. 2016, 117, 215–225. [Google Scholar] [CrossRef]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef]
- Carnesecchi, J.; Forcet, C.; Zhang, L.; Tribollet, V.; Barenton, B.; Boudra, R.; Cerutti, C.; Billas, I.M.; Serandour, A.A.; Carroll, J.S.; et al. ERRalpha induces H3K9 demethylation by LSD1 to promote cell invasion. Proc. Natl Acad Sci. USA 2017, 114, 3909–3914. [Google Scholar] [CrossRef]
- Kim, D.; Nam, H.J.; Lee, W.; Yim, H.Y.; Ahn, J.Y.; Park, S.W.; Shin, H.R.; Yu, R.; Won, K.J.; Bae, J.S.; et al. PKCα-LSD1-NF-κB-Signaling Cascade Is Crucial for Epigenetic Control of the Inflammatory Response. Mol. Cell 2018, 69, 398–411.e6. [Google Scholar] [CrossRef]
- Ahmed, M.; Streit, A. Lsd1 interacts with cMyb to demethylate repressive histone marks and maintain inner ear progenitor identity. Development 2018, 145, dev160325. [Google Scholar] [CrossRef]
- Hu, X.; Xiang, D.; Xie, Y.; Tao, L.; Zhang, Y.; Jin, Y.; Pinello, L.; Wan, Y.; Yuan, G.C.; Li, Z. LSD1 suppresses invasion, migration and metastasis of luminal breast cancer cells via activation of GATA3 and repression of TRIM37 expression. Oncogene 2019, 38, 7017–7034. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Hong, S.; Kang, M.; Jeong, C.W.; Ku, J.H.; Kim, H.H.; Kwak, C. Histone demethylase KDM7A controls androgen receptor activity and tumor growth in prostate cancer. Int. J. Cancer 2018, 143, 2849–2861. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-H.; Kim, B.-C.; Jeong, S.-H.; Jeong, C.W.; Ku, J.H.; Kwak, C.; Kim, H.H. Histone Demethylase LSD1 Regulates Kidney Cancer Progression by Modulating Androgen Receptor Activity. Int. J. Mol. Sci. 2020, 21, 6089. https://doi.org/10.3390/ijms21176089
Lee K-H, Kim B-C, Jeong S-H, Jeong CW, Ku JH, Kwak C, Kim HH. Histone Demethylase LSD1 Regulates Kidney Cancer Progression by Modulating Androgen Receptor Activity. International Journal of Molecular Sciences. 2020; 21(17):6089. https://doi.org/10.3390/ijms21176089
Chicago/Turabian StyleLee, Kyoung-Hwa, Byung-Chan Kim, Seung-Hwan Jeong, Chang Wook Jeong, Ja Hyeon Ku, Cheol Kwak, and Hyeon Hoe Kim. 2020. "Histone Demethylase LSD1 Regulates Kidney Cancer Progression by Modulating Androgen Receptor Activity" International Journal of Molecular Sciences 21, no. 17: 6089. https://doi.org/10.3390/ijms21176089
APA StyleLee, K.-H., Kim, B.-C., Jeong, S.-H., Jeong, C. W., Ku, J. H., Kwak, C., & Kim, H. H. (2020). Histone Demethylase LSD1 Regulates Kidney Cancer Progression by Modulating Androgen Receptor Activity. International Journal of Molecular Sciences, 21(17), 6089. https://doi.org/10.3390/ijms21176089