The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents




Abstract
1. Introduction
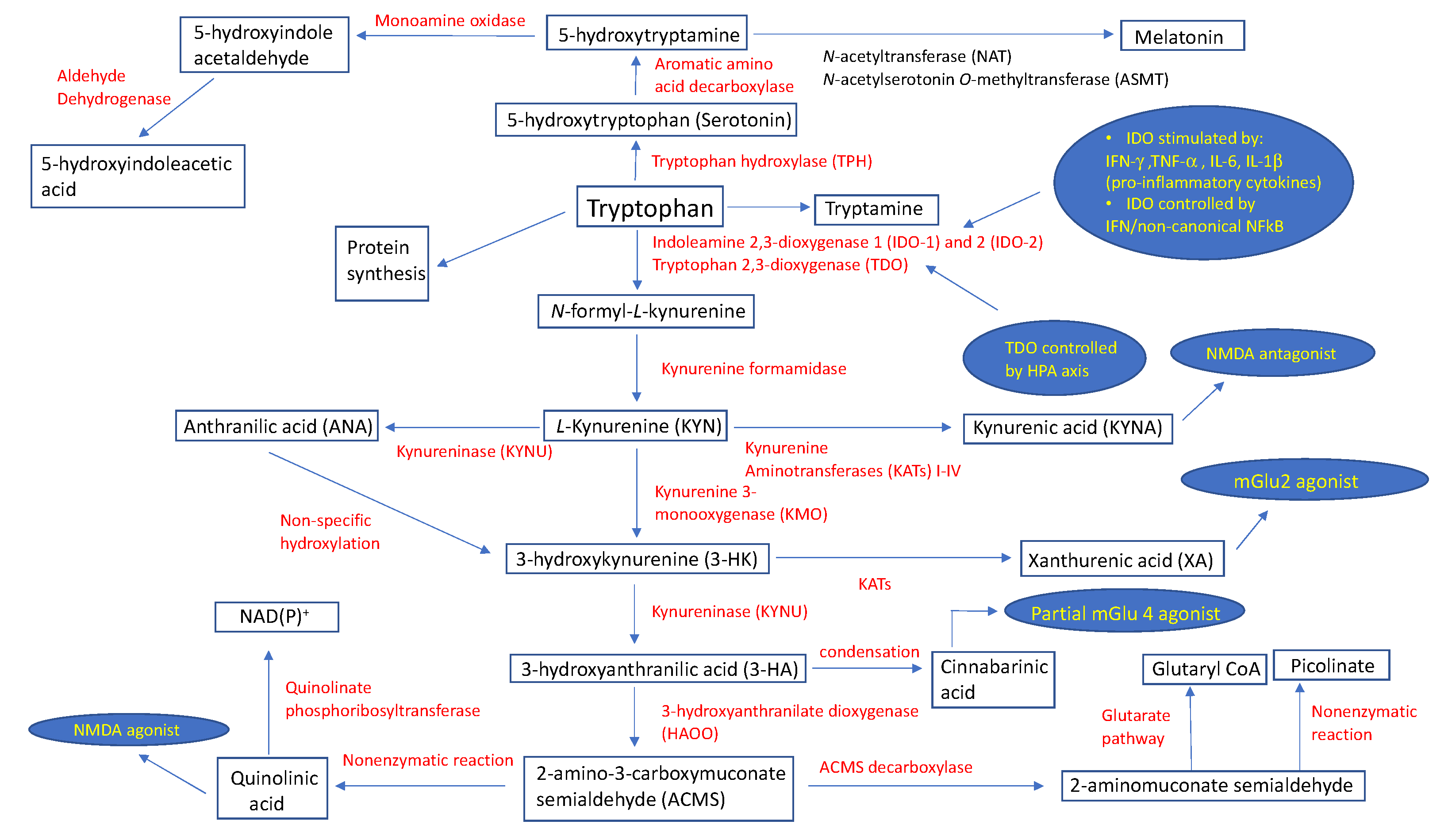
2. The Catabolic Pathway of Tryptophan
3. Variability of Kynurenine Metabolism between Human and Non-Human Species
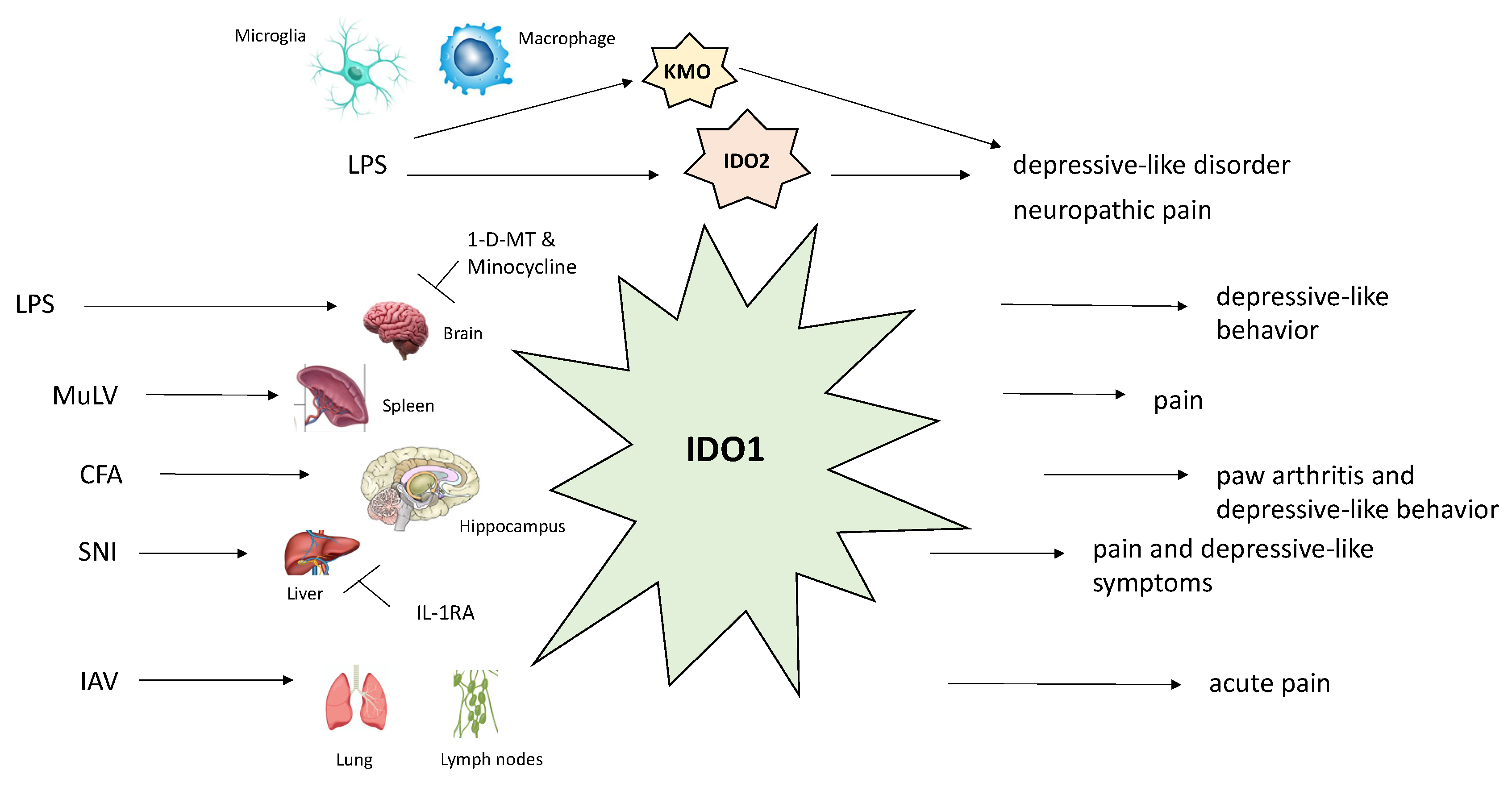
4. Kynurenine Pathway in the Pathogenesis of Neuropathic Pain
5. Kynurenine Pathway and Association with Chronic Pain and Depression
6. Kynurenine Pathway and Headaches
7. Kynurenine Pathway and Other Conditions
8. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hopkins, F.G.; Cole, S.W. A contribution to the chemistry of proteids: Part I. A preliminary study of a hitherto undescribed product of tryptic digestion. J. Physiol. 1901, 27, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Hoglund, E.; Overli, O.; Winberg, S. Tryptophan Metabolic Pathways and Brain Serotonergic Activity: A Comparative Review. Front. Endocrinol. 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Dharmshaktu, P.; Tayal, V.; Kalra, B.S. Efficacy of antidepressants as analgesics: A review. J. Clin. Pharmacol. 2012, 52, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yu, Y.; Shen, Y.; Liu, Q.; Zhao, Z.; Sharma, R.; Reiter, R.J. Melatonin Synthesis and Function: Evolutionary History in Animals and Plants. Front. Endocrinol. 2019, 10, 249. [Google Scholar] [CrossRef]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef]
- Campbell, B.M.; Charych, E.; Lee, A.W.; Möller, T. Kynurenines in CNS disease: Regulation by inflammatory cytokines. Front. Neurosci. 2014, 8, 12. [Google Scholar] [CrossRef]
- Shimizu, T.; Nomiyama, S.; Hirata, F.; Hayaishi, O. Indoleamine 2,3-dioxygenase. Purification and some properties. J. Biol. Chem. 1978, 253, 4700–4706. [Google Scholar]
- Takikawa, O.; Kuroiwa, T.; Yamazaki, F.; Kido, R. Mechanism of interferon-gamma action. Characterization of indoleamine 2,3-dioxygenase in cultured human cells induced by interferon-gamma and evaluation of the enzyme-mediated tryptophan degradation in its anticellular activity. J. Biol. Chem. 1988, 263, 2041–2048. [Google Scholar]
- Jeong, Y.I.; Kim, S.W.; Jung, I.D.; Lee, J.S.; Chang, J.H.; Lee, C.M.; Chun, S.H.; Yoon, M.S.; Kim, G.T.; Ryu, S.W.; et al. Curcumin suppresses the induction of indoleamine 2,3-dioxygenase by blocking the Janus-activated kinase-protein kinase Cdelta-STAT1 signaling pathway in interferon-gamma-stimulated murine dendritic cells. J. Biol. Chem. 2009, 284, 3700–3708. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Ogbechi, J.; Clanchy, F.I.; Williams, R.O.; Stone, T.W. IDO and Kynurenine Metabolites in Peripheral and CNS Disorders. Front. Immunol. 2020, 11, 388. [Google Scholar] [CrossRef]
- Metz, R.; DuHadaway, J.B.; Kamasani, U.; Laury-Kleintop, L.; Muller, A.J.; Prendergast, G.C. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007, 67, 7082–7087. [Google Scholar] [CrossRef] [PubMed]
- Green, A.R.; Sourkes, T.; Young, S. Liver and brain tryptophan metabolism following hydrocortisone administration to rats and gerbils. Br. J. Pharmacol. 1975, 53, 287–292. [Google Scholar] [CrossRef] [PubMed][Green Version]
- O’Farrell, K.; Harkin, A. Stress-related regulation of the kynurenine pathway: Relevance to neuropsychiatric and degenerative disorders. Neuropharmacology 2017, 112 Pt B, 307–323. [Google Scholar] [CrossRef]
- Kim, J.; Hong, H.; Heo, A.; Park, W. Indole toxicity involves the inhibition of adenosine triphosphate production and protein folding in Pseudomonas putida. FEMS Microbiol. Lett. 2013, 343, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Gargaro, M.; Vacca, C.; Massari, S.; Scalise, G.; Manni, G.; Mondanelli, G.; Mc Mazza, E.; Bicciato, S.; Pallotta, M.T.; Orabona, C.; et al. Engagement of Nuclear Coactivator 7 by 3-Hydroxyanthranilic Acid Enhances Activation of Aryl Hydrocarbon Receptor in Immunoregulatory Dendritic Cells. Front. Immunol. 2019, 10, 1973. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Cai, T.; Tagle, D.A.; Li, J. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cell. Mol. Life Sci. 2010, 67, 353–368. [Google Scholar] [CrossRef]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J. Neurochem. 2020, 152, 627–649. [Google Scholar] [CrossRef]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef]
- Rózsa, É.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-face kynurenic acid. J. Neural Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef]
- Braidy, N.; Grant, R.; Brew, B.J.; Adams, S.; Jayasena, T.; Guillemin, G.J. Effects of Kynurenine Pathway Metabolites on Intracellular NAD Synthesis and Cell Death in Human Primary Astrocytes and Neurons. Int. J. Tryptophan Res. 2009, 2, 61–69. [Google Scholar] [CrossRef]
- Ocampo, J.G.R.; Huitrón, R.L.; González-Esquivel, D.; Ugalde-Muñiz, P.; Jiménez-Anguiano, A.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; De La Cruz, V.P. Kynurenines with neuroactive and redox properties: Relevance to aging and brain diseases. Oxid. Med. Cell. Longev. 2014, 2014, 646909. [Google Scholar]
- Fazio, F.; Lionetto, L.; Molinaro, G.; Bertrand, H.O.; Acher, F.; Ngomba, R.; Notartomaso, S.; Curini, M.; Rosati, O.; Scarselli, P.; et al. Cinnabarinic acid, an endogenous metabolite of the kynurenine pathway, activates type 4 metabotropic glutamate receptors. Mol. Pharmacol. 2012, 81, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Lionetto, L.; Curto, M.; Iacovelli, L.; Cavallari, M.; Zappulla, C.; Ulivieri, M.; Napoletano, F.; Capi, M.; Corigliano, V.; et al. Xanthurenic Acid Activates mGlu2/3 Metabotropic Glutamate Receptors and is a Potential Trait Marker for Schizophrenia. Sci. Rep. 2015, 5, 17799. [Google Scholar] [CrossRef] [PubMed]
- Pucci, L.; Perozzi, S.; Cimadamore, F.; Orsomando, G.; Raffaelli, N. Tissue expression and biochemical characterization of human 2-amino 3-carboxymuconate 6-semialdehyde decarboxylase, a key enzyme in tryptophan catabolism. FEBS J. 2007, 274, 827–840. [Google Scholar] [CrossRef]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef]
- Phillips, R.S. Structure, mechanism, and substrate specificity of kynureninase. Biochim. Biophys. Acta 2011, 1814, 1481–1488. [Google Scholar] [CrossRef]
- Murakami, Y.; Saito, K. Species and cell types difference in tryptophan metabolism. Int. J. Tryptophan Res. 2013, 6 (Suppl. 1), 47–54. [Google Scholar] [CrossRef]
- Fujigaki, S.; Saito, K.; Takemura, M.; Fujii, H.; Wada, H.; Noma, A.; Seishima, M. Species differences in L-tryptophan-kynurenine pathway metabolism: Quantification of anthranilic acid and its related enzymes. Arch. Biochem. Biophys. 1998, 358, 329–335. [Google Scholar] [CrossRef]
- Fujigaki, S.; Saito, K.; Sekikawa, K.; Tone, S.; Takikawa, O.; Fujii, H.; Wada, H.; Noma, A.; Seishima, M. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-gamma-independent mechanism. Eur. J. Immunol. 2001, 31, 2313–2318. [Google Scholar] [CrossRef]
- Heyes, M.P.; Saito, K.; Chen, C.Y.; Proescholdt, M.G.; Nowak, T.S.; Li, J.; Beagles, K.E.; Proescholdt, M.A.; Zito, M.A.; Kawai, K.; et al. Species heterogeneity between gerbils and rats: Quinolinate production by microglia and astrocytes and accumulations in response to ischemic brain injury and systemic immune activation. J. Neurochem. 1997, 69, 1519–1529. [Google Scholar] [CrossRef]
- Heyes, M.P.; Chen, C.Y.; Major, E.O.; Saito, K. Different kynurenine pathway enzymes limit quinolinic acid formation by various human cell types. Biochem. J. 1997, 326 Pt 2, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Leitner, K.L.; Meyer, M.; Leimbacher, W.; Peterbauer, A.; Hofer, S.; Heufler, C.; Müller, A.; Heller, R.; Werner, E.R.; Thöny, B.; et al. Low tetrahydrobiopterin biosynthetic capacity of human monocytes is caused by exon skipping in 6-pyruvoyl tetrahydropterin synthase. Biochem. J. 2003, 373 Pt 3, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Schneemann, M.; Schoedon, G. Species differences in macrophage NO production are important. Nat. Immunol. 2002, 3, 102. [Google Scholar] [CrossRef]
- Samelson-Jones, B.J.; Yeh, S.R. Interactions between nitric oxide and indoleamine 2,3-dioxygenase. Biochemistry 2006, 45, 8527–8538. [Google Scholar] [CrossRef]
- Pires, A.S.; Tan, V.X.; Heng, B.; Guillemin, G.J.; Latini, A. Kynurenine and Tetrahydrobiopterin Pathways Crosstalk in Pain Hypersensitivity. Front. Neurosci. 2020, 14, 620. [Google Scholar] [CrossRef]
- Pickert, G.; Myrczek, T.; Rückert, S.; Weigert, A.; Häussler, A.; Ferreirós, N.; Brüne, B.; Lotsch, J.; Tegeder, I. Inhibition of GTP cyclohydrolase reduces cancer pain in mice and enhances analgesic effects of morphine. J. Mol. Med. 2012, 90, 1473–1486. [Google Scholar] [CrossRef]
- International Association for the Study of Pain. Available online: https://www.iasp-pain.org/Education/Content.aspx?ItemNumber=1698#Neuropathicpain (accessed on 11 July 2020).
- Colburn, R.W.; Rickman, A.; DeLeo, J. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp. Neurol. 1999, 157, 289–304. [Google Scholar] [CrossRef]
- Teng, Y.; Zhang, Y.; Yue, S.; Chen, H.; Qu, Y.-J.; Wei, H.; Jia, X. Intrathecal injection of bone marrow stromal cells attenuates neuropathic pain via inhibition of P2X4R in spinal cord microglia. J. Neuroinflamm. 2019, 16, 271. [Google Scholar] [CrossRef]
- Woolf, C.J.; Mannion, R.J. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999, 353, 1959–1964. [Google Scholar] [CrossRef]
- Rojewska, E.; Ciapała, K.; Piotrowska, A.; Makuch, W.; Mika, J. Pharmacological Inhibition of Indoleamine 2,3-Dioxygenase-2 and Kynurenine 3-Monooxygenase, Enzymes of the Kynurenine Pathway, Significantly Diminishes Neuropathic Pain in a Rat Model. Front. Pharmacol. 2018, 9, 724. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Farias, J.B.; Perez-Severiano, F.; Gonzalez-Esquivel, D.F.; Barragan-Iglesias, P.; Bravo-Hernandez, M.; Cervantes-Duran, C.; Aguilera, P.; Ríos, C.; Granados-Soto, V. The L-kynurenine-probenecid combination reduces neuropathic pain in rats. Eur. J. Pain 2013, 17, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Laumet, G.; Zhou, W.; Dantzer, R.; Edralin, J.D.; Huo, X.; Budac, D.P.; O’Connor, J.C.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Upregulation of neuronal kynurenine 3-monooxygenase mediates depression-like behavior in a mouse model of neuropathic pain. Brain Behav. Immun. 2017, 66, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Rojewska, E.; Piotrowska, A.; Makuch, W.; Przewlocka, B.; Mika, J. Pharmacological kynurenine 3-monooxygenase enzyme inhibition significantly reduces neuropathic pain in a rat model. Neuropharmacology 2016, 102, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Dantzer, R.; Budac, D.P.; Walker, A.K.; Mao-Ying, Q.-L.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Peripheral indoleamine 2,3-dioxygenase 1 is required for comorbid depression-like behavior but does not contribute to neuropathic pain in mice. Brain Behav. Immun. 2015, 46, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Kita, T.; Morrison, P.F.; Heyes, M.P.; Markey, S. Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the L-kynurenine and quinolinic acid pools in brain. J. Neurochem. 2002, 82, 258–268. [Google Scholar] [CrossRef]
- Bair, M.J.; Robinson, R.L.; Katon, W.; Kroenke, K. Depression and pain comorbidity: A literature review. Arch. Intern. Med. 2003, 163, 2433–2445. [Google Scholar] [CrossRef]
- Meerwijk, E.L.; Ford, J.M.; Weiss, S.J. Brain regions associated with psychological pain: Implications for a neural network and its relationship to physical pain. Brain Imaging Behav. 2013, 7, 1–14. [Google Scholar] [CrossRef]
- Patel, A. Review: The role of inflammation in depression. Psychiatr. Danub. 2013, 25 (Suppl. 2), S216–S223. [Google Scholar]
- Tanaka, M.; Bohar, Z.; Vecsei, L. Are Kynurenines Accomplices or Principal Villains in Dementia? Maintenance of Kynurenine Metabolism. Molecules 2020, 25, 564. [Google Scholar] [CrossRef]
- Kinney, J.W.; BeMiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Brosseron, F.; Krauthausen, M.; Kummer, M.P.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Fujii, T.; Koga, N.; Hori, H.; Teraishi, T.; Hattori, K.; Noda, T.; Higuchi, T.; Motohashi, N.; Kunugi, H. Plasma L-tryptophan concentration in major depressive disorder: New data and meta-analysis. J. Clin. Psychiatry 2014, 75, e906–e915. [Google Scholar] [CrossRef]
- Ogyu, K.; Kubo, K.; Noda, Y.; Iwata, Y.; Tsugawa, S.; Omura, Y.; Wada, M.; Tarumi, R.; Plitman, E.; Moriguchi, S.; et al. Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2018, 90, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Réus, G.Z.; Jansen, K.; Titus, S.; Carvalho, A.F.; Gabbay, V.; Quevedo, J. Kynurenine pathway dysfunction in the pathophysiology and treatment of depression: Evidences from animal and human studies. J. Psychiatr. Res. 2015, 68, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Fuchs, D.; Widner, B.; Glover, C.; Henderson, D.C.; Allen-Mersh, T.G. Serum tryptophan decrease correlates with immune activation and impaired quality of life in colorectal cancer. Br. J. Cancer 2002, 86, 1691–1696. [Google Scholar] [CrossRef]
- Capuron, L.; Moranis, A.; Combe, N.; Cousson-Gélie, F.; Fuchs, D.; De Smedt-Peyrusse, V.; Barberger-Gateau, P.; Layé, S. Vitamin E status and quality of life in the elderly: Influence of inflammatory processes. Br. J. Nutr. 2009, 102, 1390–1394. [Google Scholar] [CrossRef]
- Lawson, M.A.; Parrott, J.M.; McCusker, R.H.; Dantzer, R.; Kelley, K.W.; O’Connor, J.C. Intracerebroventricular administration of lipopolysaccharide induces indoleamine-2,3-dioxygenase-dependent depression-like behaviors. J. Neuroinflamm. 2013, 10, 875. [Google Scholar] [CrossRef]
- O’Connor, J.C.; Lawson, M.; André, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 2009, 14, 511–522. [Google Scholar] [CrossRef]
- Deng, Y.-T.; Zhao, M.-G.; Xu, T.-J.; Hou, J.-; Li, X.-H. Gentiopicroside abrogates lipopolysaccharide-induced depressive-like behavior in mice through tryptophan-degrading pathway. Metab. Brain Dis. 2018, 33, 1413–1420. [Google Scholar] [CrossRef]
- Farghaly, H.S.; Mahmoud, A.M.; Abdel-Sater, K.A. Effect of dexmedetomidine and cold stress in a rat model of neuropathic pain: Role of interleukin-6 and tumor necrosis factor-alpha. Eur. J. Pharmacol. 2016, 776, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Feng, Y.-B.; Wang, L.; Li, Y.; Fan, C.; Song, Q.; Yu, S.Y. Interleukin-6: Its role and mechanisms in rescuing depression-like behaviors in rat models of depression. Brain Behav. Immun. 2019, 82, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chen, L.; Lim, G.; Sung, B.; Wang, S.; McCabe, M.F.; Rusanescu, G.; Yang, L.; Tian, Y.; Mao, J. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J. Clin. Investig. 2012, 122, 2940–2954. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Ou, R.; De Souza, G.R.; Cunha, T.M.; Lemos, H.; Mohamed, E.; Li, L.; Pacholczyk, G.; Randall, J.; Munn, D.H.; et al. Virus Infections Incite Pain Hypersensitivity by Inducing Indoleamine 2,3 Dioxygenase. PLoS Pathog. 2016, 12, e1005615. [Google Scholar] [CrossRef]
- Huang, L.; Li, L.; Klonowski, K.D.; Tompkins, S.M.; Tripp, R.A.; Mellor, A.L. Induction and role of indoleamine 2,3 dioxygenase in mouse models of influenza a virus infection. PLoS ONE 2013, 8, e66546. [Google Scholar] [CrossRef]
- Walker, A.K.; Budac, D.P.; Bisulco, S.; Lee, A.W.; Smith, R.A.; Beenders, B.; Kelley, K.W.; Dantzer, R. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology 2013, 38, 1609–1616. [Google Scholar] [CrossRef]
- Parrott, J.M.; Redus, L.; Santana-Coelho, D.; Morales, J.; Gao, X.; O’Connor, J.C. Neurotoxic kynurenine metabolism is increased in the dorsal hippocampus and drives distinct depressive behaviors during inflammation. Transl. Psychiatry 2016, 6, e918. [Google Scholar] [CrossRef]
- Pan, W.; Zhang, G.-F.; Li, H.-H.; Ji, M.-H.; Zhou, Z.-Q.; Li, K.-Y.; Yang, J.-J. Ketamine differentially restores diverse alterations of neuroligins in brain regions in a rat model of neuropathic pain-induced depression. Neuroreport 2018, 29, 863–869. [Google Scholar] [CrossRef]
- Rogachov, A.; Bhatia, A.; Cheng, J.C.; Bosma, R.L.; Kim, J.A.; Osborne, N.R.; Hemington, K.S.; Venkatraghavan, L.; Davis, K.D. Plasticity in the dynamic pain connectome associated with ketamine-induced neuropathic pain relief. Pain 2019, 160, 1670–1679. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Collins, S.; Sigtermans, M.J.; Dahan, A.; Zuurmond, W.W.A.; Perez, R.S. NMDA receptor antagonists for the treatment of neuropathic pain. Pain Med. 2010, 11, 1726–1742. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yan, Q.; Liu, F.; Jing, C.; Ding, L.; Zhang, L.; Pang, C. Chronic trans-astaxanthin treatment exerts antihyperalgesic effect and corrects co-morbid depressive like behaviors in mice with chronic pain. Neurosci. Lett. 2018, 662, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; May, A. Diagnosis, pathophysiology, and management of cluster headache. Lancet Neurol. 2018, 17, 75–83. [Google Scholar] [CrossRef]
- Anttila, V.; Stefansson, H.; Kallela, M.; Todt, U.; Terwindt, G.M.; Calafato, M.S.; Freilinger, T.; Müller-Myhsok, B.; Artto, V. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat. Genet. 2010, 42, 869–873. [Google Scholar]
- Anttila, V.; Winsvold, B.S.; Gormley, P.; Kurth, T.; Bettella, F.; McMahon, G.; Kallela, M.; Malik, R.; De Vries, B.; Terwindt, G.; et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat. Genet. 2013, 45, 912–917. [Google Scholar] [CrossRef]
- Chasman, D.I.; Schürks, M.; Anttila, V.; De Vries, B.; Schminke, U.; Launer, L.J.; Terwindt, G.M.; van den Maagdenberg, A.M.; Fendrich, K.; Völzke, H.; et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat. Genet. 2011, 43, 695–698. [Google Scholar] [CrossRef]
- Ferrari, M.D.; Klever, R.R.; Terwindt, G.M.; Ayata, C.; van den Maagdenberg, A.M. Migraine pathophysiology: Lessons from mouse models and human genetics. Lancet Neurol. 2015, 14, 65–80. [Google Scholar] [CrossRef]
- Freilinger, T.; Anttila, V.; De Vries, B.; Malik, R.; Kallela, M.; Terwindt, G.M.; Pozo-Rosich, P.; Winsvold, B.S.; Nyholt, D.R.; Van Oosterhout, W.P.J.; et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat. Genet. 2012, 44, 777–782. [Google Scholar] [CrossRef]
- Kallela, M.; Wessman, M.; Havanka, H.; Palotie, A.; Farkkila, M. Familial migraine with and without aura: Clinical characteristics and co-occurrence. Eur. J. Neurol. 2001, 8, 441–449. [Google Scholar] [CrossRef]
- Nyholt, D.R.; Gillespie, N.G.; Heath, A.C.; Merikangas, K.R.; Duffy, D.; Martin, N. Latent class and genetic analysis does not support migraine with aura and migraine without aura as separate entities. Genet. Epidemiol. 2004, 26, 231–244. [Google Scholar] [CrossRef]
- Akerman, S.; Romero-Reyes, M.; Holland, P.R. Current and novel insights into the neurophysiology of migraine and its implications for therapeutics. Pharmacol. Ther. 2017, 172, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, S.D.; Msc, M.B.D.; Snoer, A.H.; Deen, M.; Edvinsson, L. Serotonin and Neuropeptides in Blood From Episodic and Chronic Migraine and Cluster Headache Patients in Case-Control and Case-Crossover Settings: A Systematic Review and Meta-Analysis. Headache 2020, 60, 1132–1164. [Google Scholar] [CrossRef] [PubMed]
- Samsam, M.; Coveñas, R.; Ahangari, R.; Yajeya, J.; Narvaez, J.A.; Tramu, G. Simultaneous depletion of neurokinin A, substance P and calcitonin gene-related peptide from the caudal trigeminal nucleus of the rat during electrical stimulation of the trigeminal ganglion. Pain 2000, 84, 389–395. [Google Scholar] [CrossRef]
- Tuka, B.; Helyes, Z.; Markovics, A.; Bagoly, T.; Németh, J.; Mark, L.; Brubel, R.; Reglodi, R.; Párdutz, Á.; Szolcsányi, J.; et al. Peripheral and central alterations of pituitary adenylate cyclase activating polypeptide-like immunoreactivity in the rat in response to activation of the trigeminovascular system. Peptides 2012, 33, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Messlinger, K.; Fischer, M.J.M.; Lennerz, J.K. Neuropeptide effects in the trigeminal system: Pathophysiology and clinical relevance in migraine. Keio J. Med. 2011, 60, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, L.; Tajti, J.; Szalardy, L.; Vecsei, L. PACAP and its role in primary headaches. J. Headache Pain 2018, 19, 21. [Google Scholar] [CrossRef]
- Tuka, B.; Helyes, Z.; Markovics, A.; Bagoly, T.; Szolcsányi, J.; Szabó, N.; Tóth, E.; Kincses, Z.T.; Vécsei, L.; Tajti, J. Alterations in PACAP-38-like immunoreactivity in the plasma during ictal and interictal periods of migraine patients. Cephalalgia 2013, 33, 1085–1095. [Google Scholar] [CrossRef]
- Tuka, B.; Szabó, N.; Tóth, E.; Kincses, Z.T.; Párdutz, Á.; Szok, D.; Körtési, T.; Bagoly, T.; Helyes, Z.; Edvinsson, L.; et al. Release of PACAP-38 in episodic cluster headache patients—An exploratory study. J. Headache Pain 2016, 17, 69. [Google Scholar] [CrossRef]
- Schytz, H.W.; Birk, S.; Wienecke, T.; Kruuse, C.; Olesen, J.; Ashina, M. PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain 2009, 132 Pt 1, 16–25. [Google Scholar] [CrossRef]
- Körtési, T.; Tuka, B.; Tajti, J.; Bagoly, T.; Fülöp, F.; Helyes, Z.; Vécsei, L. Kynurenic Acid Inhibits the Electrical Stimulation Induced Elevated Pituitary Adenylate Cyclase-Activating Polypeptide Expression in the TNC. Front. Neurol. 2017, 8, 745. [Google Scholar] [CrossRef]
- Greco, R.; DeMartini, C.; Zanaboni, A.M.; Redavide, E.; Pampalone, S.; Toldi, J.; Fülöp, F.; Blandini, F.; Nappi, G.; Sandrini, G.; et al. Effects of kynurenic acid analogue 1 (KYNA-A1) in nitroglycerin-induced hyperalgesia: Targets and anti-migraine mechanisms. Cephalalgia 2017, 37, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Baba, H.; Brenner, G.J.; Woolf, C.J. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat. Neurosci. 1999, 2, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, M.; A Haanes, K.; Majláth, Z.; Tajti, J.; Vécsei, L.; Warfvinge, K.; Edvinsson, L. Dural administration of inflammatory soup or Complete Freund’s Adjuvant induces activation and inflammatory response in the rat trigeminal ganglion. J. Headache Pain 2015, 16, 564. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Aou, S.; Hori, T. Intracerebroventricular injection of interleukin-1 beta enhances nociceptive neuronal responses of the trigeminal nucleus caudalis in rats. Brain Res. 1994, 656, 236–244. [Google Scholar] [CrossRef]
- Lukács, M.; Warfvinge, K.; Kruse, L.S.; Tajti, J.; Fülöp, F.; Toldi, J.; Vécsei, L.; Edvinsson, L. KYNA analogue SZR72 modifies CFA-induced dural inflammation-regarding expression of pERK1/2 and IL-1beta in the rat trigeminal ganglion. J. Headache Pain 2016, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Grocz, G.; Laborc, K.F.; Veres, G.; Bajtai, A.; Bohar, Z.; Zadori, D.; Fejes-Szabó, A.; Spekker, E.; Vécsei, L.; Párdutz, Á. The Effect of Systemic Nitroglycerin Administration on the Kynurenine Pathway in the Rat. Front. Neurol. 2017, 8, 278. [Google Scholar] [CrossRef]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered kynurenine pathway metabolites in serum of chronic migraine patients. J. Headache Pain 2015, 17, 47. [Google Scholar] [CrossRef]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Perugino, F.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered serum levels of kynurenine metabolites in patients affected by cluster headache. J. Headache Pain 2015, 17, 27. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Holland, P.R.; Martins-Oliveira, M.; Hoffmann, J.; Schankin, C.; Akerman, S. Pathophysiology of Migraine: A Disorder of Sensory Processing. Physiol. Rev. 2017, 97, 553–622. [Google Scholar] [CrossRef]
- Campos, F.; Sobrino, T.; Pérez-Mato, M.; Rodríguez-Osorio, X.; Leira, R.; Blanco, M.; Mirelman, D.; Castillo, J. Glutamate oxaloacetate transaminase: A new key in the dysregulation of glutamate in migraine patients. Cephalalgia 2013, 33, 1148–1154. [Google Scholar] [CrossRef]
- Cosentino, G.; Fierro, B.; Brighina, F. From different neurophysiological methods to conflicting pathophysiological views in migraine: A critical review of literature. Clin. Neurophysiol. 2014, 125, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Zielman, R.; Wijnen, J.; Webb, A.; Onderwater, G.L.J.; Ronen, I.; Ferrari, M.D.; Kan, H.E.; Terwindt, G.M.; Kruit, M.C. Cortical glutamate in migraine. Brain 2017, 140, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Volta, G.D.; Zavarise, P.; Perego, L.; Savi, L.; Pezzini, A. Comparison of the Effect of Tanacethum Parthenium, 5-Hydroxy Tryptophan, and Magnesium (Aurastop) versus Magnesium Alone on Aura Phenomenon and Its Evolution. Pain Res. Manag. 2019, 2019, 6320163. [Google Scholar] [CrossRef] [PubMed]
- Lukács, M.; Warfvinge, K.; Tajti, J.; Fülöp, F.; Toldi, J.; Vécsei, L.; Edvinsson, L. Topical dura mater application of CFA induces enhanced expression of c-fos and glutamate in rat trigeminal nucleus caudalis: Attenuated by KYNA derivate (SZR72). J. Headache Pain 2017, 18, 39. [Google Scholar] [CrossRef]
- Ren, C.; Liu, J.; Zhou, J.; Liang, H.; Wang, Y.; Sun, Y.; Ma, B.; Yin, Y. Low levels of serum serotonin and amino acids identified in migraine patients. Biochem. Biophys. Res. Commun. 2018, 496, 267–273. [Google Scholar] [CrossRef]
- Rossi, C.; Pini, L.A.; Cupini, M.L.; Calabresi, P.; Sarchielli, P. Endocannabinoids in platelets of chronic migraine patients and medication-overuse headache patients: Relation with serotonin levels. Eur. J. Clin. Pharmacol. 2007, 64, 1–8. [Google Scholar] [CrossRef]
- Barbanti, P.; Aurilia, C.; Egeo, G.; Fofi, L.; Palmirotta, R. Serotonin receptor targeted therapy for migraine treatment: An overview of drugs in phase I and II clinical development. Expert Opin. Investig. Drugs 2017, 26, 269–277. [Google Scholar] [CrossRef]
- Cseh, E.K.; Veres, G.; Körtési, T.; Polyák, H.; Nánási, N.; Tajti, J.; Párdutz, Á.; Klivényi, P.; Vécsei, L.; Zádori, D. Neurotransmitter and tryptophan metabolite concentration changes in the complete Freund’s adjuvant model of orofacial pain. J. Headache Pain 2020, 21, 35. [Google Scholar] [CrossRef]
- Noseda, R.; Borsook, D.; Burstein, R. Neuropeptides and Neurotransmitters That Modulate Thalamo-Cortical Pathways Relevant to Migraine Headache. Headache 2017, 57, 97–111. [Google Scholar] [CrossRef]
- Bigal, M.E.; Walter, S.; Rapoport, A.M. Therapeutic antibodies against CGRP or its receptor. Br. J. Clin. Pharmacol. 2015, 79, 886–895. [Google Scholar] [CrossRef]
- Castle, D.; Robertson, N.P. Monoclonal antibodies for migraine: An update. J. Neurol. 2018, 265, 1491–1492. [Google Scholar] [CrossRef] [PubMed]
- Raffaelli, B.; Reuter, U. The Biology of Monoclonal Antibodies: Focus on Calcitonin Gene-Related Peptide for Prophylactic Migraine Therapy. Neurotherapeutics 2018, 15, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Vollesen, A.L.; Benemei, S.; Cortese, F.; Labastida-Ramirez, A.; Marchese, F.; Pellesi, L.; Romoli, M.; Ashina, M.; Lampl, C. Migraine and cluster headache—The common link. J. Headache Pain 2018, 19, 89. [Google Scholar] [CrossRef] [PubMed]
- Barjandi, G.; Jounger, S.L.; Löfgren, M.; Bileviciute-Ljungar, I.; Kosek, E.; Ernberg, M. Plasma tryptophan and kynurenine in females with temporomandibular disorders and fibromyalgia-An exploratory pilot study. J. Oral Rehabil. 2020, 47, 150–157. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Condition | The Role of the Kynurenic Pathway |
|---|---|
| Neuropathic pain | Activation of microglia/macrophages with simultaneous upregulation of IDO-2/KMO at spinal cord level; Increased expression of KMO, KYNU, and HAOO in NeuN-positive neurons in contralateral hippocampal dentate gyrus; Increased liver IDO-1/IL-1β mRNAs expression; increased spinal cord IL-1β levels. |
| Possible therapeutic agents: I.t. 1-d-MT (IDO-2 inhibitor) and UPF 648 (KMO inhibitor) administration ↓ mechanical and thermal hypersensitivity; I.p. minocycline injection ↓ IDO-2/KMO and ↓ tactile and thermal hypersensitivity; IL-1RA i.t. administration attenuated both pain and depression through ↓ liver IDO-1/IL-1β mRNAs expression; I.t. (L-KYN) and i.p. (L-KYN and probenecid) injection reversed tactile allodynia; this was mediated by KYNA on NMDARs. | |
| Depression | CFA induces both depression and arthritis through IDO-1 upregulation (↓ serotonin, ↑ QA); LPS-induced behavior mediated by activating NMDARs from increased QA levels; SNI-induced mechanical allodynia and depression are mediated by IL-1β signaling pathway in the spinal cord and IDO-1 in the liver. |
| Possible therapeutic agents: Oral trans-astaxanthin ↓ pain and depression by ↓ pro-inflammatory cytokines in the spinal cord and ↑ 5-HT/5-HIAA and ↓ KYN/TRP ratios in the spinal cord and hippocampus. I.p. P-chlorophenylalanine administration terminated these changes; Ro 61-8048 (KMO inhibitor) or IL-1RA i.c.v. administration attenuated depression in the SNI-induced model; LPS administration (i.c.v.) induced depression mediated by increased IDO-1 and KYN levels in the CN; however, 1-d-MT (IDO-1 inhibitor) administered in the same fashion prevented depression; I.p. gentiopicroside prevented depression and IDO-1 overactivation by downregulating LPS-induced GluN2B-containing NMDARs in the prefrontal cortex; 1-d-MT decreased both pain and depression in the CFA model. | |
| Headache | ↑ XA and ANA in migraine patients; XA represents endogenous analgesic metabolite acting upon mGlu2 receptors; CFA whisker pad injection led to ↑ KYN, KYNA, and glutamate in TNC as well as ↑ 5-HT and KYNA in the somatosensory cortex; I.p. injection of NTG produces a neurogenic inflammation by downregulating IDO-1, TDO-2, KMO, and KYNU enzymes in TNC; Reduction in levels of KYN, KYNA, 3-HK, 3-HA, 5-HIAA, and QA in migraine and cluster headaches; 5-HT, KYN, and KYNA may serve to diminish glutamate sensitization and trigeminovascular activation; PACAP1-38 levels are elevated during ictal phases of migraine and CH; i.v. PACAP1-38 induced headache in both migraineurs (without aura) and the control group, as well as delaying migraine-like headache in migraineurs. |
| Possible therapeutic agents: KYNA-A1 i.p. application prevents expression of nNOS, CGRP, and proinflammatory cytokines in TNC and trigeminal ganglia in migraine; Aurastop (tanacetum parthenium, 5-HT, and magnesium) resulted in significant reduction in aura and aura-related pain disability consistent with ↑ KYNA; I.v. pretreatment with KYN, KYNA-a, and MK-801 decreased TNC PACAP1-38 levels by modulation of glutamatergic transmission. | |
| Other conditions | Temporomandibular disorders myalgia: positive correlation between KYN/TRP and average/worst pain intensity; negative correlation between TRP and worst pain; Fibromyalgia: ↓ TRP; negative correlation between KYN/TRP and anxiety. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jovanovic, F.; Candido, K.D.; Knezevic, N.N. The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 6045. https://doi.org/10.3390/ijms21176045
Jovanovic F, Candido KD, Knezevic NN. The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. International Journal of Molecular Sciences. 2020; 21(17):6045. https://doi.org/10.3390/ijms21176045
Chicago/Turabian StyleJovanovic, Filip, Kenneth D. Candido, and Nebojsa Nick Knezevic. 2020. "The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents" International Journal of Molecular Sciences 21, no. 17: 6045. https://doi.org/10.3390/ijms21176045
APA StyleJovanovic, F., Candido, K. D., & Knezevic, N. N. (2020). The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. International Journal of Molecular Sciences, 21(17), 6045. https://doi.org/10.3390/ijms21176045