Mitochondrial Dysfunction in Diabetic Cardiomyopathy: The Possible Therapeutic Roles of Phenolic Acids



Abstract
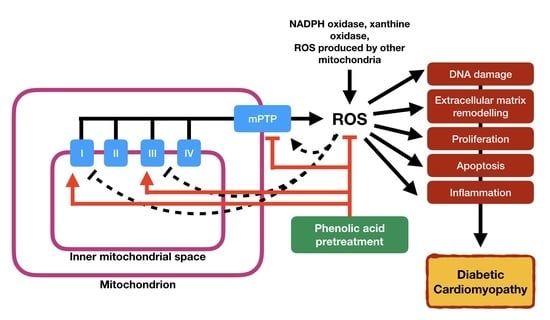
1. Introduction
2. Diabetic Cardiomyopathy
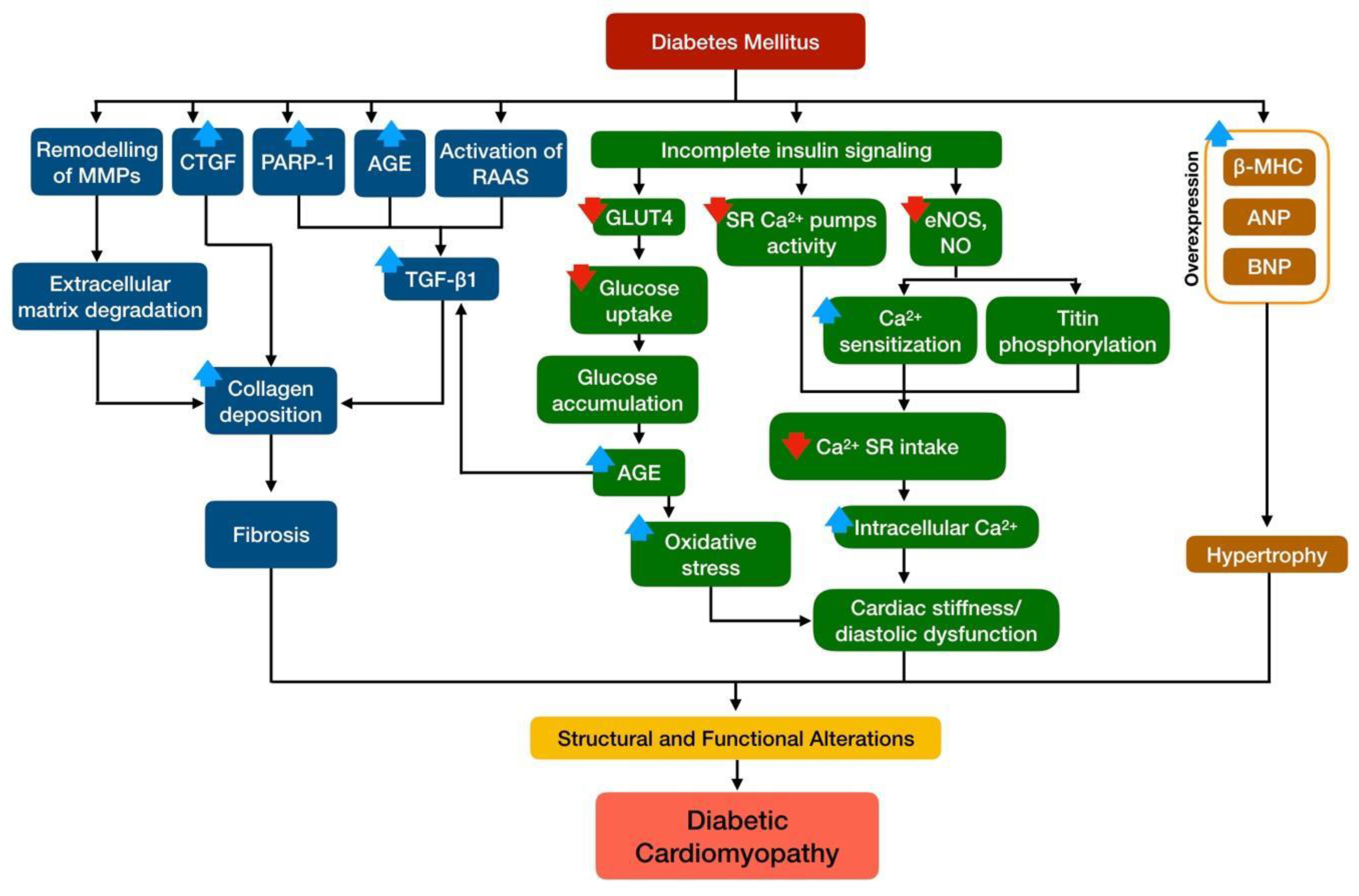
3. Structural and Functional Alterations in DCM
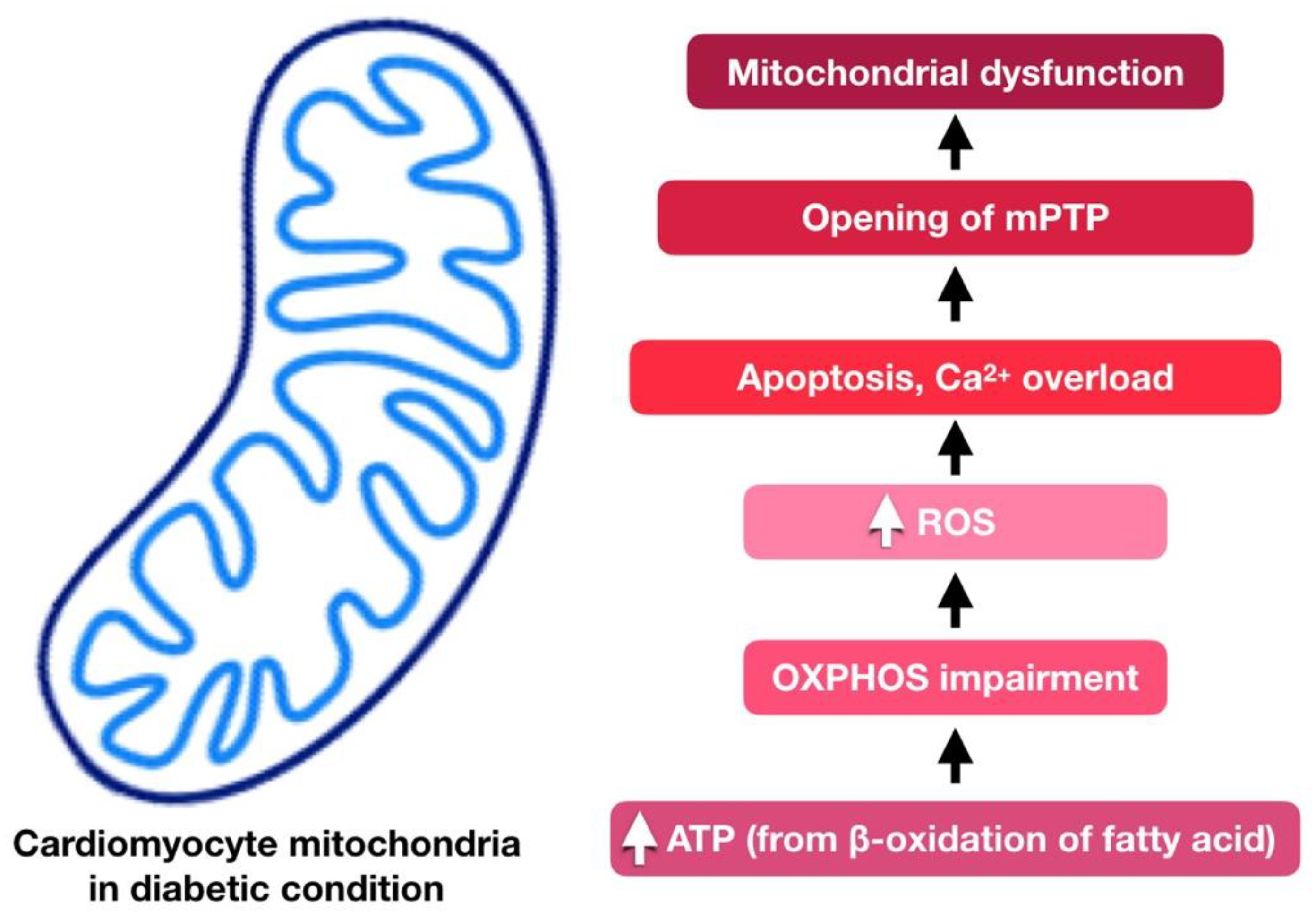
4. Cardiac Mitochondria in Diabetic Cardiomyopathy
5. Involvement of Mitochondria Dysfunction in Diabetic Cardiomyopathy
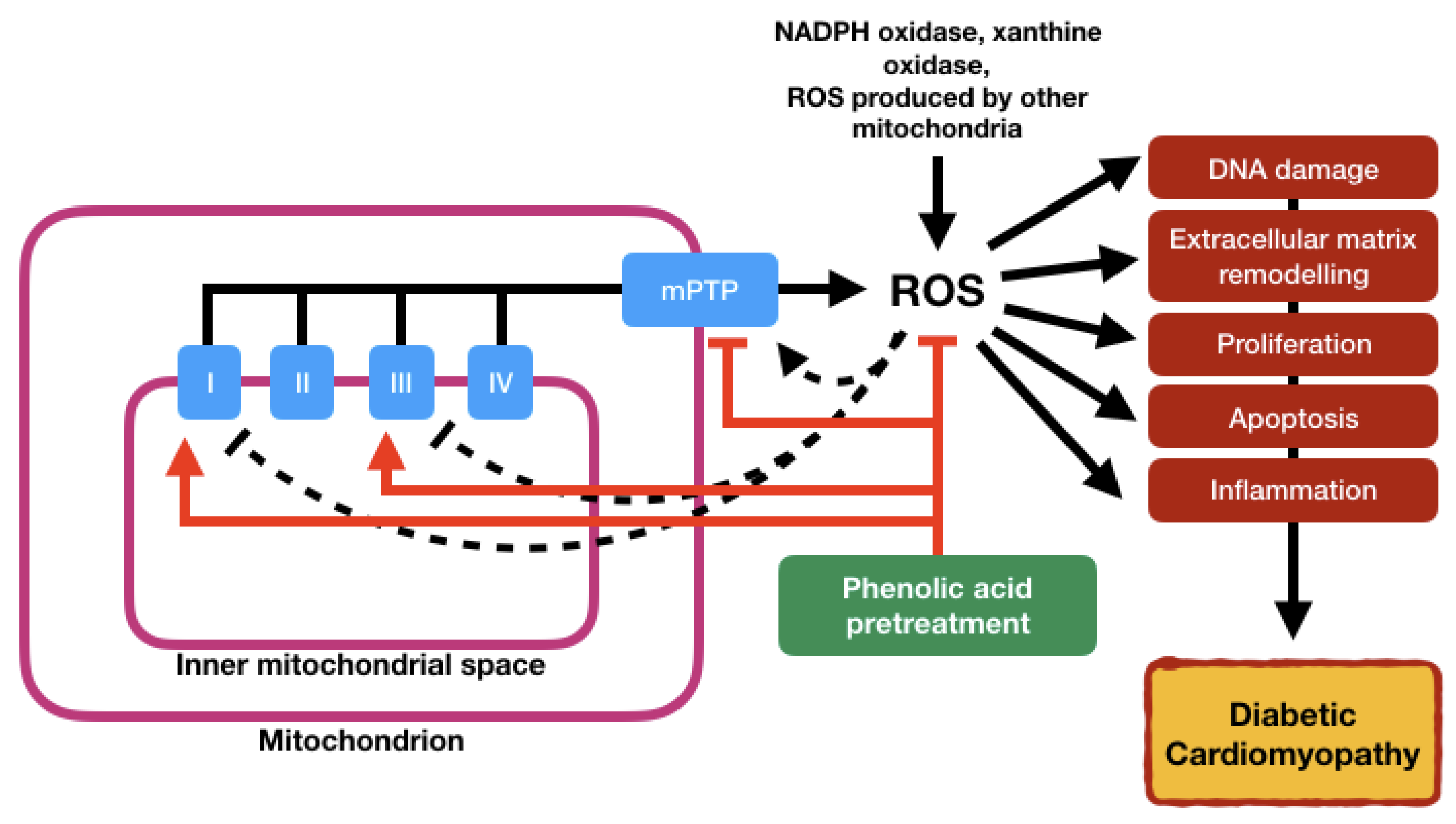
5.1. Oxidative Stress
5.2. Inflammation
5.3. Apoptosis
5.4. Reduced Mitochondrial ATP Production
5.5. Insulin Resistance
6. Phenolic Acids: Therapeutic Implications for Mitochondrial Dysfunction
6.1. Ferulic Acid
6.2. Ellagic Acid
6.3. Gallic Acid
6.4. Salvianolic Acid
6.5. Chlorogenic Acid
6.6. Rosmarinic Acid
6.7. Vanillic Acid
6.8. Caffeic Acid
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Abdollahpour, I.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 17360–17884. [Google Scholar] [CrossRef]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF diabetes atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Hamby, R.I.; Zoneraich, S.; Sherman, L. Diabetic cardiomyopathy. JAMA 1974, 229, 1749–1754. [Google Scholar] [CrossRef] [PubMed]
- Regan, T.J.; Lyons, M.M.; Ahmed, S.S.; Levinson, G.E.; Oldewurtel, H.A.; Ahmad, M.R.; Haider, B. Evidence for cardiomyopathy in familial diabetes mellitus. J. Clin. Investig. 1977, 60, 884–899. [Google Scholar] [CrossRef]
- Wilmot, E.G.; Edwardson, C.L.; Biddle, S.J.; Gorely, T.; Henson, J.; Khunti, K.; Nimmo, M.A.; Yates, M.; Davies, M.J. Prevalence of diabetes and impaired glucose metabolism in younger ‘at risk’ UK adults: Insights from the STAND programme of research. Diabet. Med. 2013, 30, 671–675. [Google Scholar] [CrossRef]
- Haines, L.; Wan, K.C.; Lynn, R.; Barrett, T.G.; Shield, J.P.H. Rising incidence of type 2 diabetes in children in the UK. Diabetes Care 2007, 30, 1097–1101. [Google Scholar] [CrossRef]
- Shen, X.; Zheng, S.; Thongboonkerd, V.; Xu, M.; Pierce, W.M.; Klein, J.B.; Epstein, P.N. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E896–E905. [Google Scholar] [CrossRef]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef]
- Diamant, M.; Lamb, H.J.; Groeneveld, Y.; Endert, E.L.; Smit, J.W.; Bax, J.J.; Romijn, J.A.; de Roos, A.; Radder, J.K. Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well-controlled type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2003, 42, 328–335. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Mitochondria in the diabetic heart. Cardiovasc. Res. 2010, 88, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.; Rolo, A.P.; Palmeira, C.M.; Reis, F. Diabetic Cardiomyopathy: Focus on Oxidative Stress, Mitochondrial Dysfunction and Inflammation. In Cardiomyopathies-Types and Treatments; Kirali, K., Ed.; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Garikipati, V.N.S.; Kishore, R. Mitochondrial dysfunction and its impact on diabetic heart. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2017, 1863, 1098–1105. [Google Scholar] [CrossRef]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar]
- Miki, T.; Yuda, S.; Kouzu, H.; Miura, T. Diabetic cardiomyopathy: Pathophysiology and clinical features. Heart Fail. Rev. 2013, 18, 149–166. [Google Scholar] [CrossRef]
- Shaver, A.; Nichols, A.; Thompson, E.; Mallick, A.; Payne, K.; Jones, C.; Manne, N.D.P.K.; Sundaram, S.; Shapiro, J.I.; Sodhi, K. Role of serum biomarkers in early detection of diabetic cardiomyopathy in the West Virginian population. Int. J. Med. Sci. 2016, 13, 161–168. [Google Scholar] [CrossRef]
- Zamora, M.; Villena, J.A. Contribution of Impaired Insulin Signaling to the Pathogenesis of Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 2833. [Google Scholar] [CrossRef]
- Bhagani, H.; Nasser, S.A.; Dakroub, A.; El-Yazbi, A.F.; Eid, A.A.; Kobeissy, F.; Pintus, G.; Eid, A.H. The Mitochondria: A Target of Polyphenols in the Treatment of Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 4962. [Google Scholar] [CrossRef]
- Faria, A.; Persaud, S.J. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacol. Ther. 2017, 172, 50–62. [Google Scholar] [CrossRef]
- Diaz-Juarez, J.; Suarez, J.; Cividini, F.; Scott, B.T.; Diemer, T.; Dai, A.; Dillmann, W.H. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. Am. J. Physiol. Cell. Physiol. 2016, 311, C1005–C1013. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, W. The Sodium-Glucose Co-Transporter 2 Inhibitor, Empagliflozin, Protects against Diabetic Cardiomyopathy by Inhibition of the Endoplasmic Reticulum Stress Pathway. Cell. Physiol. Biochem. 2017, 41, 2503–2512. [Google Scholar] [CrossRef]
- Yamazaki, D.; Hitomi, H.; Nishiyama, A. Hypertension with diabetes mellitus complications. Hypertens. Res. 2018, 41, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Michelli, A.; Zuolo, G.; Candido, R.; Fabris, B. Update on RAAS Modulation for the Treatment of Diabetic Cardiovascular Disease. J. Diabetes. Res. 2016, 2016, 8917578. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.V.; Hill, J.A. Diabetic cardiomyopathy catabolism driving metabolism. Circulation 2015, 131, 771–773. [Google Scholar] [CrossRef] [PubMed]
- Negishi, K. Echocardiographic feature of diabetic cardiomyopathy: Where are we now? Cardiovasc. Diagn. Ther. 2018, 8, 47–56. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, in press. [Google Scholar] [CrossRef]
- Mohammed Yusof, N.L.M.; Zainalabidin, S.; Mohd Fauzi, N.; Budin, S.B. Cardioprotective Effects of Roselle (Hibiscus Sabdariffa Linn.) Polyphenol-Rich Extract in Streptozotocin-Induced Diabetic Rats. Int. J. Cardiol. 2017, 249, S4–S15. [Google Scholar] [CrossRef]
- Khalil, H.; Alzahrani, T. Cardiomyopathy Imaging. In StatPearls [Internet]; StatPearls Publishing: Petersburg, FL, USA; Treasure Island, FL, USA, 2020. [Google Scholar]
- Hamblin, M.; Friedman, D.B.; Hill, S.; Caprioli, R.M.; Smith, H.M.; Hill, M.F. Alterations in the diabetic myocardial proteome coupled with increased myocardial oxidative stress underlies diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2007, 42, 884–895. [Google Scholar] [CrossRef]
- Von Bibra, H.; St John Sutton, M. Diastolic dysfunction in diabetes and the metabolic syndrome: Promising potential for diagnosis and prognosis. Diabetologia 2010, 53, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Finkel, T. Cardiac mitochondria: A surprise about size. J. Mol. Cell. Cardiol. 2015, 82, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Sowers, J.R.; Bender, S.B.; Nistala, R.; Garro, M.; Mugerfeld, I.; Hayden, M.R.; Johnson, M.S.; Salam, M.; Whaley-Connell, A.; et al. Dipeptidylpeptidase inhibition is associated with improvement in blood pressure and diastolic function in insulin-resistant male Zucker obese rats. Endocrinology 2013, 154, 2501–2513. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and thera-peutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef]
- Mohammed Yusof, N.L.; Zainalabidin, S.; Mohd Fauzi, N.; Budin, S.B. Hibiscus sabdariffa (roselle) polyphenol-rich extract averts cardiac functional and structural abnormalities in type 1 diabetic rats. Appl. Physiol. Nutr. Metab. 2018, 43, 1224–1232. [Google Scholar] [CrossRef]
- Qin, W.D.; Liu, G.L.; Wang, J.; Wang, H.; Zhang, J.N.; Zhang, F.; Ma, Y.; Ji, X.Y.; Li, C.; Zhang, M.X. Poly(ADP-ribose) polymerase 1 inhibition protects cardiomyocytes from inflammation and apoptosis in diabetic cardiomyopathy. Oncotarget 2016, 7, 35618–35631. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef]
- Eguchi, M.; Xu, G.; Li, R.K.; Sweeney, G. Diabetes influences cardiac extracellular matrix remodelling after myocardial infarction and subsequent development of cardiac dysfunction. J. Cell. Mol. Med. 2012, 16, 2925–2934. [Google Scholar] [CrossRef]
- D’Souza, A.; Howarth, F.C.; Yanni, J.; Dobrzynski, H.; Boyett, M.R.; Adeghate, E.; Bidasee, K.R.; Singh, J. Chronic effects of mild hyperglycaemia on left ventricle transcriptional profile and structural remodelling in the spontaneously type 2 diabetic Goto-Kakizaki rat. Heart Fail. Rev. 2014, 19, 65–74. [Google Scholar] [CrossRef]
- Eckhouse, S.R.; Spinale, F.G. Changes in the myocardial interstitium and contribution to the progression of heart failure. Heart Fail. Clin. 2012, 8, 7–20. [Google Scholar] [CrossRef]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, A.C.; Hood, S.G.; Woods, R.L.; Dusting, G.J.; Ritchie, R.H. B-type natriuretic pep- tide prevents acute hypertrophic responses in the diabetic rat heart: Importance of cyclic GMP. Diabetes 2003, 52, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.; Soares, E.; Fernandes, J.; Viana, S.; Carvalho, E.; Pereira, F.C.; Reis, F. Early cardiac changes in a rat model of prediabetes: Brain natriuretic peptide overexpression seems to be the best marker. Cardiovasc. Diabetol. 2013, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J. Hypertens. 2003, 21, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef]
- Hamdani, N.; Herwig, M.; Linke, W.A. Tampering with springs: Phosphorylation of titin affecting the mechanical function of cardiomyocytes. Biophys. Rev. 2017, 9, 225–237. [Google Scholar] [CrossRef]
- Dorn, G.W., II. Mitochondrial dynamics in heart disease. Biochim. Biophys. Acta 2013, 1833, 233–241. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial metabolism in aging heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Deshpande, O.A.; Mohiuddin, S.S. Biochemistry, Oxidative Phophorylation. [Updated 18 December 2019]; In StatPearls [Internet]; StatPearls Publishing: Petersburg, FL, USA; Treasure Island, FL, USA, 2020. [Google Scholar]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Electron Transport and Oxidative Phosphorylation. In Molecular Cell Biology, 4th ed.; W.H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Ramsay, R.R. Electron carriers and energy conservation in mitochondrial respiration. ChemTexts 2019, 5, 9. [Google Scholar] [CrossRef]
- Berry, B.J.; Trewin, A.J.; Amitrano, A.M.; Kim, M.; Wojtovich, A.P. Use the Protonmotive Force: Mitochondrial Uncoupling and Reactive Oxygen Species. J. Mol. Biol. 2018, 430, 3873–3891. [Google Scholar] [CrossRef] [PubMed]
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschütz, W.; Lipke, C.; Köstler, H.; von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol. 2002, 40, 1267–1274. [Google Scholar] [CrossRef]
- Galloway, C.A.; Yoon, Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid. Redox Signal. 2015, 22, 1545–1562. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Habibi, J.; DeMarco, V.G.; Martinez-Lemus, L.A.; Ma, L.; Whaley-Connell, A.T.; Aroor, A.R.; Domeier, T.L.; Zhu, Y.; Meininger, G.A.; et al. Endothelial Mineralocorticoid Receptor Deletion Prevents Diet-Induced Cardiac Diastolic Dysfunction in Females. Hypertension 2015, 66, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.Y.; Ruiz-Velasco, A.; Bui, T.; Collins, L.; Wang, X.; Liu, W. Mitochondrial function in the heart: The insight into mechanisms and therapeutic potentials. Br. J. Pharmacol. 2019, 176, 4302–4318. [Google Scholar] [CrossRef]
- Gorski, P.A.; Ceholski, D.K.; Hajjar, R.J. Altered myocardial calcium cycling and energetics in heart failure—A rational approach for disease treatment. Cell Metab. 2015, 21, 183–194. [Google Scholar] [CrossRef]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef]
- Ghosh, N.; Das, A.; Chaffee, S.; Roy, S.; Sen, C.K. Chapter 4: Reactive Oxygen Species, Oxidative Damage and Cell Death. In Immunity and Inflammation in Health and Disease; Academic Press: Cambridge, MA, USA, 2018; pp. 45–55. [Google Scholar]
- Brondani, A.L.; Assmann, T.S.; Duarte, G.C.; Gross, J.L.; Canani, L.H.; Crispim, D. The role of the uncoupling protein 1 (UCP1) on the development of obesity and type 2 diabetes mellitus. Arq. Bras. Endocrinol. Metabol. 2012, 56, 215–225. [Google Scholar] [CrossRef]
- Wilson, A.J.; Gill, E.K.; Abudalo, R.A.; Edgar, K.S.; Watson, C.J.; Grieve, D.J. Reactive oxygen species signalling in the diabetic heart: Emerging prospect for therapeutic targeting. Heart 2018, 104, 293–299. [Google Scholar] [CrossRef]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- Yusof, N.L.M.; Zainalabidin, S.; Fauzi, N.M.; Budin, S.B. Hibiscus sabdariffa (roselle) polyphenol-rich extract prevents the aortic oxidative damage in type 1 diabetic rats. J. Teknol. 2018, 80, 1–8. [Google Scholar]
- Dymkowska, D.; Drabarek, B.; Podszywalow-Bartnicka, P.; Szczepanowska, J.; Zablocki, K. Hyperglycaemia modifies energy metabolism and reactive oxygen species formation in endothelial cells in vitro. Arch. Biochem. Biophys. 2014, 542, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed]
- Teshima, Y.; Takahashi, N.; Nishio, S.; Saito, S.; Kondo, H.; Fukui, A.; Aoki, K.; Yufu, K.; Nakagawa, M.; Saikawa, T. Production of reactive oxygen species in the diabetic heart. Roles of mitochondria and NADPH oxidase. Circ. J. 2014, 78, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Laddha, A.P.; Kulkarni, Y.A. NADPH oxidase: A membrane-bound enzyme and its inhibitors in diabetic complications. Eur. J. Pharmacol. 2020, 881, 173206. [Google Scholar] [CrossRef]
- Murdoch, C.E.; Chaubey, S.; Zeng, L.; Yu, B.; Ivetic, A.; Walker, S.J.; Vanhoutte, D.; Heymans, S.; Grieve, D.J.; Cave, A.C.; et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J. Am. Coll. Cardiol. 2014, 63, 2734–2741. [Google Scholar] [CrossRef]
- Lee, S.J.; Kang, J.G.; Ryu, O.H.; Kim, C.S.; Ihm, S.H.; Choi, M.G.; Yoo, H.J.; Kim, D.S.; Kim, T.W. Effects of alpha-lipoic acid on transforming growth factor beta1-p38 mitogen-activated protein kinase-fibronectin pathway in diabetic nephropathy. Metabolism 2009, 58, 616–662. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Yusof, N.L.M.; Zainalabidin, S.; Fauzi, N.M.; Budin, S.B. Attenuation of Cardiac Functional and Structural Abnormalities by Hibiscus Sabdariffa Polyphenol-Rich Extract in Type-1-Induced Diabetic Rats. Int. J. Cardiol. 2018, 273, 19. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filado, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional Mitochondria in Health and Disease. Front. Endocrinol. 2017, 8, 296. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders —A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Smail, M.M.A.; Howarth, C.F.; Singh, J.; Ismail, A.M. Inflammation and Diabetic Cardiomyopathy. In Inflammatory Heart Diseases; Aronow, W.S., Murashita, T., Eds.; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Westermann, D.; Van Linthout, S.; Dhayat, S.; Dhayat, N.; Escher, F.; Bucker-Gartner, C.; Spillmann, F.; Noutsias, M.; Riad, A.; Schultheiss, H.P.; et al. Cardioprotective and anti-inflammatory effects of interleukin converting enzyme inhibition in experimental diabetic cardiomyopathy. Diabetes 2007, 56, 1834–1841. [Google Scholar] [CrossRef]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar]
- Miliara, X.; Garnett, J.A.; Tatsuta, T.; Abid Ali, F.; Baldie, H.; Pérez-Dorado, I.; Simpson, P.; Yague, E.; Langer, T.; Matthews, S. Intra-mitochondrial signaling: Interactions among mitoKATP, PKCε, ROS, and MPT. EMBO Rep. 2015, 16, 824–835. [Google Scholar] [CrossRef]
- Bajpai, A.; Tilley, D.G. The Role of Leukocytes in Diabetic Cardiomyopathy. Front. Physiol. 2018, 9, 1547. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation—A two-way street. Mol. Med. 2017, 23, 188–195. [Google Scholar] [CrossRef]
- Luo, B.; Huang, F.; Liu, Y.; Liang, Y.; Wei, Z.; Ke, H.; Zeng, Z.; Huang, W.; He, Y. NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Front. Physiol. 2017, 25, 519. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Katare, P.B.; Bagul, P.K.; Dinda, A.K.; Banerjee, S.K. Toll-Like Receptor 4 Inhibition Improves Oxidative Stress and Mitochondrial Health in Isoproterenol-Induced Cardiac Hypertrophy in Rats. Front. Immunol. 2017, 8, 719. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Li, T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front. Immunol. 2018, 9, 230583. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 7, 179–188. [Google Scholar] [CrossRef]
- Weber, K.; Schilling, J.D. Lysosomes integrate metabolic-inflammatory crosstalk in primary macrophage inflammasome activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef]
- Riswana, N.A.; Bhuvaneshwari, P. Apoptotic cell death in heart failure associated with diabetes. Int. J. Clinicopathol. Correl. 2019, 3, 12–18. [Google Scholar]
- Marunouchi, T.; Tanonaka, K. Cell Death in the Cardiac Myocyte. Biol. Pharm. Bull. 2015, 38, 1094–1097. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Mughal, W.; Dhingra, R.; Kirshenbaum, L.A. Striking a balance: Autophagy, apoptosis, and necrosis in a normal and failing heart. Curr. Hypertens. Rep. 2012, 14, 540–547. [Google Scholar] [CrossRef]
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1-caspase-9 apoptosome. J. Cell. Sci. 2010, 123, 3209–3214. [Google Scholar] [CrossRef] [PubMed]
- Kuethe, F.; Sigusch, H.H.; Bornstein, S.R.; Hilbig, K.; Kamvissi, V.; Figulla, H.R. Apoptosis in patients with dilated cardiomyopathy and diabetes: A feature of diabetic cardiomyopathy? Horm. Metab. Res. 2007, 39, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Bai, T.; Xu, Z.; Liu, Q.; Zheng, Y.; Cai, L. Pathophysiological fundamentals of diabetic cardiomyopathy. Compr. Physiol. 2017, 7, 693–711. [Google Scholar] [PubMed]
- Ramalingam, A.; Budin, S.B.; Mohd Fauzi, N.; Ritchie, R.H.; Zainalabidin, S. Angiotensin II Type I Receptor Antagonism Attenuates Nicotine-Induced Cardiac Remodeling, Dysfunction, and Aggravation of Myocardial Ischemia-Reperfusion Injury in Rats. Front. Pharmacol. 2019, 10, 1493. [Google Scholar] [CrossRef]
- Liu, M.; Chen, Z.; Chen, L. Endoplasmic reticulum stress: A novel mechanism and therapeutic target for cardiovascular diseases. Acta Pharmacol. Sin. 2016, 37, 425–443. [Google Scholar] [CrossRef]
- Zhang, Y.; Cui, L.; Guan, G.; Wang, J.; Qiu, C.; Yang, T.; Liu, Z. Matrine suppresses cardiac fibrosis by inhibiting the TGF-β/Smad pathway in experimental diabetic cardiomyopathy. Mol. Med. Rep. 2017, 17, 1775–1781. [Google Scholar] [CrossRef]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Cai, L.; Wang, Y.; Zhou, G.; Chen, T.; Song, Y.; Li, X.; Kang, Y.J. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 1688–1697. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, T.; Dai, H.; Liu, G.; Wang, H.; Sun, Y.; Zhang, Y.; Ge, Z. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin-induced diabetic rats. J. Clin. Biochem. Nutr. 2007, 41, 58–67. [Google Scholar] [CrossRef]
- Wu, W.; Liu, X.; Han, L. Apoptosis of cardiomyocytes in diabetic cardiomyopathy involves overexpression of glycogen synthase kinase-3β. Biosci. Rep. 2019, 39, BSR20171307. [Google Scholar] [CrossRef]
- Xu, K.; Liu, X.F.; Ke, Z.Q.; Yao, Q.; Guo, S.; Liu, C. Resveratrol Modulates Apoptosis and Autophagy Induced by High Glucose and Palmitate in Cardiac Cells. Cell Physiol. Biochem. 2018, 46, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.L.; Dabkowski, E.R.; Baseler, W.A.; Croston, T.L.; Always, S.E.; Hollander, J.M. Enhanced apoptotic propensity in diabetic cardiac mitochondria: Influence of subcellular spatial location. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H633–H642. [Google Scholar] [CrossRef] [PubMed]
- Dabkowski, E.R.; Baseler, W.A.; Williamson, C.L.; Powell, M.; Razunguzwa, T.T.; Frisbee, J.C.; Hollander, J.M. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H529–H540. [Google Scholar] [CrossRef] [PubMed]
- Dabkowski, E.R.; Williamson, C.L.; Bukowski, V.C.; Chapman, R.S.; Leonard, S.S.; Peer, C.J.; Callery, P.S.; Hollander, J.M. Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H359–H369. [Google Scholar] [CrossRef] [PubMed]
- Timón-Gómez, A.; Barrientos, A. Mitochondrial respiratory chain composition and organization in response to changing oxygen levels. J. Life Sci. (Westlake Village) 2020, 2. [Google Scholar] [CrossRef]
- Guzun, R.; Timohhina, N.; Tepp, K.; Monge, C.; Kaambre, T.; Sikk, P.; Kuznetsov, A.V.; Pison, C.; Saks, V. Regulation of respiration controlled by mitochondrial creatine kinase in permeabilized cardiac cells in situ. Importance of system level properties. Biochim. Biophys. Acta 2009, 1787, 1089–1105. [Google Scholar] [CrossRef]
- Guimarães-Ferreira, L. Role of the phosphocreatine system on energetic homeostasis in skeletal and cardiac muscles. Einstein 2014, 12, 126–131. [Google Scholar] [CrossRef]
- Kuzmiak-Glancy, S.; Covian, R.; Femnou, A.N.; Glancy, B.; Jaimes, R., III; Wengrowski, A.M.; Garrott, K.; French, S.A.; Balaban, R.S.; Kay, M.W. Cardiac performance is limited by oxygen delivery to the mitochondria in the crystalloid-perfused working heart. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H704–H715. [Google Scholar] [CrossRef]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G.D. Role of fatty acid uptake and fatty acid beta-oxidation in media- ting insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta 2010, 1801, 1–22. [Google Scholar] [CrossRef]
- Stanley, W.C. Rationale for a metabolic approach in diabetic coronary patients. Coron. Artery Dis. 2005, 16 (Suppl. 1), S11–S15. [Google Scholar] [CrossRef]
- Neglia, D.; De Caterina, A.; Marraccini, P.; Natali, A.; Ciardetti, M.; Vecoli, C.; Gastaldelli, A.; Ciociaro, D.; Pellegrini, P.; Testa, R.; et al. Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3270–H3278. [Google Scholar] [CrossRef] [PubMed]
- Cleland, S.J.; Fisher, B.M.; Colhoun, H.M.; Sattar, N.; Petrie, J.R. Insulin resistance in type 1 diabetes: What is ‘double diabetes’ and what are the risks? Diabetologia 2013, 56, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Wilkin, T.J. The accelerator hypothesis: A review of the evidence for insulin resistance as the basis for type 1 as well as type II diabetes. Int. J. Obes. 2009, 33, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.; Soares, E.; Pereira, F.; Reis, F. The role of inflammation in diabetic cardiomyopathy. Int. J. Infereron Cytokine Mediat. Res. 2012, 4, 59–73. [Google Scholar]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef]
- Bruce, C.R.; Risis, S.; Babb, J.R.; Yang, C.; Kowalski, G.M.; Selathurai, A.; Lee-Young, R.S.; Weir, J.M.; Yoshioka, K.; Takuwa, Y. Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high-fat diet-fed mice. Diabetes 2012, 61, 3148–3155. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Pierce, G.N.; Innes, I.R.; Beamish, R.E. Pathogenesis of cardiac dysfunction in diabetes mellitus. Can. J. Cardiol. 1985, 1, 263–281. [Google Scholar]
- Jia, G.; Habibi, J.; Bostick, B.P.; Ma, L.; DeMarco, V.G.; Aroor, A.R.; Hayden, M.R.; Whaley-Connell, A.T.; Sowers, J.R. Uric acid promotes left ventricular diastolic dysfunction in mice fed a Western diet. Hypertension 2015, 65, 531–539. [Google Scholar] [CrossRef]
- Kumar, N.; Goel, N. Phenolic acids: Natural versatile molecules with promising therapeutic applications. Biotechnol. Rep. (Amst.) 2019, 24, e00370. [Google Scholar] [CrossRef] [PubMed]
- Saibabu, V.; Fatima, Z.; Khan, L.A.; Hameed, S. Therapeutic Potential of Dietary Phenolic Acids. Adv. Pharmacol. Sci. 2015, 2015, 823539. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.Z.; Yang, J.; Zhao, G.R.; Yuan, Y.J. Ferulic acid inhibits vascular smooth muscle cell proliferation induced by angiotensin II. Eur. J. Pharmacol. 2004, 499, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Yogeeta, S.K.; Gnanapragasam, A.; Kumar, S.S.; Subhashini, R.; Sathivel, A.; Devaki, T. Synergistic interactions of ferulic acid with ascorbic acid: Its cardioprotective role during isoproterenol induced myocardial infarction in rats. Mol. Cell. Biochem. 2006, 283, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Sudheer, A.R.; Menon, V.P. Ferulic Acid: Therapeutic potential through its antioxidant property. J. Clin. Biochem. Nutr. 2007, 40, 92–100. [Google Scholar] [CrossRef]
- Perez-Ternero, C.; Werner, C.M.; Nickel, A.G.; Herrera, M.D.; Motilva, M.J.; Böhm, M.; Alvarez de Sotomayor, M.; Laufs, U. Ferulic acid, a bioactive component of rice bran, improves oxidative stress and mitochondrial biogenesis and dynamics in mice and in human mononuclear cells. J. Nutr. Biochem. 2017, 48, 51–61. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Kao, S.T.; Lee, Y.C. Ferulic Acid Exerts Anti-apoptotic Effects against Ischemic Injury by Activating HSP70/Bcl-2- and HSP70/Autophagy-Mediated Signaling after Permanent Focal Cerebral Ischemia in Rats. Am. J. Chin. Med. 2019, 47, 39–61. [Google Scholar] [CrossRef]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef]
- Salin Raj, P.; Swapna, S.U.S.; Raghu, K.G. High glucose induced calcium overload via impairment of SERCA/PLN pathway and mitochondrial dysfunction leads to oxidative stress in H9c2 cells and amelioration with ferulic acid. Fundam. Clin. Pharmacol. 2019, 33, 412–425. [Google Scholar] [CrossRef]
- Song, Y.; Wen, L.; Sun, J.; Bai, W.; Jiao, R.; Hu, Y.; Peng, X.; He, Y.; Ou, S. Cytoprotective mechanism of ferulic acid against high glucose induced oxidative stress in cardiomyocytes and hepatocytes. Food Nutr. Res. 2016, 60, 30323. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Wang, C.C.; Lai, T.Y.; Tsu, H.N.; Wang, C.H.; Liang, H.Y. Antioxidant effects of diallyl trisulfide on high glucose-induced apoptosis are mediated by the PI3K/Akt-dependent activation of Nrf2 in cardiomyocytes. Int. J. Cardiol. 2013, 168, 1286–1297. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Yagishita, Y.; Yamamoto, M. The Keap1-Nrf2 system and diabetes mellitus. Arch. Biochem. Biophys. 2015, 566, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Cho, H.-Y.; Kleeberger, S.R. Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: A potential role for Nrf2. Antioxid. Redox Signal. 2008, 10, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wu, T.; Yang, Q.; Chen, X.; Wang, M.; Wang, Y. Ferulic acid alleviates the symptoms of diabetes in obese rats. J. Funct. Foods 2014, 9, 141–147. [Google Scholar] [CrossRef]
- He, X.Q.; Kan, H.; Cai, L.; Ma, Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J. Mol. Cell. Cardiol. 2009, 46, 47–58. [Google Scholar] [CrossRef]
- Ahmed, T.; Setzer, W.N.; Nabavi, S.F.; Orhan, I.E.; Braidy, N.; Sobarzo-Sanchez, E.; Nabavi, S.M. Insights into Effects of Ellagic Acid on the Nervous System: A Mini Review. Curr. Pharm. Des. 2016, 22, 1350–1360. [Google Scholar] [CrossRef]
- Alasalvar, C.; Bolling, B.W. Review of nut phytochemicals, fat-soluble bioactives, antioxidant components and health effects. Br. J. Nutr. 2015, 113, S68–S78. [Google Scholar] [CrossRef]
- Dhingra, A.; Jayas, R.; Afshar, P. Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radic. Biol. Med. 2017, 112, 411–422. [Google Scholar] [CrossRef]
- Dhingra, R.; Margulets, V.; Chowdhury, S.R.; Thliveris, J.; Jassal, D.; Fernyhough, P.; Dorn, G.W., II; Kirshenbaum, L.A. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E5537–E5544. [Google Scholar] [CrossRef]
- Mohammad Khanlou, E.; Atashbar, S.; Kahrizi, F.; Shokouhi Sabet, N.; Salimi, A. Bevacizumab as a monoclonal antibody inhibits mitochondrial complex II in isolated rat heart mitochondria: Ameliorative effect of ellagic acid. Drug Chem. Toxicol. 2020, 23, 1–8. [Google Scholar] [CrossRef]
- Keshtzar, E.; Khodayar, M.J.; Javadipour, M.; Ghaffari, M.A.; Bolduc, D.L.; Rezaei, M. Ellagic acid protects against arsenic toxicity in isolated rat mitochondria possibly through the maintaining of complex II. Hum. Exp. Toxicol. 2016, 35, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.H.; Salgado, H.R. Gallic Acid: Review of the Methods of Determination and Quantification. Crit. Rev. Anal. Chem. 2016, 46, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Komolafe, K.; Olaleye, T.M.; Omotuyi, O.I.; Boligon, A.A.; Athayde, M.L.; Akindahunsi, A.A.; Teixeira da Rocha, J.B. In vitro antioxidant activity and effect of Parkia biglobosa bark extract on mitochondrial redox status. J. Acupunct. Meridian Stud. 2014, 7, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Frey, C.; Pavani, M.; Cordano, G.; Munoz, S.; Rivera, E.; Medina, J.; Morello, A.; Diego Maya, J.; Ferreira, J. Comparative cytotoxicity of alkyl gallates on mouse tumor cell lines and isolated rat hepatocytes. Comp. Biochem. Physiol. Mol. Integr. Physiol. 2007, 146, 520–527. [Google Scholar] [CrossRef]
- Dianat, M.; Sadeghi, N.; Badavi, M.; Panahi, M.; Taheri Moghadam, M. Protective effects of co-administration of gallic Acid and cyclosporine on rat myocardial morphology against ischemia/reperfusion. Jundishapur J. Nat. Pharm. Prod. 2014, 9, e17186. [Google Scholar] [CrossRef]
- Khurana, S.; Hollingsworth, A.; Piche, M.; Venkataraman, K.; Kumar, A.; Ross, G.M.; Tai, T.C. Antiapoptotic actions of methyl gallate on neonatal rat cardiac myocytes exposed to H2O2. Oxid. Med. Cell. Longev. 2014, 2014, 657512. [Google Scholar] [CrossRef]
- Miltonprabu, S.; Thangapandiyan, S. Epigallocatechin gallate potentially attenuates Fluoride induced oxidative stress mediated cardiotoxicity and dyslipidemia in rats. J. Trace Elem. Med. Biol. 2015, 29, 321–335. [Google Scholar] [CrossRef]
- Liu, J.; Tang, Y.; Feng, Z.; Long, J. (-)-Epigallocatechin-3-gallate attenuated myocardial mitochondrial dysfunction and autophagy in diabetic Goto-Kakizaki rats. Free Radic. Res. 2014, 48, 898–906. [Google Scholar] [CrossRef]
- Ho, J.H.; Hong, C.Y. Salvianolic acids: Small compounds with multiple mechanisms for cardiovascular protection. J. Biomed. Sci. 2011, 18, 30. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, H.; Wang, J.; Liu, Z.; Bi, J. Isolation and purification of salvianolic acid A and salvianolic acid B from Salvia miltiorrhiza by high-speed counter-current chromatography and comparison of their antioxidant activity. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 733–777. [Google Scholar] [CrossRef]
- Zhao, G.R.; Zhang, H.M.; Ye, T.X.; Xiang, Z.J.; Yuan, Y.J.; Guo, Z.X.; Zhao, L.B. Characterization of the radical scavenging and antioxidant activities of danshensu and salvianolic acid B. Food Chem. Toxicol. 2008, 46, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, Y.G.; Zhang, F.; Yang, L.; Chou, G.X.; Wang, Z.T.; Hu, Z.B. Chromatographic fingerprinting analysis of Danshen root (Salvia miltiorrhiza Radix et Rhizoma) and its preparations using high performance liquid chromatography with diode array detection and electrospray mass spectrometry (HPLC-DAD-ESI/MS). J. Sep. Sci. 2007, 30, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.B.; Tian, S.; Yang, F.; Yang, H.G.; Yang, X.Y.; Du, G.H. Cardioprotective effect of salvianolic acid A on isoproterenol-induced myocardial infarction in rats. Eur. J. Pharmacol. 2009, 615, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.J.; Liu, G.T.; Liu, Y.; Xu, G.Z. Protection by salvianolic acid A against adriamycin toxicity on rat heart mitochondria. Free Radic. Biol. Med. 1992, 12, 347–351. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, L.; Li, M.; Wu, W.; Yang, M.; Wang, J.; Guo, D.A. Salvianolic acids prevent acute doxorubicin cardiotoxicity in mice through suppression of oxidative stress. Food Chem. Toxicol. 2008, 46, 1510–1515. [Google Scholar] [CrossRef]
- Niggeweg, R.; Michael, A.J.; Martin, C. Engineering plants with increased levels of the antioxidant chlorogenic acid. Nat. Biotechnol. 2004, 22, 746–754. [Google Scholar] [CrossRef]
- Akila, P.; Asaikumar, L.; Vennila, L. Chlorogenic acid ameliorates isoproterenol-induced myocardial injury in rats by stabilizing mitochondrial and lysosomal enzymes. Biomed. Pharmacother. 2017, 85, 582–591. [Google Scholar] [CrossRef]
- Capetenaki, Y. Desmin cytoskeleton: A potential regulator of muscle mitochondria behavior and function. Trends Cardiovasc. Med. 2002, 12, 339–348. [Google Scholar] [CrossRef]
- Li, Y.; Ren, X.; Lio, C.; Sun, W.; Lai, K.; Liu, Y.; Zhang, Z.; Liang, J.; Zhou, H.; Liu, L.; et al. A chlorogenic acid-phospholipid complex ameliorates post-myocardial infarction inflammatory response mediated by mitochondrial reactive oxygen species in SAMP8 mice. Pharmacol. Res. 2018, 130, 110–122. [Google Scholar] [CrossRef]
- Shekarchi, M.; Hajimehdipoor, H.; Saeidnia, S.; Gohari, A.R.; Hamedani, M.P. Comparative study of rosmarinic acid content in some plants of Labiatae family. Pharmacogn. Mag. 2012, 8, 37. [Google Scholar]
- Diao, J.; Wei, J.; Yan, R.; Liu, X.; Li, Q.; Lin, L.; Zhu, Y.; Li, H. Rosmarinic Acid suppressed high glucose-induced apoptosis in H9c2 cells by ameliorating the mitochondrial function and activating STAT3. Biochem. Biophys. Res. Commun. 2016, 477, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Chlopcíková, S.; Psotová, J.; Miketová, P.; Sousek, J.; Lichnovský, V.; Simánek, V. Chemoprotective effect of plant phenolics against anthracycline-induced toxicity on rat cardiomyocytes. Part II. caffeic, chlorogenic and rosmarinic acids. Phytother. Res. 2004, 18, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Kiokias, S.; Proestos, C.; Oreopoulou, V. Phenolic Acids of Plant Origin-A Review on Their Antioxidant Activity In Vitro (O/W Emulsion Systems) Along with Their in Vivo Health Biochemical Properties. Foods 2020, 9, 534. [Google Scholar] [CrossRef]
- Kumar, S.; Prahalathan, P.; Raja, B. Antihypertensive and antioxidant potential of vanillic acid, a phenolic compound in L-NAME-induced hypertensive rats: A dose-dependence study. Redox Rep. 2011, 16, 208–215. [Google Scholar] [CrossRef]
- Prince, P.S.; Dhanasekar, K.; Rajakumar, S. Preventive effects of vanillic acid on lipids, bax, bcl-2 and myocardial infarct size on isoproterenol-induced myocardial infarcted rats: A biochemical and in vitro study. Cardiovas. Toxicol. 2011, 11, 58–66. [Google Scholar] [CrossRef]
- Chou, T.H.; Ding, H.Y.; Hung, W.J.; Liang, C.H. Antioxidative characteristics and inhibition of alpha-melanocyte-stimulating hormone-stimulated melanogenesis of vanillin and vanillic acid from Origanum vulgare. Exp. Dermatol. 2010, 19, 742–750. [Google Scholar] [CrossRef]
- Baniahmad, B.; Safaeian, L.; Vaseghi, G.; Rabbani, M.; Mohammadi, B. Cardioprotective effect of vanillic acid against doxorubicin-induced cardiotoxicity in rat. Res. Pharm. Sci. 2020, 15, 87–96. [Google Scholar]
- Yao, X.; Jiao, S.; Qin, M.; Hu, W.; Yi, B.; Liu, D. Vanillic Acid Alleviates Acute Myocardial Hypoxia/Reoxygenation Injury by Inhibiting Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 1–12. [Google Scholar]
- Castellari, M.; Sartini, E.; Fabiani, A.; Arfelli, G.; Amati, A. Analysis of wine phenolics by high performance liquid chromatography using a monolithic type column. J. Chromatogr. A 2002, 973, 221–227. [Google Scholar] [CrossRef]
- Lee, S.Y.; Ku, H.C.; Kuo, Y.H.; Chiu, H.L.; Su, M.J. Pyrrolidinyl caffeamide against ischemia/reperfusion injury in cardiomyocytes through AMPK/AKT pathways. J. Biomed. Sci. 2015, 22, 18. [Google Scholar] [CrossRef] [PubMed]
- Ku, H.C.; Lee, S.Y.; Chen, C.H.; Wang, Y.H.; Lin, C.T.; Lee, S.S. TM-1-1DP exerts protective effect against myocardial ischemia reperfusion injury via AKT-eNOS pathway. Naunyn Schmiedebergs Arch. Pharmacol. 2015, 388, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, K.S.; Prince, P.S. Caffeic acid protects rat heart mitochondria against isoproterenol-induced oxidative damage. Cell Stress Chaperones 2010, 15, 791–806. [Google Scholar] [CrossRef] [PubMed]
- Dudylina, A.L.; Ivanova, M.V.; Shumaev, K.B.; Ruuge, E.K. Superoxide Formation in Cardiac Mitochondria and Effect of Phenolic Antioxidants. Cell Biochem. Biophys. 2019, 77, 99–107. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenolic Acid | Dose/Concentration | Results | References |
|---|---|---|---|
| Ferulic acid | In vitro: 5.5 μM | Improves expression of mitochondrial biogenesis and dynamics markers and reduces oxidative stress. | Perez-Ternero et al., 2017 |
| In vitro: 10 and 25 μM | Protects H9c2 cardiomyoblasts from hyperglycemia-induced oxidative stress damages via maintenance of Ca2+ homeostasis by safeguarding the SERCA/PLN pathway and mitochondrial function. | Salin Raj et al., 2019 | |
| In vitro: 1, 5, 10 µg/mL | Mitigated oxidative damage via antioxidative protection by heme oxygenase-1 upregulation. | Song et al., 2016 | |
| Ellagic acid | In vitro: 1–20 μM | Suppress oxidative mitochondrial injury induced by doxorubicin and hypoxia via inhibition of Bnip3. Prevents cell necrosis by suppressing LDH release and loss of high mobility group protein B1. | Dhingra et al., 2017 |
| In vitro: 10–100 µM | Protects mitochondria against toxicity induced by bevacizumab, an anthracycline agent, by either its antioxidant properties or indirectly via maintenance of mitochondrial complex II activity. | Khanlou et al., 2020 | |
| Gallic acid | In vitro: 50 μM | Methyl gallate improves mitochondrial functions and increases glutathione synthesis. | Khurana et al., 2014 |
| In vivo: 7.5, 15, 30 mg/kg/day | Gallic acid in synergy with cyclosporine protects myocardial infarction and associated necrotic cell death. | Dianat et al., 2014 | |
| In vivo: 40 mg/kg/day | Epigallocatechin gallate, a gallic acid ester, protects cardiomyocytes by improving the functions of several mitochondrial enzymes involved in respiration and metabolism, consequently down-regulating the oxidative stress-induced apoptosis. | Miltonprabu and Thangapandiyan 2015 | |
| Salvianolic acid | In vitro: 10 mM | Sal B prevents mitochondrial dysfunction by scavenging ROS, prohibits mitochondrial fragmentation by decreasing mitochondrial fission under oxidative stress conditions. | Liu et al., 2017 |
| In vivo: 0.3–3 mg/kg | Sal A attenuates isoproterenol-induced cardiac dysfunction and myocardial injury and improves mitochondrial respiratory function. | Wang et al., 2009 | |
| In vivo: 40 mg/kg/day | Sal B protects myocardium through reducing oxidative stress. | Jiang et al., 2008 | |
| Chlorogenic acid | In vivo: 40 mg/kg | Improved lysosomal stability of the heart of isoproterenol-induced rats. | Akila et al., 2017 |
| In vivo: 20 mg/kg | Reduce mitochondrial ROS production following myocardial infarction. | Li et al., 2018 | |
| Rosmarinic acid | In vitro: 5, 20, 50 mM | Reduces intracellular ROS production of H9c2 cells by suppressing the opening of the mPTP and inhibition of apoptotic factors from mitochondria. | Diao et al., 2016 |
| In vitro: 100, 200 µm | Protect cardiac cells against doxorubicin-induced damage by suppressing energy deprivation and ATP degradation. | Chlopcíková et al., 2004 | |
| Vanilic acid | In vivo: 10, 20, and 40 mg/kg | Suppressed Toll-like receptor 4 signaling, consequently disrupting the inflammation pathway. | Baniahmad et al., 2020 |
| In vitro: 1.00 mM | Reducing of ROS generation, stabilizing mitochondrial membrane potential, limiting the mPTP opening, decreasing caspase-3 activity and inhibiting cardiomyocyte apoptosis. Exerts its cardioprotective effects via the AMPK signaling pathway. | Yao et al., 2020 | |
| Caffeic acid | Ex vivo: 1 μM to 1 mM | Caffeic acid scavenges mitochondria-produced superoxides. | Dudylina et al., 2018 |
| In vivo: 15 mg/kg | Pre-treatment of caffeic acid in rats reduces cytochrome-C-oxidase and NADH dehydrogenase, demonstrating that caffeic acid is able to prevent membrane phospholipid degradation. | Kumaran and Prince 2010 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jubaidi, F.F.; Zainalabidin, S.; Mariappan, V.; Budin, S.B. Mitochondrial Dysfunction in Diabetic Cardiomyopathy: The Possible Therapeutic Roles of Phenolic Acids. Int. J. Mol. Sci. 2020, 21, 6043. https://doi.org/10.3390/ijms21176043
Jubaidi FF, Zainalabidin S, Mariappan V, Budin SB. Mitochondrial Dysfunction in Diabetic Cardiomyopathy: The Possible Therapeutic Roles of Phenolic Acids. International Journal of Molecular Sciences. 2020; 21(17):6043. https://doi.org/10.3390/ijms21176043
Chicago/Turabian StyleJubaidi, Fatin Farhana, Satirah Zainalabidin, Vanitha Mariappan, and Siti Balkis Budin. 2020. "Mitochondrial Dysfunction in Diabetic Cardiomyopathy: The Possible Therapeutic Roles of Phenolic Acids" International Journal of Molecular Sciences 21, no. 17: 6043. https://doi.org/10.3390/ijms21176043
APA StyleJubaidi, F. F., Zainalabidin, S., Mariappan, V., & Budin, S. B. (2020). Mitochondrial Dysfunction in Diabetic Cardiomyopathy: The Possible Therapeutic Roles of Phenolic Acids. International Journal of Molecular Sciences, 21(17), 6043. https://doi.org/10.3390/ijms21176043