Abstract
Leptin is a hormone discovered almost 30 years ago with important implications in metabolism. It is primarily produced by white adipose tissue (WAT) in proportion to the amount of fat. The discovery of leptin was a turning point for two principle reasons: on one hand, it generated promising expectations for the treatment of the obesity, and on the other, it changed the classical concept that white adipose tissue was simply an inert storage organ. Thus, adipocytes in WAT produce the majority of leptin and, although its primary role is the regulation of fat stores by controlling lipolysis and lipogenesis, this hormone also has implications in other physiological processes within WAT, such as apoptosis, browning and inflammation. Although a massive number of questions related to leptin actions have been answered, the necessity for further clarification facilitates constantly renewing interest in this hormone and its pathways. In this review, leptin actions in white adipose tissue will be summarized in the context of obesity.
1. Introduction
Overweightness and obesity are medical conditions that are rising globally in which excess body fat is accumulated. Among the various health effects, this condition can increase the risk of other diseases, such as type 2 diabetes, cardiovascular disease, hypertension, dyslipidemia, fatty liver, obstructive sleep apnea, musculoskeletal disorders or certain types of cancer, many of which reduce life expectancy [1]. The fundamental cause of obesity is a positive energy imbalance such that energy intake exceeds energy expenditure (energy consumed by the body to maintain life and to perform physical activity). This energy balance is predominantly coordinated by the central nervous system (CNS), which receives neural and chemical input regarding the body’s energy status from the peripheral organs, integrates them and generates a proper response to preserve homeostasis [2]. Some of those signals consist of adipocytokines that are produced by the adipose tissue. Thus, in addition to simply serving as an energetic reservoir, the adipose tissue is also an active endocrine organ that regulates many metabolic processes [3]. Leptin is one of those adipocytokines, and its levels are linked with the nutritional status and energy storage. Consequently, leptin acts as an energy level signal, interacting with the nervous and immune system to trigger responses that regulate body weight and energy homeostasis. Ultimately, leptin reduces food intake and body weight [4]; a lack of leptin signaling in humans and rodents, either due to mutation in leptin or its receptor, leads to an increase in food intake, reduction in energy expenditure and other neuroendocrine problems, such as hypothyroidism, infertility and decreased growth [5,6]. However, abnormal or excessive fat accumulation in the context of obesity can lead to increased leptin levels, producing a phenomenon called “leptin resistance”, in which leptin signaling is attenuated. So, during this physiological resistance, leptin loses the capacity to depress feeding, increase energy expenditure and decrease body weight/adiposity [7]. Several mechanisms have been proposed to explain resistance to the catabolic effects of leptin: alterations in leptin receptor expression, problems with transport of leptin across the blood–brain barrier and/or alterations in development programming [8,9,10].
Leptin is produced primarily by the adipocytes in white adipose tissue (WAT), where it plays important physiological roles both indirectly (primarily via the nervous system) and directly (in an autocrine action). In recent years, we have accepted the adipose tissue as an endocrine organ and adipocytes, along with other constitutive cells, as important regulators of whole-body homeostasis. WAT is capable of expanding or altering adipokine production and release, which serves as a powerful crosstalk between different tissues to modulate metabolic homeostasis. Several processes occur in adipose tissue: when an excess of lipids necessitates WAT expansion, the number or size of adipocytes can increase in a phenomenon called adipogenesis; in contrast, through an apoptosis process, fat loss can occur via reduction of the size or number of adipocytes. During fasting or exercise, lipids stored in adipocytes are mobilized to provide energy to the body. Also, in specific circumstances, white adipocytes are able to burn fat in a process called browning, which has drawn interest as a new possibility for obesity treatment. Finally, WAT is also composed of other cells, such as immune cells, and it is strongly linked to the immune system. Leptin contributes to the regulation of all these processes.
2. The Beginning of the Journey: White Adipose Tissue
Adipose tissue is one of the largest endocrine organs in the body; it is involved in a large number of physiological processes. There are two types of adipose tissue: WAT and brown adipose tissue (BAT), which exhibit differences in morphology and functions. Brown adipocytes consist of multiple small lipid droplets packed with a large number of mitochondria. In mammals, BAT is a thermogenic tissue that produces heat to burn fat, and is responsible for adaptative, non-shivering thermogenesis [11]. This unique capacity is attributed to the high number of mitochondria and the expression of uncoupling protein 1 (UCP1) in the inner mitochondrial membrane. When activated, this proton channel protein enables the transport of protons across the membrane, transforming chemical energy (fatty acids) into heat (thermogenesis) [12,13]. BAT is especially important in newborns and hibernating mammals to maintain body temperature and survive cold temperatures. In humans, BAT was primarily considered in newborns and infants, as it was generally accepted that this tissue disappeared or lost functional relevance in adulthood. However, studies have more recently demonstrated BAT existence in adult humans [14], as well as UCP1 expression [15,16]. Since then, countless studies have revealed its functional metabolic role and the implications of this tissue in energy homeostasis and obesity [17,18,19]. In rodents and humans, BAT depots are mainly distributed in the interscapular, axillary, cervical and perirenal areas [20,21], and thermogenic activity can be stimulated in response to cold exposure or noradrenergic stimulation [22].
On the other hand, WAT was long considered an inert tissue to store energy in the form of fat to be mobilized during periods of food deprivation, as well as to provide thermal insulation and mechanical protection [23,24]. White adipocytes contain a single large lipid droplet (90% of the cell volume) with a small number of mitochondria. Anatomically, WAT is widely distributed in different depots; broadly, these depots can be divided into intra-abdominal or visceral (around organs: mesenteric, perigonadal, omental) and subcutaneous (under the skin) compartments [25]. Additional endocrine functions have been attributed to it after noticing that WAT is capable of producing a large number of compounds that can regulate energy balance and other physiological processes. This change was made possible by the discovery of leptin in the ‘90s [26], and, little by little, additional WAT-derived active molecules have been discovered. These active peptides or proteins, called adipocytokines, have actions in a huge number of physiological processes, such as energy metabolism, vascular hemostasis, angiogenesis, immunity and inflammation [27,28]. WAT-derived peptides include leptin, adiponectin, vesfatin and resistin, as well as some pro-inflammatory adipocytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), transforming growth factor-β (TGF-β) and plasminogen activator inhibitor-1 (PAI-1) [29,30]. Depending on its location, different WAT depots exhibit diverse cellular composition, the capacity to secret various adipocytokines, and different innervation and vascularization. This differential distribution of adipose tissue is also related to specific functions. Subcutaneous adipose tissue is implicated in temperature regulation and isolation as well as in sexually dimorphic body compositions. Visceral adipose tissue is located around the organs to serve a structural function [31].
Recently, a third type of adipocyte located in WAT was reported that exhibits characteristics associated with both white and brown adipocytes. They are called beige or brite (brown-in-white) adipocytes and most are located in subcutaneous fat depots [32]. The origin of beige adipocytes has not yet been fully elucidated. In normal conditions, beige adipocytes function as white adipocytes. However, when WAT is stimulated, such as by cold exposure, they can acquire a brown-like phenotype (termed “browning”), evinced by expression of thermogenic genes such as UCP1 and PR domain containing 16 (PRDM16), although the genetic expression patterns are distinct from WAT and BAT [32,33]. Indeed, PRDM16 is necessary for maintaining the beige phenotype, even though UCP1 undoubtedly facilitates thermogenesis [34,35]. Furthermore, adrenergic stimulation plays an essential role in triggering browning, which is facilitated by adjusted sympathetic tone to WAT originating in the brain. In this sense, several neuronal populations in the hypothalamus (the primary regulator of the autonomic nervous system) have been implicated in the control of browning [36,37,38]. Other factors implicated in the activation of browning are immune cells [39,40,41], the metabolic regulator fibroblast growth factor 21 (FGF21) [42], thyroid hormones [43], bone morphogenetic proteins (BMPs) [44,45], nicotine [46,47], cold exposure [48,49] and fasting [50,51]. Leptin also affects browning and will be discussed below.
3. Leptin Production
Leptin is a hormone that was identified in mice in 1994 [26] and in humans in 1995 [52], as a product of the ob gene. Its function was described as a protector against obesity because the ob/ob mice (leptin-deficient) and db/db mice (leptin-resistant) were obese, among other signs and symptoms. However, the literature has shown that leptin can also modulate the neuroendocrine axes, autonomic nervous system, neural plasticity and memory. Leptin is produced and secreted primarily from adipose tissue into circulation to have effects in the CNS and peripheral organs. Relatively low levels of leptin are produced by other tissues, such as skeletal muscle, brain, stomach, pituitary gland, mammary epithelium or placenta, but this appears to result in local, as opposed to systemic, actions [53]. Circulating leptin concentration is directly related to adipose tissue size; this informs the brain about available energy storage [54,55]. More leptin is released by subcutaneous relative to visceral depots, and plasma levels are higher in females than males [56].
Leptin expression and circulating levels change with nutritional state, but also display a circadian oscillation with higher values in the evening and early morning [57,58,59]. Consistent with this, during a fasting period or weight loss, leptin levels decrease, whereas during feeding or in obese conditions, levels increase [60]. However, several metabolic and endocrine factors can also regulate leptin expression and secretion. Importantly, in relation to reduced leptin levels, the adrenergic system plays a clear role. Thus, activation of beta 3 adrenergic receptor (β3-AR) (strongly implicated in the regulation of energy balance) by agonists suppresses leptin levels via a pathway downstream of leptin [61,62]. In the same line, cold exposure is accompanied by a reduction in plasma leptin levels [63,64]. Certain hormones, such as growth hormone, thyroid hormone and androgens, appear to reduce leptin levels [65], whereas others, such as estrogens, can increase leptin production in rats and humans [66]. Furthermore, some drugs can also affect leptin production; for example, glucocorticosteroid injection induces adipose tissue ob gene expression followed by a decrease in body weight gain and food consumption [67,68,69]. Some proinflammatory cytokines might modify circulating leptin levels incrementally initially, followed by a long-term decrement [70] (Figure 1).
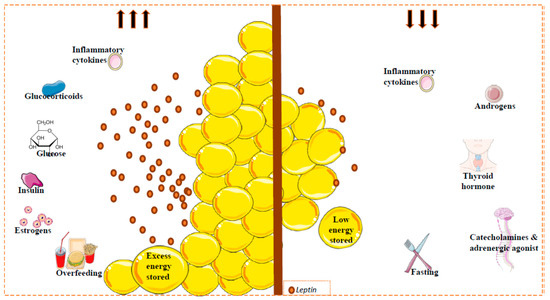
Figure 1.
Factors implicated in circulating leptin levels. Leptin is secreted mainly by white adipose tissue. Leptin levels are correlated with the amount of fat (reflecting the amount of energy stored), and with changes in caloric intake. Other factors can regulate the circulating leptin levels. Glucose, insulin, estrogens and some inflammatory cytokines (acute effect) promote leptin secretion (left); catecholamines or adrenergic agonists, thyroid hormones, androgens and inflammatory cytokines inhibit the secretion of this hormone (right). Fat mass and leptin levels are significantly affected by gender and by menopausal status, where the leptin levels can decrease; leptin levels in females are higher than in males. This sexual dimorphism is due, at least in part, to the suppressive effect of androgens on leptin [78].
It is important to mention the directly proportional relationship that exists between insulin and leptin levels. Physiological insulin infusion maintains or increases plasma leptin concentrations [71], although acute hyperinsulinemia seems have no effect [72,73]. The direct role of insulin in leptin production is supported by some in vitro and in vivo studies, though some discrepant results have been reported. In vitro, subcutaneous human adipose tissue explants incubated with insulin released more leptin compared with controls, but changes in leptin mRNA levels were not detected [74]. Nevertheless, in cultures of human adipocytes, it seems that insulin is essential for maintaining leptin mRNA levels [75]. When the direct effect of insulin was compared in explants and isolated fat cells in parallel, inhibited leptin secretion was similar in both preparations [76]. In vivo, experiments in mice have shown that levels of leptin mRNA were increased after a single insulin injection in fasted animals [57]. Nutritional regulation of leptin is directly linked to insulin levels; with feeding, when insulin levels are increased, leptin levels increase too. Insulin and leptin are important molecules which are implicated in metabolic homeostasis, and their interactions are of clear interest because of the link with obesity and diabetes. It seems clear that ob gene expression is under hormonal control. However, it is a complex scenario to study, as other factors and hormones may be implicated. Also, experimental conditions such as animal, sex, fat depot origin or incubation time [77] could affect the conclusions.
4. Leptin Receptor and Signaling
Leptin receptors (LepRs) are encoded by the db gene, and are expressed in many tissues. Six LepR isoforms (short and long forms) with different physiological roles have been identified [79]. The long isoform (LepRb) has an extracellular N-terminus ligand-binding domain (similar to other isoforms), a transmembrane domain and a complete intracellular domain required for the intracellular signaling pathways. This fully active isoform is essential for leptin action and is expressed in the CNS, mainly in the hypothalamus [80,81,82]. LepRb is implicated in energy homeostasis; in this sense, the db/db mice (which lack LepRb) have an obese phenotype, with diabetes, pubertal disorders and elevated leptin levels [83]. The majority of these receptors are localized inside the cell, and the main signaling pathway recruited by leptin is the JAK2/STAT3 pathway. When leptin binds to LepRb, the receptor changes its conformation and Janus quinasa 2 (JAK2), after translocation, becomes activated by autophosphorylation and activates specific tyrosine residues (Tyr985 and Tyr1138) in the receptor. Tyr1138 phosphorylation allows binding to occur of Signal Transducer and Activator of Transcription 3 (STAT3) (a transcription factor) to an intracellular motif. Subsequently, STAT3 migrates to the nucleus and transcriptionally regulates expression of specific genes [82,84]. Tyr985 is a binding site for the suppressor of cytokine signaling 3 (SOCS3) that exerts inhibitory effects on leptin signaling via a negative feedback loop [85]. Also, phosphorylation of another tyrosine residue, Tyr1077, recruits STAT5 and contributes to the leptin action in energy balance and reproduction [86,87]. The phosphatidylinositol 3-kinase (PI3K) pathway can be activated by leptin too. In peripheral tissues and hypothalamus, this pathway is implicated mainly in insulin actions [88], but leptin can activate PI3K and phosphodiesterase 3B (PDE3B) and provoke a reduction in cAMP levels. This PI3K-PDE3B-cAMP pathway interacts with JAK2-STAT3 pathway and constitutes another essential component of leptin signaling [89,90].
Regarding the short isoforms, LepRe is a soluble isoform that acts as a leptin-binding protein. This receptor can neutralize leptin, removing it from circulation to regulate energy homeostasis [91]. LepRa is expressed in kidney, lung, choroid plexus and in cerebral microvessels [81,92]. This short isoform may play a significant role in the transport of leptin through the blood–brain-barrier (BBB) to the brain, and can contribute to the actions of leptin in obesity [93]. Although LepRb is highly expressed in brain, specifically in areas linked to metabolic regulation (arcuate nucleus, dorsomedial nucleus and ventromedial nucleus), both long and short isoforms have been described in various peripheral tissues such as ovary, uterus, testis, lung, kidney, liver, adrenal gland, placenta, peripheral blood mononuclear cells, WAT and BAT [79,81,94,95,96]. They may be target sites for leptin actions, although their expression is lower compared with that of the CNS, especially the long isoform [95]. However, LepR isoforms have been demonstrated in adipose tissue, including the full-length isoform [97,98,99]; in the adipocyte, LepR is located on the cell membrane and in small cytoplasmic vesicles [100]. A few years after the discovery of leptin, its direct actions were confirmed; leptin treatment of white adipocytes in vitro induced activation of the JAK/STAT pathway and increased expression of target genes, suggesting the activation of the mechanism of leptin action through its receptor [101]. LepR is expressed by adipose tissue-resident immune cells and endothelial cells [100]. This suggests not only a central action, but also an autocrine and paracrine action for leptin.
5. Leptin and Adipogenesis
Adipose tissue is comprised of different cells types. Adipocytes constitute a third of the tissue and the remaining cells are preadipocytes, fibroblasts, stromal cells, T-cells, granulocytes, macrophages and monocytes [31]. The differentiation of preadipocytes to adipocytes (adipogenesis) depends on energy status; it is controlled by hormonal activity and transcription factors [102], and this capacity declines with age [103]. Adipose tissue has the ability to grow when the capacity to store lipids is insufficient. Thus, the growth can be by hyperplasia (increasing the cell number) and hypertrophy (increasing the cell size) [104]. Although one or both types of growth can occur, hyperplasia primarily occurs in early growth states, and hypertrophy occurs previous to hyperplasia during the progression of obesity in most depots [104,105]. However, in humans, these processes are still not completely understood. Depending on the fat depots, different growth patterns can occur. In obese women, it was reported that hyperplasia is predominant in the subcutaneous fat pad, and that hypertrophy occurs both in the omental and subcutaneous depots [106]. In adult humans, the increased lipid storage in adipocytes seems to be the most important process to accumulate fat mass [107,108]. Recently, Spalding et al. observed that the number of fat cells remains constant in lean and obese individuals, and that the number of adipocytes seems to be established at an early age [109]. Although adipogenesis in adult declines, it can still occur; this may be due, in part, to the expression of peroxisome proliferator-activated receptor γ2 (PPARγ2). This receptor is a key regulator of adipogenesis [110], and is more highly expressed in younger relative to older people [111].
Hyperplasia and hypertrophy in adipocytes occur in a leptin-rich environment, so it is interesting to consider the possibility that leptin could exert some paracrine or autocrine effects on adipocyte differentiation. Functional LepRa and LepRb were identified in subcutaneous preadipocytes [112]. In response to a physiological leptin dose, mitogen-activated protein kinase (MAPK) and STAT pathways were activated in rat preadipocytes, promoting adipogenesis. Also, an increase in PPARγ2 expression, lipoprotein lipase levels and more fat storage was observed in these cells [112]. However, direct exposure to high concentrations of leptin (250 and 500 ng leptin/mL) could inhibit proliferation [113]. In the same study, it was observed that leptin could indirectly inhibit the proliferation of preadipocytes by activation or inhibition of unidentified circulating factor(s) [113]. In contrast, in pigs, this inhibition was not observed; thus, leptin in porcine preadipocytes proliferation, even with high dose (1000 ng/mL)- stimulated proliferation [114]. If we compare the results from primary culture with results from 3T3-L1 cells (line derived from murine fibroblasts), some controversial data emerge. Leptin (at dose 5 to 500 ng/mL) had no effect on 3T3-L1 proliferation but suppressed lipid accumulation and increased glycerol released during the maturation process of these cells [115].
Recent studies in vitro in 3T3-L1 cells have shown that leptin (in a similar concentration to the circulating levels of leptin in obese individuals) enhances the expression of adipogenesis- and lipogenesis-related factors (included PPARγ). It also promotes the formation of lipid droplets in preadipocytes by the mTOR signaling pathway [116]. The variations in in vitro studies that have focused on the actions of leptin in this process could be explained by the various sources of leptin and the origin of the preadipocytes; nonetheless, it seems that under experimental conditions, high leptin levels could promote the differentiation of adipocytes. In overweight individuals, leptin levels are high, and an increase in preadipocyte number to facilitate the expansion of fat depots could be expected to avoid fat accumulation in other tissues. However, in natural conditions, i.e., in a healthy animal, leptin production decreases in parallel with the decrease in fat depots.
6. Leptin and Apoptosis
For the purpose of maintaining homeostasis, cells can suffer apoptosis, a normal phenomenon of cell death. Apoptosis is characterized by the activation of caspases (protease enzymes with essential role in programmed cell death); these proteases play a critical role in the execution phase of this process, and their actions lead to cell death via digestion of genomic DNA into oligonucleosomal fragments, among others. DNA fragmentation is considered a symbol of apoptosis [117]. Fat loss could be achieved by decreasing the number of adipocytes through apoptosis. Endogenous elimination of adipocytes has been observed in rats with streptozotocin-induced diabetes, in humans with malignancy and in humans with lipodystrophy [118,119,120]. It seems that adipose tissue apoptosis is linked to various conditions associated with weight loss.
The link between leptin and apoptosis has received much attention since this hormone was discovered. Early reports showed that leptin could modulate this process via CNS. The adipose tissue of leptin-intracerobroventricular (ICV) treated rats showed characteristics particular of apoptosis and a reduction in WAT mass [121]. At the intracellular level, leptin produced a significant increase in PPARγ levels, but a decrease in TNFα, probably a secondary effect of reduced fat depots [121,122]. It has been postulated that PPARγ is a mediator of leptin-induced apoptosis. However, leptin peripheral administration in mice resulted in big decreases in fat pad size, but without the presence of apoptotic markers [123]. Something similar happened with rats made hyperleptinemic by hepatic overexpression after adenoviral gene transfer, i.e., the size of the fat decreased but without changes in the DNA content and with any important decrease in PPARγ [124]. These discrepancies could be explained by the different routes of leptin administration.
Leptin supplementation in adipocytes culture induced the expression of angiopoietin-2 (Ang-2), antagonist of angiogenesis, and apoptosis in the endothelial cells [125]. A decrease in endothelial proliferation could be associated with adipose apoptosis [126]. Leptin can increase reactive oxygen species (ROS) in endothelial cells [127,128], and these species are involved in the induction of apoptosis [129], showing another possible connection. Leptin-induced adipose tissue apoptosis was also studied in ob/ob mice. Della-Fera et al. injected different leptin doses over 14 days in ob/ob and ob/? mice; the first group appeared to be more sensitive than the latter to leptin-induced adipose tissue apoptosis, but there were differences between fat depots [130].
The specific mechanisms that control the apoptotic leptin actions are not clear. However, a study showed that after blocking the neuropeptide Y (NPY) receptor, the adipose tissue apoptosis increased [131]; it is known that leptin provokes a reduction in hypothalamic NPY levels [132], so together, these factors could suggest a link between NPY and the leptin-induced adipose apoptosis. Other reports have provided the opposite idea, i.e., an anti-apoptotic role for leptin, but they were done in brown adipocytes, which is not going to be discussed here [133,134,135]. Over the years, adipocytes were considered very stable, so leptin-induced adipocyte apoptosis is a relatively new concept that needs to be explored in more depth.
7. Leptin and Lipolysis
Energy is stored mainly as triglycerides (TAG) in WAT. The process to mobilize this reservoir is known as lipolysis, i.e., sequential hydrolysis that breaks the TAG to release glycerol and non-esterified fatty acids. The first step is catalyzed by adipose triglyceride lipase (ATGL) that hydrolyzes TAG to diacylglycerol (DAG) and one fatty acid. Afterwards, DAG is transformed to monoacylglycerol (MAG) and another fatty acid by a different enzyme called hormone-sensitive lipase (HSL). Finally, by the enzyme monoacyglycerol lipase (MGL) actions, MAG is hydrolyzed to glycerol and an additional fatty acid (FA) [136,137]. When energy is required, the utilization of FAs is not the same in all fat depots; subcutaneous and some visceral depots (retroperitoneal and mesenteric) are the first to be mobilized [138]. Lipolysis is a vital process, not only providing energy for other tissues, but its intermediate and final products can act as signaling molecules to regulate several metabolic processes. This process happens under basal conditions (non-stimulated) or in conditions stimulated by hormones. For example, under different stimuli, such as periods of fasting or high energy demand, adipocytes are stimulated by hormones (primarily catecholamines) and generate intracellular reactions that activate the enzymes necessary to accelerate lipolysis [139,140]. Adrenergic receptors are widely distributed on the surface of adipocytes in WAT, indicating that the sympathetic nervous system (SNS) plays an important role regulating lipolysis. Also, the contribution of the adrenal gland to this regulation is subdominant, since the adrenodemedullation in rats does not stop lipolysis; thus, catecholaminergic stimulation is transmitted through the SNS to these adrenergic receptors in adipocytes [141]. Anatomic SNS outflow from the CNS to WAT was demonstrated using a viral transsynaptic retrograde tract trace [142]. This bidirectional connection is essential for lipid mobilization, and also has implications in the fat cell number [143,144,145,146]. Leptin exerts actions on sympathetic nerve activity that can affect appetite, energy expenditure and arterial pressure [147]. Regarding the effects in the sympathetic tone to WAT, there are several studies showing its impact. Almost 20 years ago, Sahu et al. showed in rats that, after central chronic leptin infusion, the epididymal fat weight decreased drastically without variations in food intake [148]. Later, another study with leptin infusion into the medio-basal hypothalamus (MBH) showed a suppression in the epidydimal WAT lipogenesis independently of STAT3 signaling, but where an intact autonomic innervation was required [149]. Also, peripheral administration of leptin can excite the sympathetic tone innervating WAT, regulating the lipolytic process specifically in the inguinal fat depots. The authors of this study specifically postulated that leptin travels to the brain through the BBB, and via central histamine H1 receptors, the sympathetic outflow to the WAT is triggered [150].
However, there have been some contradictory findings that are worth mentioning. Rooks et al. revealed interesting results in sympathectomized mice in just one epididymal fat pad by local injection of 6-hydroxydopamine (6OHDA). Explicitly, they observed a reduction in both fat pads, i.e., the intact and denervated, after a peripheral leptin infusion, suggesting that the SNS was not essential for leptin to decrease fat mass and the possibility of an existing communication between fat depots [151]. Similarly, the results of Penn et al. supported the same idea: leptin could reduce the fat size independent of the SNS [152]. In one of the most recent and revealing works, Zeng et al. [153] reconstructed a 3D anatomical picture of the entire subcutaneous fat pad in mouse, revealing a thick bundle of sympathetic neuronal projections in close proximity to adipocytes and in contact with some of them. This was possible using optical projection tomography together with multiphoton microscopy. The authors also showed that the optogenetic activation of these axons increased the levels of noradrenaline, activated HSL, and reduced fat mass. Interestingly, leptin actions in HSL were blocked after disrupting neuronal input surgically, genetically or pharmacologically, signifying that this local innervation is essential for the lipolysis leptin effects [153].Very recently, an important paper was published regarding the mechanisms involved in the effects of leptin on thermogenesis and lipolysis [154]. The effects of acute and chronic leptin treatment on SNS innervation, the main regulator of lipolysis, were evaluated. Low sympathetic innervation in WAT from ob/ob mice was ameliorated (anatomically and functionally) following peripheral chronic leptin treatment. They showed that Brain-Derived Neurotrophic Factor (BDNF)-expressing neurons in the paraventricular nucleus were downstream of agouti-related peptide (AGRP) and proopiomelanocortin (POMC) neurons in the arcuate nucleus of the hypothalamus (ARC), and are essential for the leptin actions restoring sympathetic innervation of WAT in ob/ob mice [154]. These findings also demonstrate that leptin regulates the architecture of the SNS in the adipose tissue.
Leptin receptor is present in adipocytes, so the study of the possible effect of this hormone directly in lipolysis, in an autocrine/paracrine action, began years ago. In vitro, leptin treatment of isolated adipocytes (pooled from different fat depots of lean mice or ob/ob mice) produced an increase in lipolytic activity [155]. Similar results were found in rat epididymal WAT explants [156] and in subcutaneous and perirenal adipocytes [101,157]. Nevertheless, in human adipocytes, this direct lipolytic effect was not observed in adipocytes from children or adults [158]. Also, obese Zucker rats and db/db mice (both possess a mutation in the leptin receptor) do not respond to leptin-induced lipolysis, so seems that the leptin receptor is indispensable for the lipolytic actions [101,159]. Most recently, Pereira et al. observed that some enzymes involved in lipolysis and lipid synthesis decreased in mice with specific deletion of LepR in perigonadal WAT, however they did not find difference in adipose tissue lipolysis [160]; also, excision of LepR specifically in adipose tissue slightly reduced body weight, suggesting that this specific LepR knockdown does not affect lipid turnover [160]. This contrast with the results obtained by Huan et al., who used antisense RNA to downregulate LepR expression in WAT, and observed a strong dysregulation in fat metabolism [161]. The different strategies used could explain the differences; notably, the first study targeted just the long isoform in both BAT and WAT, while the second targeted all isoforms only in WAT. All together, these data suggest that leptin may play roles not only in an endocrine or hypothalamic fashion, but also through an autocrine or paracrine pathway. However, some questions still need be answered, for example, the exact mechanism triggering lipid mobilization after sympathetic denervation is not clarified. Also, leptin acts in several brain areas, but the neuronal networks implicated in the regulation of lipolysis or the control of these sympathetic branches in the fat mass are still unclear.
8. Leptin and Browning
In the last years, the browning (or beiging) of WAT has continuously generated interest because of the potential to increase energy expenditure. Beige adipocytes, together with brown adipocytes, have been linked to obesity resistance in many mouse models [162,163,164], suggesting that beige adipocytes could serve as a therapeutic target to treat metabolic disease. Nevertheless, the thermogenic potential and contribution of this browning process to energy homeostasis is still controversial [165]. The predominant idea was that UCP1 was expressed exclusively in BAT, but lately, some reports showed expression of this molecule in WAT and even in skeletal muscle [166]. Commins et al. showed that peripheral leptin treatment of ob/ob mice induced UCP1 expression in WAT, and this result was considered indirect evidence of increased sympathetic nerve activity (SNA) [167,168]. Using a volume fluorescence-imaging technique, dense sympathetic arborizations were detected in mouse inguinal WAT, showing that these sympathetic fibers originated from celiac ganglia, were in close apposition to most of the adipocytes and were activated by cold exposure [169]. One year later, these observations were supported by 3D whole-tissue technique showing WAT architecture and the dense sympathetic innervation on it [170]. The role of this intra-adipose sympathetic arborization seems to be critical for the cold-induced browning, but it is not clear whether they could control the production of hormones such as leptin [169,171]. However, fasting and chemical-genetic activation of AgRP neurons in hypothalamus suppress the adipocyte browning, specifically in retroperitoneal and inguinal WAT; after activation of these orexigenic neurons, browning was suppressed by regulating sympathetic activity [36]. On the other hand, leptin production is altered by fasting and cold exposure too, suffering a reduction in ob gene expression followed by a decrease in its blood circulating levels [172,173,174]. Specifically, it was observed that in mice with overnight cold exposure, a clear stimulus for beiging, there was a suppression of ob gene expression in WAT and this was mediated by the SNS [63]. It is clear that the SNS is implicated in browning and also in leptin secretion, and both are influenced by different stimuli with diverse responses, but the pathways involved in leptin actions on beiging are not completely clear. In the last years, some new discoveries provided more information about the mechanisms involved in its actions. In 2007, Plum et al. established that PI3K activation in specific leptin-responsive neurons of the CNS regulated sympathetic nerve activity in WAT, leading to elevated mitochondria content and UCP1 expression. This had an impact on energy homeostasis with an increased energy expenditure and leanness [175]. Years later, some authors suggested that browning could be a combined melanocortin response between leptin and insulin and both were required for optimal CNS-mediated WAT browning. Specifically, they showed that the coinfusion of both hormones into the CNS promoted WAT browning acting through hypothalamic POMC neurons, and they were essential for the optimal CNS-meditated WAT browning. In this complementary leptin and insulin signaling action, two phosphatases expressed in ARC POMC neurons, T cell protein tyrosine phosphatase (TCPTP) and protein tyrosine phosphatase1B (PTP1B), seem to be involved [37]. Other proteins, as Forkhead box C2 protein (Foxc2) with important roles in the regulation of several cellular processes included energy metabolism, were linked to the leptin signal and the browning process. Thus, Foxc2 increases the activity of JAK2/STAT3 pathway [176]) leading to increased browning activity in WAT by elevating the expression of thermogenic proteins as PRDM16, UCP1 and PGC1α, but also increasing leptin transcription [177]. An indirect negative effect of leptin in browning has been shown recently. In muscle, the myokine fibronectin type III domain-containing five (FNDC5) after a cleavage process, release a soluble protein called irisin, that acts on white adipose cells to increase UCP1 expression and browning in WAT [178]. When leptin is administrated in mice, the Fndc5 transcription is downregulated in subcutaneous adipocytes, and the irisin-stimulated expression of Ucp1 and Cidec is also reduced in this tissue, although its administration increases the FNDC5 expression in muscle favoring the myogenesis [179]. This suggests a cross-talk between WAT and skeletal muscle where leptin could have a role. The Hedgehog (Hh) signaling pathway is a signal transduction pathway in several biological process and its abnormal activation can result in tumor development [180]. Recently some authors reported with in vitro studies that leptin can interact with this pathway, upregulating the expression of Gli (the key transcription factor in Hh signaling pathway) and in consequence, inhibiting the adipogenesis process [181]. However, another study showed that leptin could inhibit the Hh pathway and promote adipocyte browning [182]. Nevertheless, the specific effect and the mechanism involved should be explored in the future. As previously mentioned, leptin can also exert autocrine actions, and its receptor is expressed in adipose tissue. It does not appear that papers describing a direct/autocrine role for leptin in the browning process are not yet published. However, some papers have shown leptin effect in browning in in vitro experiments, so an autocrine effect seems that exist, but its contribution to and implication for full body homeostasis have yet to be elucidated.
9. Leptin and Inflammation
As we previously discussed, adipose tissue is a dynamic endocrine organ that produces and secretes adipokines as well as pro- and anti-inflammatory cytokines that play an important role in body homeostasis. In obesity conditions, chronic low-grade inflammation is developed and the circulating level of pro-inflammatory cytokines are increased. In this status, WAT is the major source of inflammation associated with obesity [183,184]; the adipose depots suffer an expansion and this is accompanied by the accumulation of immune cells, including infiltrated macrophages with specific characteristics [185]. Leptin, of which levels are increased during obesity, regulates the inflammatory response at several levels (development, proliferation, maturation and activation) and fundamentally, it stimulates the production of pro-inflammatory cytokines [116,186]. Also, most of the immune cells express LepR [187]. All together, these data lead us to consider leptin as a connector among the neuroendocrine and immune system [188,189]. Some reports already have described interesting actions of leptin directly in adipose tissue macrophages (ATM), the most abundant inflammatory cells in WAT. ATMs exhibit an M2 cell phenotype (alternatively activated macrophages; anti-inflammatory type) but they produce cytokines and chemokines usually produced by M1 cells (classically activated macrophages; pro-inflammatory type). Recently, Acedo et al. investigated the role of leptin in the polarization process of macrophages found in adipose tissue. After a leptin exposition, M2-phenotype markers were found in these cells, but also the capacity to produce pro-inflammatory cytokines (M1) [190]. As in previous data, they found the same specific ATM phenotype, but leptin could have a role in their polarization. However, one year later in an interesting study in vivo, other authors have shown that leptin can stimulate the cAMP signaling pathway via the histone deacetylase HDAC4 in ATM, producing a decrease in inflammatory gene expression [191]. Leptin actions in resident macrophages can also be indirectly through mast cells, a migrant cell of connective tissue that contribute to obesity and diabetes [192]. Leptin expression in mast cells from diet-induced obesity (DIO) WAT is higher than those from lean WAT.
Interestingly, leptin-deficient mast cells drive macrophage polarization from M1 towards M2 phenotypes, however do not affect T-cell differentiation [193]. Ob/ob mast cell (leptin-deficient mast cells) were transferred to WT mice and the results observed were surprising: reduction in body weight gain, WAT weight and adipocyte size [193]. Furthermore, the transfer of db/db bone marrow to WT mice placed on high fat diet reduced macrophage infiltration and adipose tissue inflammation with a reduction in adiposity [194]. Leptin has been shown to moderate obesity-related inflammation through mast cells. Some evidences in human adipocytes support the idea that leptin could initiate the recruitment of macrophages to adipose tissue. Leptin could provoke endothelial cell activation and start the recruitment of circulating blood monocytes into fat pads, but this action could be not exclusive since other adipokines could participate too [195]. One of the most recent studies which was already mentioned in previous sections, links leptin inflammation actions with Foxc2; this specific protein is also implicated in the inflammatory process, alleviating it into the adipocyte by downregulation of leptin-JAK2/STAT3-PRDM16 pathway [177]. Despite these findings, some data also support the idea that leptin has no effects in macrophage infiltration in the adipose tissue. For example, Gutierrez et al. observed that hematopoietic LepR deficiency does not affect macrophage accumulation [196] and Gove et al. showed that LepR signaling in bone marrow cells is not implicated in WAT inflammation [197]. Although it is clear that infiltrating macrophages are increased in WAT in models of genetic or high-fat-diet induced obesity and they play an active role in this pathology, mice that are leptin or LepR deficient (ob/ob and db/db respectively) present lower levels of these macrophages and low inflammatory gene expression, even with higher body weight and adiposity [198,199]. Existence of leptin actions in the immune system are clear, but more contributions regarding its role in adipose tissue inflammation, macrophages recruitment and the specific pathways involved are warranted in this novel and fascinating topic.
10. Conclusions and Future Therapeutic Perspectives
The identification of leptin and its crucial role as a central regulator in energy metabolism has opened a new door to increase our knowledge in the factors implicated in obesity and its associated metabolic disorders. Leptin gene mutations in humans are rare [200], and the treatment of obesity with leptin is only effective in a minority of individuals [201], suggesting that leptin may not be the most promising approach for obesity treatment. This effect is, in part, because of the leptin resistance observed in obese individuals, where the exogenous leptin treatment has diffident effects. For this reason, leptin resistance could be considered one of the most important risk factors for overweightness and obesity. In order to find effective treatments against obesity and its comorbidities, it is important to better understand this leptin resistance phenomenon and the underlying mechanisms. Problems in the LepR signaling pathway are predictable causations of leptin resistance; for example, a decrease in LepR trafficking to the cell surface [202,203] and/or increase in the internalization of the receptor via endocytosis [204] could contribute to leptin resistance due to a reduction in plasma membrane LepR. Also, alterations in intracellular proteins implicated in leptin signaling, such as SOCS3 [205], PTP1B and TCPTP [206,207], or Src homology 2B 1 (SH2B1) [208], may contribute to this phenomenon too. Leptin acts mainly through the CNS, so modifications in the hypothalamic neural circuitry could also contribute to the leptin resistance. Thus, inhibition of a BDNF pathway via melanocortin 4 receptor (MC4R)-dependent mechanism in ventromedial nucleus of the hypothalamus (VMH) results in leptin resistance, along with hyperphagia and obesity [209,210]. Recently, it was shown that overexpression of Pblorotein tyrosine phosphatase receptor type J (Ptprj), expressed in hypothalamic neurons together with leptin receptors, could be a factor contributing to the development of leptin resistance [211]. Most of the leptin receptors are in the CNS, and the transport of leptin into the brain is one of the mechanisms that could be implicated in this resistance phenomenon; consequently, this process is damaged with obesity in humans and mice models [9,212]. In humans, Banks et al. found that triglycerides can cross the BBB and induce leptin resistance [213]. Leptin may also exert its activity at a peripheral level, for that reason peripheral leptin resistance can occur in parallel to central leptin resistance.
It is important to mention that some factors can contribute to or cause leptin resistance under an obesity context. Hyperleptinemia itself can cause leptin resistance by downregulating cellular response to leptin [214], thus reversing the hyperleptinemia by reducing leptin expression or secretion by adipocytes could be a approach to control obesity effects [215]. Endoplasmic Reticulum (ER) stress occurs when there is an accumulation of unfolded or misfolded proteins in the ER lumen, and this is associated with several metabolic diseases including obesity [216]. Specifically, obesity cause ER stress in the hypothalamus which can lead to inhibition of leptin receptor signaling contributing to leptin resistance [217,218]. Reducing central ER stress could be an approach to improve leptin resistance and ameliorate obesity problems [219,220,221]. External factors can have implications in leptin resistance too. For example, specific diets with high content in sucrose or salt, or physical activity, can have implications in leptin sensitivity [222,223,224]. Some compounds have shown positive effects reducing leptin resistance; liraglutide (a glucagon-like peptide-1 (GLP1) analogue) reverse this problem by improving endothelial function and reducing inflammatory signals [225]. Chitosan Oligosaccharide Capsules (COSCs) seems to mitigate leptin resistance by activation of JAK2/STAT3 pathway in obese rats [226] and KBH-1 (an herbal mixture) could alleviate this phenomenon by regulation of the AMP-activated protein kinase (AMPK) pathway [227]. Finally, the neuropeptide oxytocin, when administered subcutaneously, showed positive effects reducing the acute but not the chronic leptin resistance in obese mice [228]. Leptin administration in an obesity context has an insignificant impact because of this phenomenon of leptin resistance; for that reason, it is crucial to continue studying the mechanisms underlying this metabolic event.
Likewise, leptin actions are also interesting in other pathologies because of the wide number of biological processes that is implicated (Figure 2), especially under energetic deficit. For example, in lipodystrophy, a condition with abnormal or degenerative adipose tissue unable to accumulate fat; this pathology causes lipid accumulation in other organs leading to the development of secondary problems such as insulin resistance, hepatic steatosis or dyslipidemia [229]. Lipodystrophy can be partial (adipose tissue abnormalities in one or more sites) or generalized (near-total lack of body adipose tissue), and congenital or acquired (caused by autoimmune-mediated destruction or iatrogenic-induced dysfunction of adipose tissue) [230]. Deficiency of adipose tissue can occur in different body areas as well as include different amounts of fat loss, both of which determine the levels of hyperleptinemia and insulin resistance [230].In these patients, exogenous leptin administration improves the metabolic problems associated to lipodystrophy [231,232,233,234,235]. However, some lipodystrophic patients cannot be hypoleptinemic, so the treatment can be more challenging. In caloric-restricted circumstances, leptin levels are low and this can lead to problems in the reproductive axis [236], for example, in functional hypothalamic amenorrhea. In this condition, the existence of a chronic negative energy balance because of excessive exercise or low food intake leads to a leptin deficiency [237]. Women with hypothalamic amenorrhea can benefit from leptin treatment; specifically, some studies have shown an improvement in reproductive, thyroid and growth hormone axes in these patients after receiving a 3-month recombinant leptin therapy [238,239]. Moreover, bone mineral density was improved [239,240]. Anorexia nervosa is characterized by refusal to keep body weight and it is also associated with low concentrations of leptin and neuroendocrine abnormalities [241]; in this context of energy deprivation, leptin treatment has shown positive impacts and a protective effect [242,243,244]. Finally, although in type 2 diabetes leptin treatment shows small effects (probably because of the high serum leptin levels), in type 1 diabetes the effects are more promising. Thus, leptin therapy seems to improve levels of hepatic intermediary metabolites, drops lipogenic and cholesterologenic factors, and reduces plasma and tissue lipids [245,246]. Though these studies are in animals, the results suggest a promising therapeutic potential against diabetes.
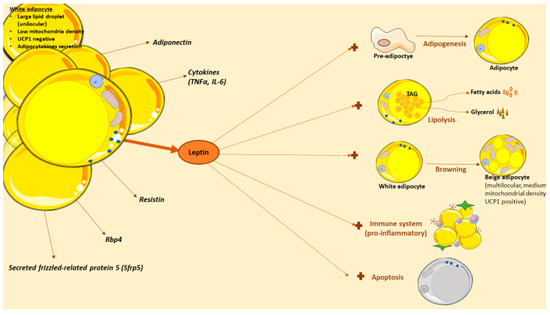
Figure 2.
Leptin actions in white adipose tissue. White adipose tissue (WAT) is composed of by adipocytes, among other cells types. White adipocytes are cells with capacity to store lipids in a large droplet. WAT adipose tissue is more than a reservoir, as it also serves as an endocrine organ that produce and release several adipocytokines with effects in obesity, including leptin. Leptin has several effects in WAT, in direct or indirect mechanisms. Thus, leptin can promote adipogenesis, lipolysis or browning in this tissue. Recently, leptin actions in immune system have sparked the interest in the field; briefly, this hormone stimulates the production of pro-inflammatory cytokines and it is implicated in adipose tissue macrophages (ATMs) polarization. Also, leptin stimulates the apoptotic adipocyte process, another promising area since fat loss could be achieved by decreasing the number of adipocytes through apoptosis.
In other diseases where WAT is altered, leptin has important implications too. In Crohn’s disease where fat-wrapping is one of the main characteristics, leptin has been implicated in the intestinal inflammation associated with these patients; in this way, hyperplastic mesenteric fat, wrapping inflamed intestinal segments, produce leptin and other adipokines that modulate the systemic immune cell composition and the intestinal cell function [247,248]. These patients can suffer at the same time acquired lipodystrophy; in this context, where leptin treatment could have benefits, it is important considerer that also could result in an aggravation of the intestinal inflammation [249]. In the same context, in inflammatory bowel disease (IBD) the mesenteric WAT is hypertrophied; these patients are characterized by suffering anorexia and altered body composition. Leptin levels are also altered in these patients showing overexpression of this adipokine in mesenteric WAT [250]. These data are really important in order to stablish therapeutic interventions for these patients.
WAT is no longer considered an inert reservoir; it can produce a wide number of molecules, as well as include different kind of cells (adipocytes, vascular cells, endothelial cells or immune cells). Leptin is the main adipokine produced by this tissue, and although it has functions in many other tissues, its functions in the same tissue that produce it are essential and have implications in several metabolic processes. Since the discovery of leptin in 1994, countless studies have provided details about its production, functions and pathways, but a better understanding of the precise mechanisms implicated may lead to new approaches and new therapeutic applications. Every year, new studies provide more details or roles about this fascinating hormone, for example the recent leptin role in immunometabolism opening a new window in the field of immunology. Also, leptin resistance is still one of the biggest problems in obesity, where more doubt should be resolved to try to find solutions to one of the biggest problems of the 21 centuries. I strongly believe that, regarding leptin, “there is much else that may be told”.
Funding
N.M.S. and this work were supported by Horizon 2020 Framework Programme H2020-Marie Sklodowska-Curie Actions Individual Fellowship 2017 (H2020-MSCA-IF-2017) (project 795891).
Acknowledgments
The author thanks Chelsea Larabee for her help with editing of this manuscript.
Conflicts of Interest
“The authors declare no conflict of interest”.
References
- Engin, A. The Definition and Prevalence of Obesity and Metabolic Syndrome. Adv. Exp. Med. Biol. 2017, 960, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.V.; Hamr, S.C.; Duca, F.A. Regulation of energy balance by a gut-brain axis and involvement of the gut microbiota. Cell. Mol. Life Sci. 2016, 73, 737–755. [Google Scholar] [CrossRef]
- Heinonen, S.; Jokinen, R.; Rissanen, A.; Pietilainen, K.H. White adipose tissue mitochondrial metabolism in health and in obesity. Obes. Rev. 2019, 21, e12958. [Google Scholar] [CrossRef] [PubMed]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Clement, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998, 392, 398–401. [Google Scholar] [CrossRef]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Munzberg, H.; Bjornholm, M.; Bates, S.H.; Myers, M.G., Jr. Leptin receptor action and mechanisms of leptin resistance. Cell. Mol. Life Sci. 2005, 62, 642–652. [Google Scholar] [CrossRef]
- El-Haschimi, K.; Pierroz, D.D.; Hileman, S.M.; Bjorbaek, C.; Flier, J.S. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Investig. 2000, 105, 1827–1832. [Google Scholar] [CrossRef]
- Munzberg, H.; Flier, J.S.; Bjorbaek, C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology 2004, 145, 4880–4889. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Symonds, M.E. Brown adipose tissue growth and development. Scientifica 2013, 2013, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Betz, M.J.; Enerback, S. Human brown adipose tissue: What we have learned so far. Diabetes 2015, 64, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Heaton, J.M. The distribution of brown adipose tissue in the human. J. Anat. 1972, 112 Pt 1, 35–39. [Google Scholar]
- Lean, M.E.; James, W.P.; Jennings, G.; Trayhurn, P. Brown adipose tissue uncoupling protein content in human infants, children and adults. Clin. Sci. 1986, 71, 291–297. [Google Scholar] [CrossRef]
- Garruti, G.; Ricquier, D. Analysis of uncoupling protein and its mRNA in adipose tissue deposits of adult humans. Int. J. Obes. Relat. Metab. Disord. 1992, 16, 383–390. [Google Scholar]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef]
- Cinti, S. The adipose organ. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 9–15. [Google Scholar] [CrossRef]
- Park, A.; Kim, W.K.; Bae, K.H. Distinction of white, beige and brown adipocytes derived from mesenchymal stem cells. World J. Stem Cells 2014, 6, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N.; Karpe, F.; Fielding, B.A.; Macdonald, I.A.; Coppack, S.W. Integrative physiology of human adipose tissue. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Schoettl, T.; Fischer, I.P.; Ussar, S. Heterogeneity of adipose tissue in development and metabolic function. J. Exp. Biol. 2018, 221 (Suppl. 1). [Google Scholar] [CrossRef]
- Pond, C.M. An evolutionary and functional view of mammalian adipose tissue. Proc. Nutr. Soc. 1992, 51, 367–377. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef]
- Wang, B.; Jenkins, J.R.; Trayhurn, P. Expression and secretion of inflammation-related adipokines by human adipocytes differentiated in culture: Integrated response to TNF-alpha. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E731–E740. [Google Scholar] [CrossRef]
- Maury, E.; Brichard, S.M. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol. Cell. Endocrinol. 2010, 314, 1–16. [Google Scholar] [CrossRef]
- Conde, J.; Scotece, M.; Gomez, R.; Lopez, V.; Gomez-Reino, J.J.; Lago, F.; Gualillo, O. Adipokines: Biofactors from white adipose tissue. A complex hub among inflammation, metabolism, and immunity. Biofactors 2011, 37, 413–420. [Google Scholar] [CrossRef]
- Vazquez-Vela, M.E.; Torres, N.; Tovar, A.R. White adipose tissue as endocrine organ and its role in obesity. Arch. Med. Res. 2008, 39, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, I.G.; Petrovic, N.; de Jong, J.M.; Kalinovich, A.V.; Cannon, B.; Nedergaard, J. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep. 2013, 5, 1196–1203. [Google Scholar] [CrossRef]
- Harms, M.J.; Ishibashi, J.; Wang, W.; Lim, H.W.; Goyama, S.; Sato, T.; Kurokawa, M.; Won, K.J.; Seale, P. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell Metab. 2014, 19, 593–604. [Google Scholar] [CrossRef]
- Ruan, H.B.; Dietrich, M.O.; Liu, Z.W.; Zimmer, M.R.; Li, M.D.; Singh, J.P.; Zhang, K.; Yin, R.; Wu, J.; Horvath, T.L.; et al. O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell 2014, 159, 306–317. [Google Scholar] [CrossRef]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Munzberg, H.; Zhang, Z.Y.; Kahn, B.B.; et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef]
- Beiroa, D.; Imbernon, M.; Gallego, R.; Senra, A.; Herranz, D.; Villarroya, F.; Serrano, M.; Ferno, J.; Salvador, J.; Escalada, J.; et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes 2014, 63, 3346–3358. [Google Scholar] [CrossRef]
- Qiu, Y.; Nguyen, K.D.; Odegaard, J.I.; Cui, X.; Tian, X.; Locksley, R.M.; Palmiter, R.D.; Chawla, A. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 2014, 157, 1292–1308. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Kim, B.S.; Saenz, S.A.; Stine, R.R.; Monticelli, L.A.; Sonnenberg, G.F.; Thome, J.J.; Farber, D.L.; Lutfy, K.; Seale, P.; et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 2015, 519, 242–246. [Google Scholar] [CrossRef]
- Lee, M.W.; Odegaard, J.I.; Mukundan, L.; Qiu, Y.; Molofsky, A.B.; Nussbaum, J.C.; Yun, K.; Locksley, R.M.; Chawla, A. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell 2015, 160, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.M.; Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes. Dev. 2012, 26, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchez, N.; Moreno-Navarrete, J.M.; Contreras, C.; Rial-Pensado, E.; Ferno, J.; Nogueiras, R.; Dieguez, C.; Fernandez-Real, J.M.; Lopez, M. Thyroid hormones induce browning of white fat. J. Endocrinol. 2017, 232, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Elsen, M.; Raschke, S.; Tennagels, N.; Schwahn, U.; Jelenik, T.; Roden, M.; Romacho, T.; Eckel, J. BMP4 and BMP7 induce the white-to-brown transition of primary human adipose stem cells. Am. J. Physiol. Cell Physiol. 2014, 306, C431–C440. [Google Scholar] [CrossRef]
- Whittle, A.J.; Carobbio, S.; Martins, L.; Slawik, M.; Hondares, E.; Vazquez, M.J.; Morgan, D.; Csikasz, R.I.; Gallego, R.; Rodriguez-Cuenca, S.; et al. BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell 2012, 149, 871–885. [Google Scholar] [CrossRef]
- Yoshida, T.; Sakane, N.; Umekawa, T.; Kogure, A.; Kondo, M.; Kumamoto, K.; Kawada, T.; Nagase, I.; Saito, M. Nicotine induces uncoupling protein 1 in white adipose tissue of obese mice. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 570–575. [Google Scholar] [CrossRef][Green Version]
- Seoane-Collazo, P.; Linares-Pose, L.; Rial-Pensado, E.; Romero-Pico, A.; Moreno-Navarrete, J.M.; Martinez-Sanchez, N.; Garrido-Gil, P.; Iglesias-Rey, R.; Morgan, D.A.; Tomasini, N.; et al. Central nicotine induces browning through hypothalamic kappa opioid receptor. Nat. Commun. 2019, 10, 4037. [Google Scholar] [CrossRef]
- Barbatelli, G.; Murano, I.; Madsen, L.; Hao, Q.; Jimenez, M.; Kristiansen, K.; Giacobino, J.P.; De Matteis, R.; Cinti, S. The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1244–E1253. [Google Scholar] [CrossRef]
- Coolbaugh, C.L.; Damon, B.M.; Bush, E.C.; Welch, E.B.; Towse, T.F. Cold exposure induces dynamic, heterogeneous alterations in human brown adipose tissue lipid content. Sci. Rep. 2019, 9, 13600. [Google Scholar] [CrossRef]
- Fabbiano, S.; Suarez-Zamorano, N.; Rigo, D.; Veyrat-Durebex, C.; Stevanovic Dokic, A.; Colin, D.J.; Trajkovski, M. Caloric restriction leads to browning of white adipose tissue through type 2 immune signaling. Cell Metab. 2016, 24, 434–446. [Google Scholar] [CrossRef]
- Li, G.; Xie, C.; Lu, S.; Nichols, R.G.; Tian, Y.; Li, L.; Patel, D.; Ma, Y.; Brocker, C.N.; Yan, T.; et al. Intermittent fasting promotes white adipose browning and decreases obesity by shaping the gut microbiota. Cell Metab. 2017, 26, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Green, E.D.; Maffei, M.; Braden, V.V.; Proenca, R.; DeSilva, U.; Zhang, Y.; Chua, S.C., Jr.; Leibel, R.L.; Weissenbach, J.; Friedman, J.M. The human obese (OB) gene: RNA expression pattern and mapping on the physical, cytogenetic, and genetic maps of chromosome 7. Genome Res. 1995, 5, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [PubMed]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Lollmann, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef]
- Klein, S.; Coppack, S.W.; Mohamed-Ali, V.; Landt, M. Adipose tissue leptin production and plasma leptin kinetics in humans. Diabetes 1996, 45, 984–987. [Google Scholar] [CrossRef] [PubMed]
- Stepien, M.; Rosniak-Bak, K.; Paradowski, M.; Misztal, M.; Kujawski, K.; Banach, M.; Rysz, J. Waist circumference, ghrelin and selected adipose tissue-derived adipokines as predictors of insulin resistance in obese patients: Preliminary results. Med. Sci. Monit. 2011, 17, PR13–PR18. [Google Scholar] [CrossRef]
- Saladin, R.; De Vos, P.; Guerre-Millo, M.; Leturque, A.; Girard, J.; Staels, B.; Auwerx, J. Transient increase in obese gene expression after food intake or insulin administration. Nature 1995, 377, 527–529. [Google Scholar] [CrossRef]
- Sinha, M.K.; Ohannesian, J.P.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Magosin, S.; Marco, C.; Caro, J.F. Nocturnal rise of leptin in lean, obese, and non-insulin-dependent diabetes mellitus subjects. J. Clin. Investig. 1996, 97, 1344–1347. [Google Scholar] [CrossRef]
- Licinio, J.; Mantzoros, C.; Negrao, A.B.; Cizza, G.; Wong, M.L.; Bongiorno, P.B.; Chrousos, G.P.; Karp, B.; Allen, C.; Flier, J.S.; et al. Human leptin levels are pulsatile and inversely related to pituitary-adrenal function. Nat. Med. 1997, 3, 575–579. [Google Scholar] [CrossRef]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef]
- Mantzoros, C.S.; Qu, D.; Frederich, R.C.; Susulic, V.S.; Lowell, B.B.; Maratos-Flier, E.; Flier, J.S. Activation of beta(3) adrenergic receptors suppresses leptin expression and mediates a leptin-independent inhibition of food intake in mice. Diabetes 1996, 45, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Duncan, J.S.; Rayner, D.V.; Hardie, L.J. Rapid inhibition of ob gene expression and circulating leptin levels in lean mice by the beta 3-adrenoceptor agonists BRL 35135A and ZD2079. Biochem. Biophys. Res. Commun. 1996, 228, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Duncan, J.S.; Rayner, D.V. Acute cold-induced suppression of ob (obese) gene expression in white adipose tissue of mice: Mediation by the sympathetic system. Biochem. J. 1995, 311 Pt 3, 729–733. [Google Scholar] [CrossRef]
- Hardie, L.J.; Rayner, D.V.; Holmes, S.; Trayhurn, P. Circulating leptin levels are modulated by fasting, cold exposure and insulin administration in lean but not Zucker (fa/fa) rats as measured by ELISA. Biochem. Biophys. Res. Commun. 1996, 223, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Alaniz, M.H.; Takada, J.; Alonso-Vale, M.I.; Lima, F.B. Adipose tissue as an endocrine organ: From theory to practice. J. Pediatr. 2007, 83 (Suppl. 5), S192–S203. [Google Scholar] [CrossRef]
- Shimizu, H.; Shimomura, Y.; Nakanishi, Y.; Futawatari, T.; Ohtani, K.; Sato, N.; Mori, M. Estrogen increases in vivo leptin production in rats and human subjects. J. Endocrinol. 1997, 154, 285–292. [Google Scholar] [CrossRef]
- De Vos, P.; Saladin, R.; Auwerx, J.; Staels, B. Induction of ob gene expression by corticosteroids is accompanied by body weight loss and reduced food intake. J. Biol. Chem. 1995, 270, 15958–15961. [Google Scholar] [CrossRef]
- Slieker, L.J.; Sloop, K.W.; Surface, P.L.; Kriauciunas, A.; LaQuier, F.; Manetta, J.; Bue-Valleskey, J.; Stephens, T.W. Regulation of expression of ob mRNA and protein by glucocorticoids and cAMP. J. Biol. Chem. 1996, 271, 5301–5304. [Google Scholar] [CrossRef]
- Masuzaki, H.; Ogawa, Y.; Hosoda, K.; Miyawaki, T.; Hanaoka, I.; Hiraoka, J.; Yasuno, A.; Nishimura, H.; Yoshimasa, Y.; Nishi, S.; et al. Glucocorticoid regulation of leptin synthesis and secretion in humans: Elevated plasma leptin levels in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1997, 82, 2542–2547. [Google Scholar] [CrossRef]
- Bruun, J.M.; Pedersen, S.B.; Kristensen, K.; Richelsen, B. Effects of pro-inflammatory cytokines and chemokines on leptin production in human adipose tissue in vitro. Mol. Cell. Endocrinol. 2002, 190, 91–99. [Google Scholar] [CrossRef]
- Malmstrom, R.; Taskinen, M.R.; Karonen, S.L.; Yki-Jarvinen, H. Insulin increases plasma leptin concentrations in normal subjects and patients with NIDDM. Diabetologia 1996, 39, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, S.; Fanelli, C.; Paramore, D.; Brothers, J.; Landt, M. Plasma leptin and insulin relationships in obese and nonobese humans. Diabetes 1996, 45, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Pratley, R.E.; Nicolson, M.; Bogardus, C.; Ravussin, E. Effects of acute hyperinsulinemia on plasma leptin concentrations in insulin-sensitive and insulin-resistant Pima Indians. J. Clin. Endocrinol. Metab. 1996, 81, 4418–4421. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.D.; Petersen, R.N.; Rao, S.P.; Ricci, M.R.; Prasad, A.; Zhang, Y.; Brolin, R.E.; Fried, S.K. Leptin expression in adipose tissue from obese humans: Depot-specific regulation by insulin and dexamethasone. Am. J. Physiol. 1998, 275, E507–E515. [Google Scholar] [CrossRef]
- Wabitsch, M.; Jensen, P.B.; Blum, W.F.; Christoffersen, C.T.; Englaro, P.; Heinze, E.; Rascher, W.; Teller, W.; Tornqvist, H.; Hauner, H. Insulin and cortisol promote leptin production in cultured human fat cells. Diabetes 1996, 45, 1435–1438. [Google Scholar] [CrossRef]
- Mick, G.J.; Wang, X.; Ling Fu, C.; McCormick, K.L. Inhibition of leptin secretion by insulin and metformin in cultured rat adipose tissue. Biochim. Biophys. Acta. (BBA)—Bioenerg. 2000, 1502, 426–432. [Google Scholar] [CrossRef]
- Montague, C.T.; Prins, J.B.; Sanders, L.; Digby, J.E.; O’Rahilly, S. Depot- and sex-specific differences in human leptin mRNA expression: Implications for the control of regional fat distribution. Diabetes 1997, 46, 342–347. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Nicolson, M.; Hirsch, J.; Heymsfield, S.B.; Gallagher, D.; Chu, F.; Leibel, R.L. Effects of gender, body composition, and menopause on plasma concentrations of leptin. J. Clin. Endocrinol. Metab. 1996, 81, 3424–3427. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef]
- Bjorbaek, C.; Uotani, S.; da Silva, B.; Flier, J.S. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J. Biol. Chem. 1997, 272, 32686–32695. [Google Scholar] [CrossRef]
- Fei, H.; Okano, H.J.; Li, C.; Lee, G.H.; Zhao, C.; Darnell, R.; Friedman, J.M. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc. Natl. Acad. Sci. USA 1997, 94, 7001–7005. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Lobo, A.M.; Donato, J., Jr. The role of leptin in health and disease. Temperature 2017, 4, 258–291. [Google Scholar] [CrossRef] [PubMed]
- Chua, S.C., Jr.; Chung, W.K.; Wu-Peng, X.S.; Zhang, Y.; Liu, S.M.; Tartaglia, L.; Leibel, R.L. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 1996, 271, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.H.; Stearns, W.H.; Dundon, T.A.; Schubert, M.; Tso, A.W.; Wang, Y.; Banks, A.S.; Lavery, H.J.; Haq, A.K.; Maratos-Flier, E.; et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 2003, 421, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G., Jr. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef]
- Patterson, C.M.; Villanueva, E.C.; Greenwald-Yarnell, M.; Rajala, M.; Gonzalez, I.E.; Saini, N.; Jones, J.; Myers, M.G., Jr. Leptin action via LepR-b Tyr1077 contributes to the control of energy balance and female reproduction. Mol. Metab. 2012, 1, 61–69. [Google Scholar] [CrossRef]
- Furigo, I.C.; Ramos-Lobo, A.M.; Frazao, R.; Donato, J., Jr. Brain STAT5 signaling and behavioral control. Mol. Cell. Endocrinol. 2016, 438, 70–76. [Google Scholar] [CrossRef]
- Niswender, K.D.; Morrison, C.D.; Clegg, D.J.; Olson, R.; Baskin, D.G.; Myers, M.G., Jr.; Seeley, R.J.; Schwartz, M.W. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: A key mediator of insulin-induced anorexia. Diabetes 2003, 52, 227–231. [Google Scholar] [CrossRef]
- Sahu, A.; Metlakunta, A.S. Hypothalamic phosphatidylinositol 3-kinase-phosphodiesterase 3B-cyclic AMP pathway of leptin signalling is impaired following chronic central leptin infusion. J. Neuroendocrinol. 2005, 17, 720–726. [Google Scholar] [CrossRef]
- Hill, J.W.; Williams, K.W.; Ye, C.; Luo, J.; Balthasar, N.; Coppari, R.; Cowley, M.A.; Cantley, L.C.; Lowell, B.B.; Elmquist, J.K. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J. Clin. Investig. 2008, 118, 1796–1805. [Google Scholar] [CrossRef]
- Zhang, J.; Scarpace, P.J. The soluble leptin receptor neutralizes leptin-mediated STAT3 signalling and anorexic responses in vivo. Br. J. Pharmacol. 2009, 158, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Bjorbaek, C.; Elmquist, J.K.; Michl, P.; Ahima, R.S.; van Bueren, A.; McCall, A.L.; Flier, J.S. Expression of leptin receptor isoforms in rat brain microvessels. Endocrinology 1998, 139, 3485–3491. [Google Scholar] [CrossRef] [PubMed]
- Hileman, S.M.; Pierroz, D.D.; Masuzaki, H.; Bjorbaek, C.; El-Haschimi, K.; Banks, W.A.; Flier, J.S. Characterizaton of short isoforms of the leptin receptor in rat cerebral microvessels and of brain uptake of leptin in mouse models of obesity. Endocrinology 2002, 143, 775–783. [Google Scholar] [CrossRef]
- Zamorano, P.L.; Mahesh, V.B.; De Sevilla, L.M.; Chorich, L.P.; Bhat, G.K.; Brann, D.W. Expression and localization of the leptin receptor in endocrine and neuroendocrine tissues of the rat. Neuroendocrinology 1997, 65, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Hoggard, N.; Mercer, J.G.; Rayner, D.V.; Moar, K.; Trayhurn, P.; Williams, L.M. Localization of leptin receptor mRNA splice variants in murine peripheral tissues by RT-PCR and in situ hybridization. Biochem. Biophys. Res. Commun. 1997, 232, 383–387. [Google Scholar] [CrossRef]
- De Matteis, R.; Dashtipour, K.; Ognibene, A.; Cinti, S. Localization of leptin receptor splice variants in mouse peripheral tissues by immunohistochemistry. Proc. Nutr. Soc. 1998, 57, 441–448. [Google Scholar] [CrossRef]
- Kielar, D.; Clark, J.S.; Ciechanowicz, A.; Kurzawski, G.; Sulikowski, T.; Naruszewicz, M. Leptin receptor isoforms expressed in human adipose tissue. Metabolism 1998, 47, 844–847. [Google Scholar] [CrossRef]
- Kutoh, E.; Boss, O.; Levasseur, F.; Giacobino, J.P. Quantification of the full length leptin receptor (OB-Rb) in human brown and white adipose tissue. Life Sci. 1998, 62, 445–451. [Google Scholar] [CrossRef]
- Lollmann, B.; Gruninger, S.; Stricker-Krongrad, A.; Chiesi, M. Detection and quantification of the leptin receptor splice variants Ob-Ra, b, and, e in different mouse tissues. Biochem. Biophys. Res. Commun. 1997, 238, 648–652. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Abu-Asab, M.; Glasow, A.; Path, G.; Hauner, H.; Tsokos, M.; Chrousos, G.P.; Scherbaum, W.A. Immunohistochemical and ultrastructural localization of leptin and leptin receptor in human white adipose tissue and differentiating human adipose cells in primary culture. Diabetes 2000, 49, 532–538. [Google Scholar] [CrossRef]
- Siegrist-Kaiser, C.A.; Pauli, V.; Juge-Aubry, C.E.; Boss, O.; Pernin, A.; Chin, W.W.; Cusin, I.; Rohner-Jeanrenaud, F.; Burger, A.G.; Zapf, J.; et al. Direct effects of leptin on brown and white adipose tissue. J. Clin. Investig. 1997, 100, 2858–2864. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.R. Transcriptional control of adipocyte formation. Cell Metab. 2006, 4, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Karagiannides, I.; Tchkonia, T.; Dobson, D.E.; Steppan, C.M.; Cummins, P.; Chan, G.; Salvatori, K.; Hadzopoulou-Cladaras, M.; Kirkland, J.L. Altered expression of C/EBP family members results in decreased adipogenesis with aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R1772–R1780. [Google Scholar] [CrossRef]
- Lemonnier, D. Effect of age, sex, and sites on the cellularity of the adipose tissue in mice and rats rendered obese by a high-fat diet. J. Clin. Investig. 1972, 51, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Faust, I.M.; Johnson, P.R.; Stern, J.S.; Hirsch, J. Diet-induced adipocyte number increase in adult rats: A new model of obesity. Am. J. Physiol. 1978, 235, E279–E286. [Google Scholar] [CrossRef]
- Drolet, R.; Richard, C.; Sniderman, A.D.; Mailloux, J.; Fortier, M.; Huot, C.; Rheaume, C.; Tchernof, A. Hypertrophy and hyperplasia of abdominal adipose tissues in women. Int. J. Obes. 2008, 32, 283–291. [Google Scholar] [CrossRef]
- Bjorntorp, P. Effects of age, sex, and clinical conditions on adipose tissue cellularity in man. Metabolism 1974, 23, 1091–1102. [Google Scholar] [CrossRef]
- Hirsch, J.; Batchelor, B. Adipose tissue cellularity in human obesity. Clin. Endocrinol. Metab. 1976, 5, 299–311. [Google Scholar] [CrossRef]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Schipper, B.M.; Marra, K.G.; Zhang, W.; Donnenberg, A.D.; Rubin, J.P. Regional anatomic and age effects on cell function of human adipose-derived stem cells. Ann. Plast. Surg. 2008, 60, 538–544. [Google Scholar] [CrossRef]
- Machinal-Quelin, F.; Dieudonne, M.N.; Leneveu, M.C.; Pecquery, R.; Giudicelli, Y. Proadipogenic effect of leptin on rat preadipocytes in vitro: Activation of MAPK and STAT3 signaling pathways. Am. J. Physiol. Cell Physiol. 2002, 282, C853–C863. [Google Scholar] [CrossRef] [PubMed]
- Wagoner, B.; Hausman, D.B.; Harris, R.B. Direct and indirect effects of leptin on preadipocyte proliferation and differentiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1557–R1564. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, T.G. Porcine preadipocyte proliferation and differentiation: A role for leptin? J. Anim. Sci. 2005, 83, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; Lee, C.Y.; Kang, M.S.; Kim, M.H.; Ryu, Y.H.; Bae, K.H.; Shin, S.J.; Lee, S.C.; Ko, Y. Effects of leptin on lipid metabolism and gene expression of differentiation-associated growth factors and transcription factors during differentiation and maturation of 3T3-L1 preadipocytes. Endocr. J. 2008, 55, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Palhinha, L.; Liechocki, S.; Hottz, E.D.; Pereira, J.; de Almeida, C.J.; Moraes-Vieira, P.M.M.; Bozza, P.T.; Maya-Monteiro, C.M. Leptin induces proadipogenic and proinflammatory signaling in adipocytes. Front. Endocrinol. 2019, 10, 841. [Google Scholar] [CrossRef]
- Sorisky, A.; Magun, R.; Gagnon, A.M. Adipose cell apoptosis: Death in the energy depot. Int. J. Obes. Relat. Metab. Disord. 2000, 24 (Suppl. 4), S3–S7. [Google Scholar] [CrossRef]
- Prins, J.B.; Walker, N.I.; Winterford, C.M.; Cameron, D.P. Human adipocyte apoptosis occurs in malignancy. Biochem. Biophys. Res. Commun. 1994, 205, 625–630. [Google Scholar] [CrossRef]
- Geloen, A.; Roy, P.E.; Bukowiecki, L.J. Regression of white adipose tissue in diabetic rats. Am. J. Physiol. 1989, 257, E547–E553. [Google Scholar] [CrossRef]
- Domingo, P.; Matias-Guiu, X.; Pujol, R.M.; Francia, E.; Lagarda, E.; Sambeat, M.A.; Vazquez, G. Subcutaneous adipocyte apoptosis in HIV-1 protease inhibitor-associated lipodystrophy. AIDS 1999, 13, 2261–2267. [Google Scholar] [CrossRef]
- Qian, H.; Azain, M.J.; Compton, M.M.; Hartzell, D.L.; Hausman, G.J.; Baile, C.A. Brain administration of leptin causes deletion of adipocytes by apoptosis. Endocrinology 1998, 139, 791–794. [Google Scholar] [CrossRef]
- Qian, H.; Hausman, G.J.; Compton, M.M.; Azain, M.J.; Hartzell, D.L.; Baile, C.A. Leptin regulation of peroxisome proliferator-activated receptor-gamma, tumor necrosis factor, and uncoupling protein-2 expression in adipose tissues. Biochem. Biophys. Res. Commun. 1998, 246, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, U.; Benson, B.; Kaufman, S.; Ross, L.; Qi, M.; Scully, S.; DiPalma, C. Morphologic and molecular changes induced by recombinant human leptin in the white and brown adipose tissues of C57BL/6 mice. Lab. Investig. 1997, 77, 243–256. [Google Scholar]
- Zhou, Y.T.; Wang, Z.W.; Higa, M.; Newgard, C.B.; Unger, R.H. Reversing adipocyte differentiation: Implications for treatment of obesity. Proc. Natl. Acad. Sci. USA 1999, 96, 2391–2395. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.; Barkan, D.; Levy, Y.; Goldberg, I.; Fridman, E.; Kopolovic, J.; Rubinstein, M. Leptin induces angiopoietin-2 expression in adipose tissues. J. Biol. Chem. 2001, 276, 7697–7700. [Google Scholar] [CrossRef] [PubMed]
- Rupnick, M.A.; Panigrahy, D.; Zhang, C.Y.; Dallabrida, S.M.; Lowell, B.B.; Langer, R.; Folkman, M.J. Adipose tissue mass can be regulated through the vasculature. Proc. Natl. Acad. Sci. USA 2002, 99, 10730–10735. [Google Scholar] [CrossRef]
- Yamagishi, S.I.; Edelstein, D.; Du, X.L.; Kaneda, Y.; Guzman, M.; Brownlee, M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J. Biol. Chem. 2001, 276, 25096–25100. [Google Scholar] [CrossRef]
- Bouloumie, A.; Marumo, T.; Lafontan, M.; Busse, R. Leptin induces oxidative stress in human endothelial cells. FASEB J. 1999, 13, 1231–1238. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Della-Fera, M.A.; Choi, Y.H.; Hartzell, D.L.; Duan, J.; Hamrick, M.; Baile, C.A. Sensitivity of ob/ob mice to leptin-induced adipose tissue apoptosis. Obes. Res. 2005, 13, 1540–1547. [Google Scholar] [CrossRef]
- Margareto, J.; Aguado, M.; Oses-Prieto, J.A.; Rivero, I.; Monge, A.; Aldana, I.; Marti, A.; Martinez, J.A. A new NPY-antagonist strongly stimulates apoptosis and lipolysis on white adipocytes in an obesity model. Life Sci. 2000, 68, 99–107. [Google Scholar] [CrossRef]
- Wang, Q.; Bing, C.; Al-Barazanji, K.; Mossakowaska, D.E.; Wang, X.M.; McBay, D.L.; Neville, W.A.; Taddayon, M.; Pickavance, L.; Dryden, S.; et al. Interactions between leptin and hypothalamic neuropeptide Y neurons in the control of food intake and energy homeostasis in the rat. Diabetes 1997, 46, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Nechad, M.; Nedergaard, J.; Cannon, B. Noradrenergic stimulation of mitochondriogenesis in brown adipocytes differentiating in culture. Am. J. Physiol. 1987, 253 Pt 1, C889–C894. [Google Scholar] [CrossRef]
- Porras, A.; Fernandez, M.; Benito, M. Adrenergic regulation of the uncoupling protein expression in foetal rat brown adipocytes in primary culture. Biochem. Biophys. Res. Commun. 1989, 163, 541–547. [Google Scholar] [CrossRef]
- Lindquist, J.M.; Rehnmark, S. Ambient temperature regulation of apoptosis in brown adipose tissue. Erk1/2 promotes norepinephrine-dependent cell survival. J. Biol. Chem. 1998, 273, 30147–30156. [Google Scholar] [CrossRef]
- Lafontan, M.; Langin, D. Lipolysis and lipid mobilization in human adipose tissue. Prog. Lipid. Res. 2009, 48, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Bolsoni-Lopes, A.; Alonso-Vale, M.I. Lipolysis and lipases in white adipose tissue—An update. Arch. Endocrinol. Metab. 2015, 59, 335–342. [Google Scholar] [CrossRef]
- Smith, S.R.; Lovejoy, J.C.; Greenway, F.; Ryan, D.; deJonge, L.; de la Bretonne, J.; Volafova, J.; Bray, G.A. Contributions of total body fat, abdominal subcutaneous adipose tissue compartments, and visceral adipose tissue to the metabolic complications of obesity. Metabolism 2001, 50, 425–435. [Google Scholar] [CrossRef]
- Mauriege, P.; De Pergola, G.; Berlan, M.; Lafontan, M. Human fat cell beta-adrenergic receptors: Beta-agonist-dependent lipolytic responses and characterization of beta-adrenergic binding sites on human fat cell membranes with highly selective beta 1-antagonists. J. Lipid Res. 1988, 29, 587–601. [Google Scholar]
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 2007, 27, 79–101. [Google Scholar] [CrossRef]
- Takahashi, A.; Shimazu, T. Hypothalamic regulation of lipid metabolism in the rat: Effect of hypothalamic stimulation on lipolysis. J. Auton. Nerv. Syst. 1981, 4, 195–205. [Google Scholar] [CrossRef]
- Bamshad, M.; Aoki, V.T.; Adkison, M.G.; Warren, W.S.; Bartness, T.J. Central nervous system origins of the sympathetic nervous system outflow to white adipose tissue. Am. J. Physiol. 1998, 275, R291–R299. [Google Scholar] [CrossRef] [PubMed]
- Youngstrom, T.G.; Bartness, T.J. White adipose tissue sympathetic nervous system denervation increases fat pad mass and fat cell number. Am. J. Physiol. 1998, 275 Pt 2, R1488–R1493. [Google Scholar] [CrossRef]
- Bowers, R.R.; Festuccia, W.T.; Song, C.K.; Shi, H.; Migliorini, R.H.; Bartness, T.J. Sympathetic innervation of white adipose tissue and its regulation of fat cell number. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R1167–R1175. [Google Scholar] [CrossRef] [PubMed]
- Cousin, B.; Casteilla, L.; Lafontan, M.; Ambid, L.; Langin, D.; Berthault, M.F.; Penicaud, L. Local sympathetic denervation of white adipose tissue in rats induces preadipocyte proliferation without noticeable changes in metabolism. Endocrinology 1993, 133, 2255–2262. [Google Scholar] [CrossRef]
- Shi, H.; Song, C.K.; Giordano, A.; Cinti, S.; Bartness, T.J. Sensory or sympathetic white adipose tissue denervation differentially affects depot growth and cellularity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1028–R1037. [Google Scholar] [CrossRef]
- Rahmouni, K. Leptin-induced sympathetic nerve activation: Signaling mechanisms and cardiovascular consequences in obesity. Curr. Hypertens. Rev. 2010, 6, 104–209. [Google Scholar] [CrossRef]
- Sahu, A. Resistance to the satiety action of leptin following chronic central leptin infusion is associated with the development of leptin resistance in neuropeptide Y neurones. J. Neuroendocrinol. 2002, 14, 796–804. [Google Scholar] [CrossRef]
- Buettner, C.; Muse, E.D.; Cheng, A.; Chen, L.; Scherer, T.; Pocai, A.; Su, K.; Cheng, B.; Li, X.; Harvey-White, J.; et al. Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat. Med. 2008, 14, 667–675. [Google Scholar] [CrossRef]
- Shen, J.; Tanida, M.; Niijima, A.; Nagai, K. In vivo effects of leptin on autonomic nerve activity and lipolysis in rats. Neurosci. Lett. 2007, 416, 193–197. [Google Scholar] [CrossRef]
- Rooks, C.R.; Penn, D.M.; Kelso, E.; Bowers, R.R.; Bartness, T.J.; Harris, R.B. Sympathetic denervation does not prevent a reduction in fat pad size of rats or mice treated with peripherally administered leptin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R92–R102. [Google Scholar] [CrossRef]
- Penn, D.M.; Jordan, L.C.; Kelso, E.W.; Davenport, J.E.; Harris, R.B. Effects of central or peripheral leptin administration on norepinephrine turnover in defined fat depots. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1613–R1621. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zeng, W.; Pirzgalska, R.M.; Pereira, M.M.; Kubasova, N.; Barateiro, A.; Seixas, E.; Lu, Y.H.; Kozlova, A.; Voss, H.; Martins, G.G.; et al. Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell 2015, 163, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Loh, K.H.; Wu, M.; Morgan, D.A.; Schneeberger, M.; Yu, X.; Chi, J.; Kosse, C.; Kim, D.; Rahmouni, K.; et al. A leptin-BDNF pathway regulating sympathetic innervation of adipose tissue. Nature 2020, 583, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeck, G.; Aguado, M.; Martinez, J.A. In vitro lipolytic effect of leptin on mouse adipocytes: Evidence for a possible autocrine/paracrine role of leptin. Biochem. Biophys. Res. Commun. 1997, 240, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Jaubert, A.-M.; Penot, G.; Niang, F.; Durant, S.; Forest, C. Rapid nitration of adipocyte phosphoenolpyruvate carboxykinase by leptin reduces glyceroneogenesis and induces fatty acid release. PLoS ONE 2012, 7, e40650. [Google Scholar] [CrossRef]
- Rodriguez, V.M.; Macarulla, M.T.; Echevarria, E.; Portillo, M.P. Lipolysis induced by leptin in rat adipose tissue from different anatomical locations. Eur. J. Nutr. 2003, 42, 149–153. [Google Scholar] [CrossRef]
- Elimam, A.; Kamel, A.; Marcus, C. In vitro effects of leptin on human adipocyte metabolism. Horm. Res. 2002, 58, 88–93. [Google Scholar] [CrossRef]
- Fruhbeck, G.; Aguado, M.; Gomez-Ambrosi, J.; Martinez, J.A. Lipolytic effect of in vivo leptin administration on adipocytes of lean and ob/ob mice, but not db/db mice. Biochem. Biophys. Res. Commun. 1998, 250, 99–102. [Google Scholar] [CrossRef]
- Pereira, S.; O’Dwyer, S.M.; Webber, T.D.; Baker, R.K.; So, V.; Ellis, C.E.; Yoon, J.S.; Mojibian, M.; Glavas, M.M.; Karunakaran, S.; et al. Metabolic effects of leptin receptor knockdown or reconstitution in adipose tissues. Sci. Rep. 2019, 9, 3307. [Google Scholar] [CrossRef]
- Huan, J.N.; Li, J.; Han, Y.; Chen, K.; Wu, N.; Zhao, A.Z. Adipocyte-selective reduction of the leptin receptors induced by antisense RNA leads to increased adiposity, dyslipidemia, and insulin resistance. J. Biol. Chem. 2003, 278, 45638–45650. [Google Scholar] [CrossRef] [PubMed]
- Kopecky, J.; Clarke, G.; Enerback, S.; Spiegelman, B.; Kozak, L.P. Expression of the mitochondrial uncoupling protein gene from the aP2 gene promoter prevents genetic obesity. J. Clin. Investig. 1995, 96, 2914–2923. [Google Scholar] [CrossRef] [PubMed]
- Cederberg, A.; Gronning, L.M.; Ahren, B.; Tasken, K.; Carlsson, P.; Enerback, S. FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell 2001, 106, 563–573. [Google Scholar] [CrossRef]
- Seale, P.; Conroe, H.M.; Estall, J.; Kajimura, S.; Frontini, A.; Ishibashi, J.; Cohen, P.; Cinti, S.; Spiegelman, B.M. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Investig. 2011, 121, 96–105. [Google Scholar] [CrossRef]
- Warner, A.; Kjellstedt, A.; Carreras, A.; Bottcher, G.; Peng, X.R.; Seale, P.; Oakes, N.; Linden, D. Activation of beta3-adrenoceptors increases in vivo free fatty acid uptake and utilization in brown but not white fat depots in high-fat-fed rats. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E901–E910. [Google Scholar] [CrossRef]
- Nagase, I.; Yoshida, T.; Kumamoto, K.; Umekawa, T.; Sakane, N.; Nikami, H.; Kawada, T.; Saito, M. Expression of uncoupling protein in skeletal muscle and white fat of obese mice treated with thermogenic beta 3-adrenergic agonist. J. Clin. Investig. 1996, 97, 2898–2904. [Google Scholar] [CrossRef]
- Commins, S.P.; Watson, P.M.; Padgett, M.A.; Dudley, A.; Argyropoulos, G.; Gettys, T.W. Induction of uncoupling protein expression in brown and white adipose tissue by leptin. Endocrinology 1999, 140, 292–300. [Google Scholar] [CrossRef]
- Commins, S.P.; Watson, P.M.; Levin, N.; Beiler, R.J.; Gettys, T.W. Central leptin regulates the UCP1 and ob genes in brown and white adipose tissue via different beta-adrenoceptor subtypes. J. Biol. Chem. 2000, 275, 33059–33067. [Google Scholar] [CrossRef]
- Jiang, H.; Ding, X.; Cao, Y.; Wang, H.; Zeng, W. Dense intra-adipose sympathetic arborizations are essential for cold-induced beiging of mouse white adipose tissue. Cell Metab. 2017, 26, 686–692. [Google Scholar] [CrossRef]
- Chi, J.; Wu, Z.; Choi, C.H.J.; Nguyen, L.; Tegegne, S.; Ackerman, S.E.; Crane, A.; Marchildon, F.; Tessier-Lavigne, M.; Cohen, P. Three-dimensional adipose tissue imaging reveals regional variation in beige fat biogenesis and PRDM16-dependent sympathetic neurite density. Cell Metab. 2018, 27, 226–236. [Google Scholar] [CrossRef]
- Zhu, Y.; Gao, Y.; Tao, C.; Shao, M.; Zhao, S.; Huang, W.; Yao, T.; Johnson, J.A.; Liu, T.; Cypess, A.M.; et al. Connexin 43 mediates white adipose tissue beiging by facilitating the propagation of sympathetic neuronal signals. Cell Metab. 2016, 24, 420–433. [Google Scholar] [CrossRef]
- Becker, D.J.; Ongemba, L.N.; Brichard, V.; Henquin, J.C.; Brichard, S.M. Diet- and diabetes-induced changes of ob gene expression in rat adipose tissue. FEBS Lett. 1995, 371, 324–328. [Google Scholar] [CrossRef][Green Version]
- MacDougald, O.A.; Hwang, C.S.; Fan, H.; Lane, M.D. Regulated expression of the obese gene product (leptin) in white adipose tissue and 3T3-L1 adipocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 9034–9037. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Duncan, J.S.; Hoggard, N.; Rayner, D.V. Regulation of leptin production: A dominant role for the sympathetic nervous system? Proc. Nutr. Soc. 1998, 57, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.; Rother, E.; Munzberg, H.; Wunderlich, F.T.; Morgan, D.A.; Hampel, B.; Shanabrough, M.; Janoschek, R.; Konner, A.C.; Alber, J.; et al. Enhanced leptin-stimulated Pi3k activation in the CNS promotes white adipose tissue transdifferentiation. Cell Metab. 2007, 6, 431–445. [Google Scholar] [CrossRef]
- Myers, M.G.; Cowley, M.A.; Munzberg, H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Liu, Z.; Feng, F.; Wu, T.; Luo, D.; Hu, C.; Sun, C. Foxc2 coordinates inflammation and browning of white adipose by leptin-STAT3-PRDM16 signal in mice. Int. J. Obes. 2018, 42, 252–259. [Google Scholar] [CrossRef]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Rodriguez, A.; Becerril, S.; Mendez-Gimenez, L.; Ramirez, B.; Sainz, N.; Catalan, V.; Gomez-Ambrosi, J.; Fruhbeck, G. Leptin administration activates irisin-induced myogenesis via nitric oxide-dependent mechanisms, but reduces its effect on subcutaneous fat browning in mice. Int. J. Obes. 2015, 39, 397–407. [Google Scholar] [CrossRef]
- Pasca di Magliano, M.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef]
- Liu, Y.; Meng, H.; Chi, S. Up-regulating effect of leptin on Hedgehog signaling pathway in the process of adipocyte differentiation and maturity. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi = Chin. J. Cell. Mol. Immunol. 2015, 31, 1602–1605. [Google Scholar]
- Wang, J.; Ge, J.; Cao, H.; Zhang, X.; Guo, Y.; Li, X.; Xia, B.; Yang, G.; Shi, X. Leptin promotes white adipocyte browning by inhibiting the hh signaling pathway. Cells 2019, 8, 372. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, I.S.; Choue, R. Obesity, inflammation and diet. Pediatr. Gastroenterol. Hepatol. Nutr. 2013, 16, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Myers, M.; Vieira-Potter, V.J. Adipose tissue inflammation and metabolic dysfunction: Role of exercise. Mo. Med. 2014, 111, 65–72. [Google Scholar]
- Surmi, B.K.; Hasty, A.H. Macrophage infiltration into adipose tissue: Initiation, propagation and remodeling. Future Lipidol. 2008, 3, 545–556. [Google Scholar] [CrossRef]
- Otero, M.; Lago, R.; Gomez, R.; Dieguez, C.; Lago, F.; Gomez-Reino, J.; Gualillo, O. Towards a pro-inflammatory and immunomodulatory emerging role of leptin. Rheumatology 2006, 45, 944–950. [Google Scholar] [CrossRef]
- Procaccini, C.; Jirillo, E.; Matarese, G. Leptin as an immunomodulator. Mol. Asp. Med. 2012, 33, 35–45. [Google Scholar] [CrossRef]
- Carlton, E.D.; Demas, G.E.; French, S.S. Leptin, a neuroendocrine mediator of immune responses, inflammation, and sickness behaviors. Horm. Behav. 2012, 62, 272–279. [Google Scholar] [CrossRef]
- Abella, V.; Scotece, M.; Conde, J.; Pino, J.; Gonzalez-Gay, M.A.; Gomez-Reino, J.J.; Mera, A.; Lago, F.; Gomez, R.; Gualillo, O. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 2017, 13, 100–109. [Google Scholar] [CrossRef]
- Acedo, S.C.; Gambero, S.; Cunha, F.G.; Lorand-Metze, I.; Gambero, A. Participation of leptin in the determination of the macrophage phenotype: An additional role in adipocyte and macrophage crosstalk. Vitr. Cell. Dev. Boil. Anim. 2013, 49, 473–478. [Google Scholar] [CrossRef]
- Luan, B.; Goodarzi, M.O.; Phillips, N.G.; Guo, X.; Chen, Y.D.; Yao, J.; Allison, M.; Rotter, J.I.; Shaw, R.; Montminy, M. Leptin-mediated increases in catecholamine signaling reduce adipose tissue inflammation via activation of macrophage HDAC4. Cell Metab. 2014, 19, 1058–1065. [Google Scholar] [CrossRef]
- Liu, J.; Divoux, A.; Sun, J.; Zhang, J.; Clement, K.; Glickman, J.N.; Sukhova, G.K.; Wolters, P.J.; Du, J.; Gorgun, C.Z.; et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 2009, 15, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yu, X.; Chen, H.; Sjoberg, S.; Roux, J.; Zhang, L.; Ivoulsou, A.H.; Bensaid, F.; Liu, C.L.; Liu, J.; et al. leptin deficiency shifts mast cells toward anti-inflammatory actions and protects mice from obesity and diabetes by polarizing M2 macrophages. Cell Metab. 2015, 22, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Dib, L.H.; Ortega, M.T.; Fleming, S.D.; Chapes, S.K.; Melgarejo, T. Bone marrow leptin signaling mediates obesity-associated adipose tissue inflammation in male mice. Endocrinology 2014, 155, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Curat, C.A.; Miranville, A.; Sengenes, C.; Diehl, M.; Tonus, C.; Busse, R.; Bouloumie, A. From blood monocytes to adipose tissue-resident macrophages: Induction of diapedesis by human mature adipocytes. Diabetes 2004, 53, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, D.A.; Hasty, A.H. Haematopoietic leptin receptor deficiency does not affect macrophage accumulation in adipose tissue or systemic insulin sensitivity. J. Endocrinol. 2012, 212, 343–351. [Google Scholar] [CrossRef]
- Gove, M.E.; Sherry, C.L.; Pini, M.; Fantuzzi, G. Generation of leptin receptor bone marrow chimeras: Recovery from irradiation, immune cellularity, cytokine expression, and metabolic parameters. Obesity 2010, 18, 2274–2281. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Farooqi, I.S. Genetic and hereditary aspects of childhood obesity. Best. Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 359–374. [Google Scholar] [CrossRef]
- Heymsfield, S.B.; Greenberg, A.S.; Fujioka, K.; Dixon, R.M.; Kushner, R.; Hunt, T.; Lubina, J.A.; Patane, J.; Self, B.; Hunt, P.; et al. Recombinant leptin for weight loss in obese and lean adults: A randomized, controlled, dose-escalation trial. JAMA 1999, 282, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Rahmouni, K.; Fath, M.A.; Seo, S.; Thedens, D.R.; Berry, C.J.; Weiss, R.; Nishimura, D.Y.; Sheffield, V.C. Leptin resistance contributes to obesity and hypertension in mouse models of Bardet-Biedl syndrome. J. Clin. Investig. 2008, 118, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Guo, D.F.; Bugge, K.; Morgan, D.A.; Rahmouni, K.; Sheffield, V.C. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum. Mol. Genet. 2009, 18, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Belouzard, S.; Delcroix, D.; Rouille, Y. Low levels of expression of leptin receptor at the cell surface result from constitutive endocytosis and intracellular retention in the biosynthetic pathway. J. Biol. Chem. 2004, 279, 28499–28508. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.S.; Unger, E.K.; Olofsson, L.E.; Piper, M.L.; Myers, M.G., Jr.; Xu, A.W. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 2010, 59, 894–906. [Google Scholar] [CrossRef]
- Loh, K.; Fukushima, A.; Zhang, X.; Galic, S.; Briggs, D.; Enriori, P.J.; Simonds, S.; Wiede, F.; Reichenbach, A.; Hauser, C.; et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 2011, 14, 684–699. [Google Scholar] [CrossRef]
- Morrison, C.D.; White, C.L.; Wang, Z.; Lee, S.Y.; Lawrence, D.S.; Cefalu, W.T.; Zhang, Z.Y.; Gettys, T.W. Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology 2007, 148, 433–440. [Google Scholar] [CrossRef]
- Ren, D.; Li, M.; Duan, C.; Rui, L. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab. 2005, 2, 95–104. [Google Scholar] [CrossRef]
- Xu, B.; Goulding, E.H.; Zang, K.; Cepoi, D.; Cone, R.D.; Jones, K.R.; Tecott, L.H.; Reichardt, L.F. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 2003, 6, 736–742. [Google Scholar] [CrossRef]
- Liao, G.Y.; An, J.J.; Gharami, K.; Waterhouse, E.G.; Vanevski, F.; Jones, K.R.; Xu, B. Dendritically targeted Bdnf mRNA is essential for energy balance and response to leptin. Nat. Med. 2012, 18, 564–571. [Google Scholar] [CrossRef]
- Shintani, T.; Higashi, S.; Suzuki, R.; Takeuchi, Y.; Ikaga, R.; Yamazaki, T.; Kobayashi, K.; Noda, M. PTPRJ inhibits leptin signaling, and induction of PTPRJ in the hypothalamus is a cause of the development of leptin resistance. Sci. Rep. 2017, 7, 11627. [Google Scholar] [CrossRef] [PubMed]
- Caro, J.F.; Kolaczynski, J.W.; Nyce, M.R.; Ohannesian, J.P.; Opentanova, I.; Goldman, W.H.; Lynn, R.B.; Zhang, P.L.; Sinha, M.K.; Considine, R.V. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: A possible mechanism for leptin resistance. Lancet 1996, 348, 159–161. [Google Scholar] [CrossRef]
- Banks, W.A.; Farr, S.A.; Salameh, T.S.; Niehoff, M.L.; Rhea, E.M.; Morley, J.E.; Hanson, A.J.; Hansen, K.M.; Craft, S. Triglycerides cross the blood-brain barrier and induce central leptin and insulin receptor resistance. Int. J. Obes. 2018, 42, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Hannan, K.S.; Greenberg, M.L.; Friedman, J.M. Hyperleptinemia is required for the development of leptin resistance. PLoS ONE 2010, 5, e11376. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Cinar, R.; Liu, J.; Godlewski, G.; Wesley, D.; Jourdan, T.; Szanda, G.; Mukhopadhyay, B.; Chedester, L.; Liow, J.S.; et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012, 16, 167–179. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef]
- Hosoi, T.; Sasaki, M.; Miyahara, T.; Hashimoto, C.; Matsuo, S.; Yoshii, M.; Ozawa, K. Endoplasmic reticulum stress induces leptin resistance. Mol. Pharmacol. 2008, 74, 1610–1619. [Google Scholar] [CrossRef]
- Contreras, C.; Gonzalez-Garcia, I.; Martinez-Sanchez, N.; Seoane-Collazo, P.; Jacas, J.; Morgan, D.A.; Serra, D.; Gallego, R.; Gonzalez, F.; Casals, N.; et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep. 2014, 9, 366–377. [Google Scholar] [CrossRef]
- Contreras, C.; Gonzalez-Garcia, I.; Seoane-Collazo, P.; Martinez-Sanchez, N.; Linares-Pose, L.; Rial-Pensado, E.; Ferno, J.; Tena-Sempere, M.; Casals, N.; Dieguez, C.; et al. Reduction of hypothalamic endoplasmic reticulum stress activates browning of white fat and ameliorates obesity. Diabetes 2017, 66, 87–99. [Google Scholar] [CrossRef]
- Won, J.C.; Jang, P.G.; Namkoong, C.; Koh, E.H.; Kim, S.K.; Park, J.Y.; Lee, K.U.; Kim, M.S. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity 2009, 17, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Ropelle, E.R.; Flores, M.B.; Cintra, D.E.; Rocha, G.Z.; Pauli, J.R.; Morari, J.; de Souza, C.T.; Moraes, J.C.; Prada, P.O.; Guadagnini, D.; et al. IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biol. 2010, 8, e1000465. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B.S. Source of dietary sucrose influences development of leptin resistance in male and female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R598–R610. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Kuwabara, M.; Andres-Hernando, A.; Li, N.; Cicerchi, C.; Jensen, T.; Orlicky, D.J.; Roncal-Jimenez, C.A.; Ishimoto, T.; Nakagawa, T.; et al. High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 3138–3143. [Google Scholar] [CrossRef]
- Li, N.; Zhao, Y.; Yue, Y.; Chen, L.; Yao, Z.; Niu, W. Liraglutide ameliorates palmitate-induced endothelial dysfunction through activating AMPK and reversing leptin resistance. Biochem. Biophys. Res. Commun. 2016, 478, 46–52. [Google Scholar] [CrossRef]
- Pan, H.; Fu, C.; Huang, L.; Jiang, Y.; Deng, X.; Guo, J.; Su, Z. Anti-obesity effect of chitosan oligosaccharide capsules (COSCs) in obese rats by ameliorating leptin resistance and adipogenesis. Mar. Drugs 2018, 16, 198. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.J.; Cho, W.K.; Yim, N.H.; Kim, H.K.; Yun, B.; Ma, J.Y. KBH-1, an herbal composition, improves hepatic steatosis and leptin resistance in high-fat diet-induced obese rats. BMC Complement. Altern. Med. 2016, 16, 355. [Google Scholar] [CrossRef]
- Labyb, M.; Chretien, C.; Caillon, A.; Rohner-Jeanrenaud, F.; Altirriba, J. Oxytocin administration alleviates acute but not chronic leptin resistance of diet-induced obese mice. Int. J. Mol. Sci. 2018, 20, 88. [Google Scholar] [CrossRef]
- Akinci, B.; Sahinoz, M.; Oral, E. Lipodystrophy syndromes: Presentation and treatment. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Garg, A. Lipodystrophies: Genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 2011, 96, 3313–3325. [Google Scholar] [CrossRef]
- Shimomura, I.; Hammer, R.E.; Ikemoto, S.; Brown, M.S.; Goldstein, J.L. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 1999, 401, 73–76. [Google Scholar] [CrossRef]
- Oral, E.A.; Simha, V.; Ruiz, E.; Andewelt, A.; Premkumar, A.; Snell, P.; Wagner, A.J.; DePaoli, A.M.; Reitman, M.L.; Taylor, S.I.; et al. Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med. 2002, 346, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Oral, E.A.; Dufour, S.; Befroy, D.; Ariyan, C.; Yu, C.; Cline, G.W.; DePaoli, A.M.; Taylor, S.I.; Gorden, P.; et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J. Clin. Investig. 2002, 109, 1345–1350. [Google Scholar] [CrossRef]
- Javor, E.D.; Cochran, E.K.; Musso, C.; Young, J.R.; Depaoli, A.M.; Gorden, P. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes 2005, 54, 1994–2002. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Chong, A.Y.; Cochran, E.K.; Kleiner, D.E.; Haller, M.J.; Schatz, D.A.; Gorden, P. Type 1 diabetes associated with acquired generalized lipodystrophy and insulin resistance: The effect of long-term leptin therapy. J. Clin. Endocrinol. Metab. 2008, 93, 26–31. [Google Scholar] [CrossRef]
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. [Google Scholar] [CrossRef] [PubMed]
- Shufelt, C.L.; Torbati, T.; Dutra, E. Hypothalamic amenorrhea and the long-term health consequences. Semin. Reprod. Med. 2017, 35, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Welt, C.K.; Chan, J.L.; Bullen, J.; Murphy, R.; Smith, P.; DePaoli, A.M.; Karalis, A.; Mantzoros, C.S. Recombinant human leptin in women with hypothalamic amenorrhea. N. Engl. J. Med. 2004, 351, 987–997. [Google Scholar] [CrossRef]
- Chou, S.H.; Chamberland, J.P.; Liu, X.; Matarese, G.; Gao, C.; Stefanakis, R.; Brinkoetter, M.T.; Gong, H.; Arampatzi, K.; Mantzoros, C.S. Leptin is an effective treatment for hypothalamic amenorrhea. Proc. Natl. Acad. Sci. USA 2011, 108, 6585–6590. [Google Scholar] [CrossRef]
- Sienkiewicz, E.; Magkos, F.; Aronis, K.N.; Brinkoetter, M.; Chamberland, J.P.; Chou, S.; Arampatzi, K.M.; Gao, C.; Koniaris, A.; Mantzoros, C.S. Long-term metreleptin treatment increases bone mineral density and content at the lumbar spine of lean hypoleptinemic women. Metabolism 2011, 60, 1211–1221. [Google Scholar] [CrossRef]
- Chan, J.L.; Mantzoros, C.S. Role of leptin in energy-deprivation states: Normal human physiology and clinical implications for hypothalamic amenorrhoea and anorexia nervosa. Lancet 2005, 366, 74–85. [Google Scholar] [CrossRef]
- Ahima, R.S.; Prabakaran, D.; Mantzoros, C.; Qu, D.; Lowell, B.; Maratos-Flier, E.; Flier, J.S. Role of leptin in the neuroendocrine response to fasting. Nature 1996, 382, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.L.; Heist, K.; DePaoli, A.M.; Veldhuis, J.D.; Mantzoros, C.S. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J. Clin. Investig. 2003, 111, 1409–1421. [Google Scholar] [CrossRef] [PubMed]
- Exner, C.; Hebebrand, J.; Remschmidt, H.; Wewetzer, C.; Ziegler, A.; Herpertz, S.; Schweiger, U.; Blum, W.F.; Preibisch, G.; Heldmaier, G.; et al. Leptin suppresses semi-starvation induced hyperactivity in rats: Implications for anorexia nervosa. Mol. Psychiatry 2000, 5, 476–481. [Google Scholar] [CrossRef]
- Wang, M.Y.; Chen, L.; Clark, G.O.; Lee, Y.; Stevens, R.D.; Ilkayeva, O.R.; Wenner, B.R.; Bain, J.R.; Charron, M.J.; Newgard, C.B.; et al. Leptin therapy in insulin-deficient type I diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 4813–4819. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, T.; Chuang, J.C.; Sakata, I.; Ramadori, G.; Coppari, R. Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 17391–17396. [Google Scholar] [CrossRef]
- Sheehan, A.L.; Warren, B.F.; Gear, M.W.; Shepherd, N.A. Fat-wrapping in Crohn’s disease: Pathological basis and relevance to surgical practice. Br. J. Surg. 1992, 79, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, C.; Ziegler, J.F.; Letizia, M.; Schmidt, F.; Siegmund, B. Adipokines and their role in intestinal inflammation. Front. Immunol. 2018, 9, 1974. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Bottcher, C.; Letizia, M.; Yerinde, C.; Wu, H.; Freise, I.; Rodriguez-Sillke, Y.; Stoyanova, A.K.; Kreis, M.E.; Asbach, P.; et al. Leptin induces TNFalpha-dependent inflammation in acquired generalized lipodystrophy and combined Crohn’s disease. Nat. Commun. 2019, 10, 5629. [Google Scholar] [CrossRef]
- Barbier, M.; Vidal, H.; Desreumaux, P.; Dubuquoy, L.; Bourreille, A.; Colombel, J.F.; Cherbut, C.; Galmiche, J.P. Overexpression of leptin mRNA in mesenteric adipose tissue in inflammatory bowel diseases. Gastroenterol. Clin. Biol. 2003, 27, 987–991. [Google Scholar] [CrossRef]


© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).