Characterization of the Relationship between the Chaperone and Lipid-Binding Functions of the 70-kDa Heat-Shock Protein, HspA1A

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
2.1. Rational of Lipids Used in the Present Study
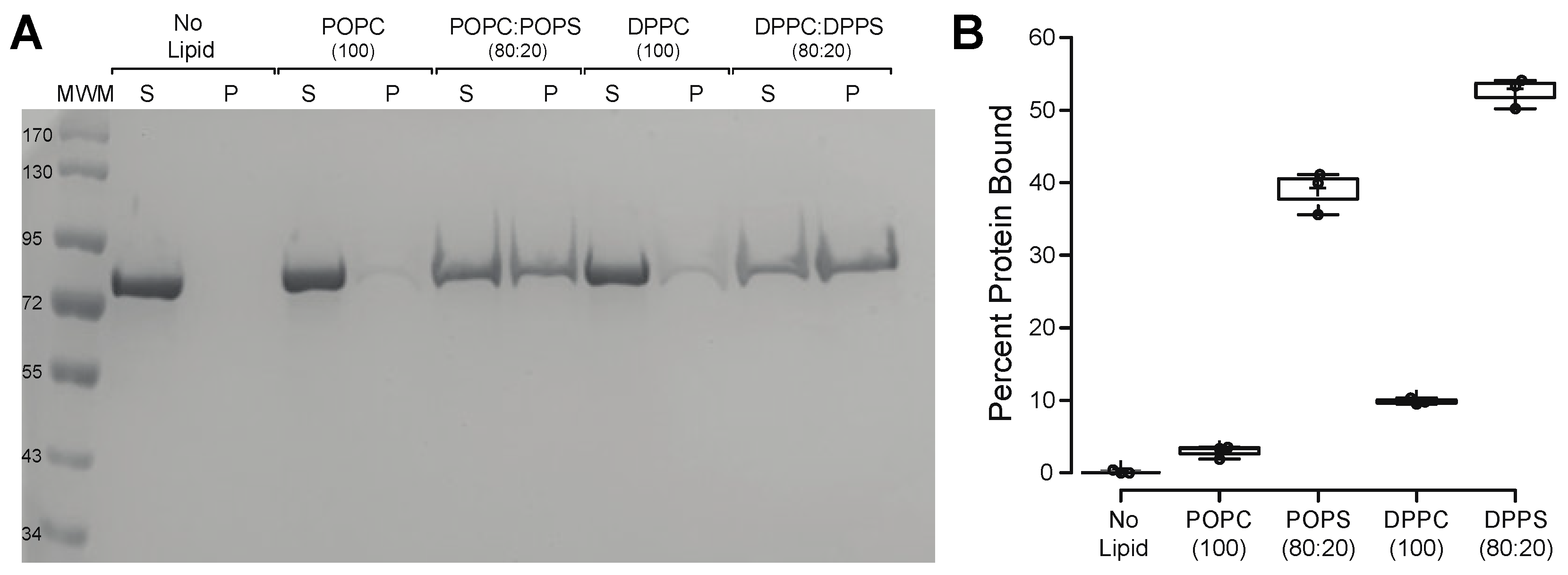
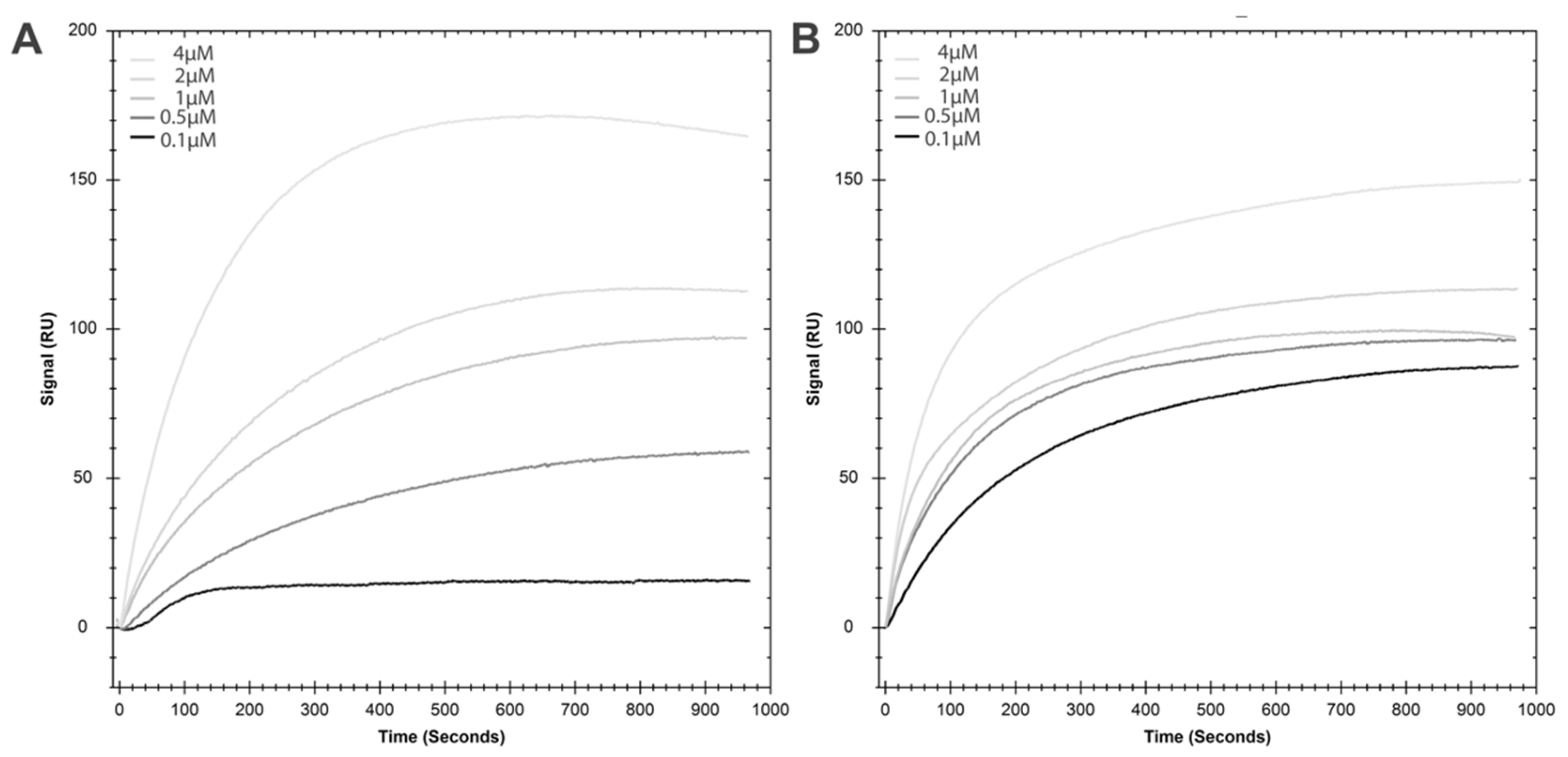
2.2. HspA1A Lipid-Binding Properties
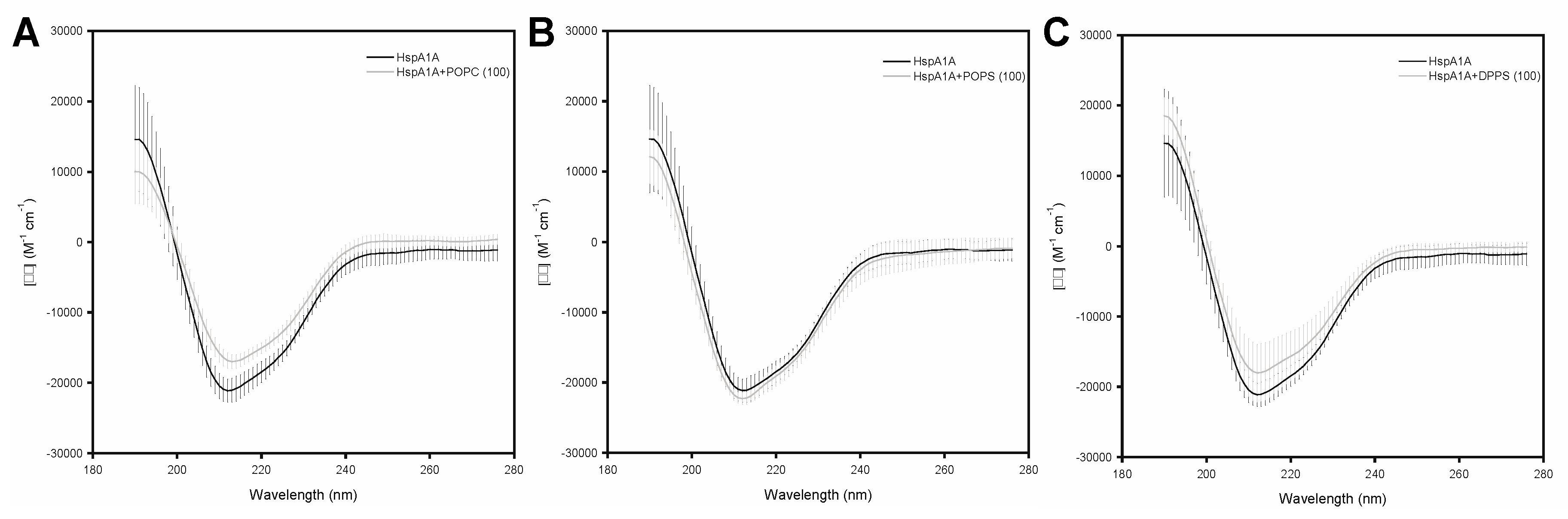
2.3. Effects of Lipids on the Secondary Structure of HspA1A
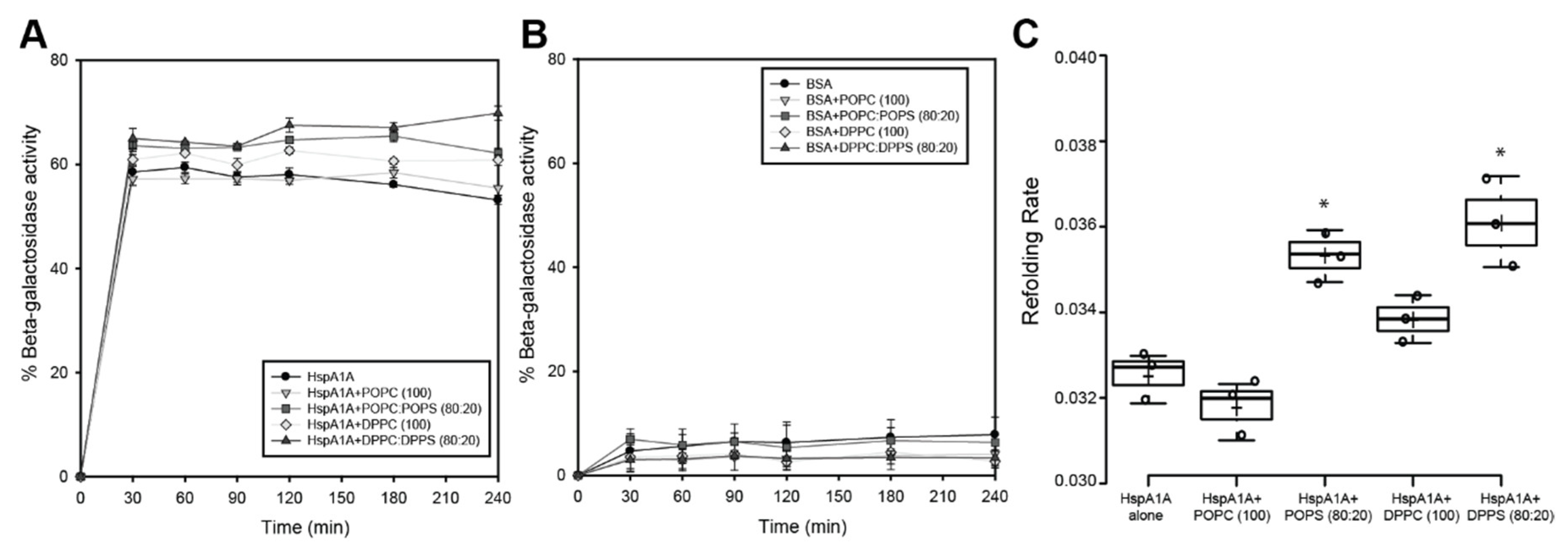
2.4. Effects of Lipids on the ATPase Activity of HspA1A
2.5. Effects of Lipids on the Refolding Activity of HspA1A
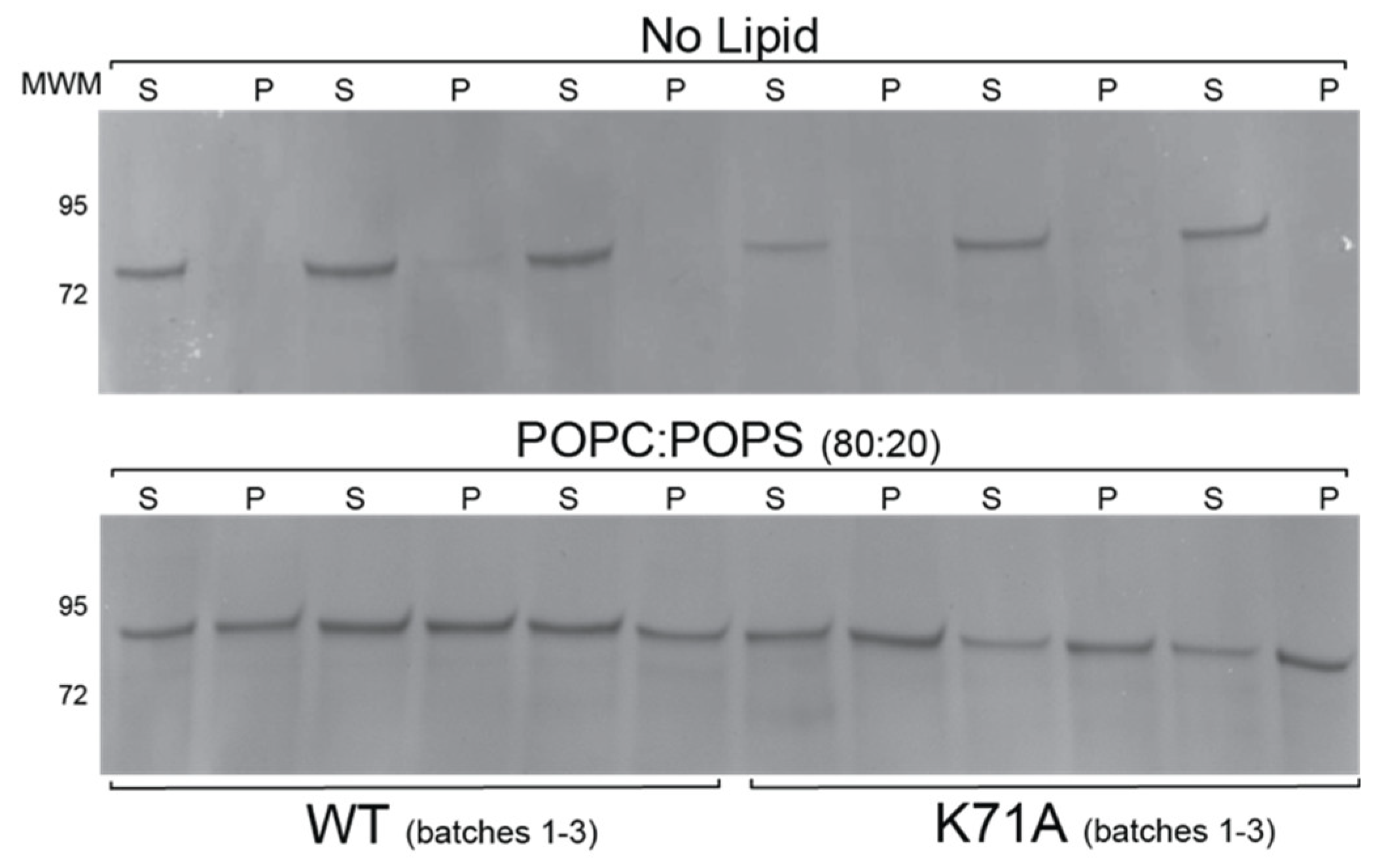
2.6. Lipid Binding Properties of the K71A Mutation of HspA1A
3. Discussion
4. Materials and Methods
4.1. Lipids, Chemicals, and Reagents
4.2. Generation of Recombinant Wild-Type and Mutated Clones, Proteins, and Protein Purification
4.3. Generation of Liposomes
4.4. Lipid Binding Assays
4.5. Circular Dichroism (CD) Spectra
4.6. ATP Hydrolysis Assay
4.7. β-Galactosidase Refolding Assay
4.8. Statistical Tests
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMP | bis-(monoacylglycerol)-phosphate [sn-(3-oleoyl-2-hydroxy)-glycerol-1-phospho-sn-3’-(1’-oleoyl-2’-hydroxy)-glycerol)] |
| DPPC | 1,2-dipalmitoyl-sn-glycero-3-phosphocholine |
| DPPS | 1,2-dipalmitoyl-sn-glycero-3-phosphoserine (sodium salt) |
| Hsp70 | seventy-kilodalton heat shock protein |
| GB3 | globotriaoslyceramide |
| LVS | liposomal vesicle sedimentation |
| NBD | nucleotide-binding domain |
| PM | plasma membrane |
| POPC | phosphatidylcholine [1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPS | phosphatidylserine [1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (sodium salt) |
| PS | Phosphatidylserine |
| SBD | substrate-binding domain |
| SGC | sulfatide (3-O-sulfo-d-galactosyl-β1-1’-N-lignoceroyl-d-erythro-sphingosine) |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
References
- Daugaard, M.; Rohde, M.; Jaattela, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 chaperone machines. Cell 1998, 92, 351–366. [Google Scholar] [CrossRef]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015, 6, 8308. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef] [PubMed]
- Balogi, Z.; Multhoff, G.; Jensen, T.K.; Lloyd-Evans, E.; Yamashima, T.; Jaattela, M.; Harwood, J.L.; Vigh, L. Hsp70 interactions with membrane lipids regulate cellular functions in health and disease. Prog. Lipid Res. 2019, 74, 18–30. [Google Scholar] [CrossRef]
- Schilling, D.; Gehrmann, M.; Steinem, C.; De Maio, A.; Pockley, A.G.; Abend, M.; Molls, M.; Multhoff, G. Binding of heat shock protein 70 to extracellular phosphatidylserine promotes killing of normoxic and hypoxic tumor cells. FASEB J. 2009, 23, 2467–2477. [Google Scholar] [CrossRef]
- Bilog, A.D.; Smulders, L.; Oliverio, R.; Labanieh, C.; Zapanta, J.; Stahelin, R.V.; Nikolaidis, N. Membrane Localization of HspA1A, a Stress Inducible 70-kDa Heat-Shock Protein, Depends on Its Interaction with Intracellular Phosphatidylserine. Biomolecules 2019, 9, 152. [Google Scholar] [CrossRef]
- McCallister, C.; Kdeiss, B.; Nikolaidis, N. Biochemical characterization of the interaction between HspA1A and phospholipids. Cell Stress Chaperones 2015, 21, 41–53. [Google Scholar] [CrossRef]
- McCallister, C.; Kdeiss, B.; Nikolaidis, N. HspA1A, a 70-kDa heat shock protein, differentially interacts with anionic lipids. Biochem. Biophys. Res. Commun. 2015, 467, 835–840. [Google Scholar] [CrossRef]
- McCallister, C.; Siracusa, M.C.; Shirazi, F.; Chalkia, D.; Nikolaidis, N. Functional diversification and specialization of cytosolic 70-kDa heat shock proteins. Sci. Rep. 2015, 5, 9363. [Google Scholar] [CrossRef]
- Armijo, G.; Okerblom, J.; Cauvi, D.M.; Lopez, V.; Schlamadinger, D.E.; Kim, J.; Arispe, N.; De Maio, A. Interaction of heat shock protein 70 with membranes depends on the lipid environment. Cell Stress Chaperones 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.; Oliverio, R.; Nguyen, P.; Le, D.; Ellis, J.; Kdeiss, B.; Ord, S.; Chalkia, D.; Nikolaidis, N. Concurrent action of purifying selection and gene conversion results in extreme conservation of the major stress-inducible Hsp70 genes in mammals. Sci. Rep. 2018, 8, 5082. [Google Scholar] [CrossRef] [PubMed]
- Oliverio, R.; Nguyen, P.; Kdeiss, B.; Ord, S.; Daniels, A.J.; Nikolaidis, N. Functional characterization of natural variants found on the major stress inducible 70-kDa heat shock gene, HSPA1A, in humans. Biochem. Biophys. Res. Commun. 2018, 506, 799–804. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.C.; Flaherty, K.M.; McKay, D.B. Lysine 71 of the chaperone protein Hsc70 Is essential for ATP hydrolysis. J. Biol. Chem. 1996, 271, 15874–15878. [Google Scholar] [CrossRef]
- Barthel, T.K.; Zhang, J.; Walker, G.C. ATPase-defective derivatives of Escherichia coli DnaK that behave differently with respect to ATP-induced conformational change and peptide release. J. Bacteriol. 2001, 183, 5482–5490. [Google Scholar] [CrossRef]
- Broquet, A.H.; Thomas, G.; Masliah, J.; Trugnan, G.; Bachelet, M. Expression of the molecular chaperone Hsp70 in detergent-resistant microdomains correlates with its membrane delivery and release. J. Biol. Chem. 2003, 278, 21601–21606. [Google Scholar] [CrossRef]
- Chen, S.; Bawa, D.; Besshoh, S.; Gurd, J.W.; Brown, I.R. Association of heat shock proteins and neuronal membrane components with lipid rafts from the rat brain. J. Neurosci. Res. 2005, 81, 522–529. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Febbraio, M.A. Exosome-dependent trafficking of HSP70: A novel secretory pathway for cellular stress proteins. J. Biol. Chem. 2005, 280, 23349–23355. [Google Scholar] [CrossRef]
- Nimmervoll, B.; Chtcheglova, L.A.; Juhasz, K.; Cremades, N.; Aprile, F.A.; Sonnleitner, A.; Hinterdorfer, P.; Vigh, L.; Preiner, J.; Balogi, Z. Cell surface localised Hsp70 is a cancer specific regulator of clathrin-independent endocytosis. FEBS Lett. 2015, 589, 2747–2753. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Pockley, A.G.; Schmid, T.E.; Schilling, D. The role of heat shock protein 70 (Hsp70) in radiation-induced immunomodulation. Cancer Lett. 2015, 368, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Breuninger, S.; Stangl, S.; Werner, C.; Sievert, W.; Lobinger, D.; Foulds, G.A.; Wagner, S.; Pickhard, A.; Piontek, G.; Kokowski, K.; et al. Membrane Hsp70-A Novel Target for the Isolation of Circulating Tumor Cells After Epithelial-to-Mesenchymal Transition. Front. Oncol. 2018, 8, 497. [Google Scholar] [CrossRef]
- Hantschel, M.; Pfister, K.; Jordan, A.; Scholz, R.; Andreesen, R.; Schmitz, G.; Schmetzer, H.; Hiddemann, W.; Multhoff, G. Hsp70 plasma membrane expression on primary tumor biopsy material and bone marrow of leukemic patients. Cell Stress Chaperones 2000, 5, 438–442. [Google Scholar] [CrossRef]
- Vega, V.L.; Rodriguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef] [PubMed]
- McCallister, C.; Kdeiss, B.; Oliverio, R.; Nikolaidis, N. Characterization of the binding between a 70-kDa heat shock protein, HspA1A, and phosphoinositides. Biochem. Biophys. Res. Commun. 2016, 472, 270–275. [Google Scholar] [CrossRef]
- Mahalka, A.K.; Kirkegaard, T.; Jukola, L.T.; Jaattela, M.; Kinnunen, P.K. Human heat shock protein 70 (Hsp70) as a peripheral membrane protein. Biochim. Biophys. Acta 2014, 1838, 1344–1361. [Google Scholar] [CrossRef]
- Arispe, N.; De Maio, A. ATP and ADP modulate a cation channel formed by Hsc70 in acidic phospholipid membranes. J. Biol. Chem. 2000, 275, 30839–30843. [Google Scholar] [CrossRef]
- Harada, Y.; Sato, C.; Kitajima, K. Sulfatide-Hsp70 interaction promotes Hsp70 clustering and stabilizes binding to unfolded protein. Biomolecules 2015, 5, 958–973. [Google Scholar] [CrossRef]
- Borges, J.C.; Ramos, C.H. Spectroscopic and thermodynamic measurements of nucleotide-induced changes in the human 70-kDa heat shock cognate protein. Arch. Biochem. Biophys. 2006, 452, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, D.; Li, P.; Sun, C.; Xu, R.; Geng, Z.; Xu, W.; Dai, Z. Interaction of Hsp90 with phospholipid model membranes. Biochim. Biophys. Acta Biomembr. 2018, 1860, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.R.; Chaturvedi, D.; Mahalakshmi, R. Modulating lipid dynamics and membrane fluidity to drive rapid folding of a transmembrane barrel. Sci. Rep. 2013, 3, 1989. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, M.; Mileykovskaya, E.; Dowhan, W. Lipids in the assembly of membrane proteins and organization of protein supercomplexes: Implications for lipid-linked disorders. Subcell. Biochem. 2008, 49, 197–239. [Google Scholar] [CrossRef]
- Jensen, M.O.; Mouritsen, O.G. Lipids do influence protein function-the hydrophobic matching hypothesis revisited. Biochim. Biophys. Acta 2004, 1666, 205–226. [Google Scholar] [CrossRef]
- Lauwers, E.; Wang, Y.C.; Gallardo, R.; Van der Kant, R.; Michiels, E.; Swerts, J.; Baatsen, P.; Zaiter, S.S.; McAlpine, S.R.; Gounko, N.V.; et al. Hsp90 Mediates Membrane Deformation and Exosome Release. Mol. Cell 2018, 71, 689–702. [Google Scholar] [CrossRef]
- Del Vecchio, K.; Stahelin, R.V. Using Surface Plasmon Resonance to Quantitatively Assess Lipid-Protein Interactions. Methods Mol. Biol. (Clifton N.J.) 2016, 1376, 141–153. [Google Scholar] [CrossRef]
- Wiedemann, C.; Bellstedt, P.; Gorlach, M. CAPITO—A web server-based analysis and plotting tool for circular dichroism data. Bioinformatics 2013, 29, 1750–1757. [Google Scholar] [CrossRef]
- Freeman, B.C.; Michels, A.; Song, J.; Kampinga, H.H.; Morimoto, R.I. Analysis of molecular chaperone activities using in vitro and in vivo approaches. Methods Mol. Biol. (Clifton N.J.) 2000, 99, 393–419. [Google Scholar] [CrossRef]
- Spitzer, M.; Wildenhain, J.; Rappsilber, J.; Tyers, M. BoxPlotR: A web tool for generation of box plots. Nat. Methods 2014, 11, 121–122. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Rmax (RU) | ka1 (1/Ms) | kd1 (1/s) | ka2 (1/s) | kd2 (1/s) | KD (M) |
| WT1 | 186.73 | 3.31 × 103 | 1.61 × 10−4 | 3.48 × 10−3 | 2.81 × 10−3 | 1.22 × 10−6 |
| WT2 | 176.98 | 2.84 × 103 | 3.03 × 10−4 | 2.53 × 10−3 | 4.59 × 10−3 | 2.41 × 10−6 |
| WT3 | 166.47 | 2.80 × 103 | 1.26 × 10−4 | 2.13 × 10−3 | 5.47 × 10−3 | 1.60 × 10−6 |
| K71A1 | 150.69 | 1.06 × 104 | 2.67 × 10−6 | 2.23 × 10−2 | 2.14 × 10−2 | 2.94 × 10−7 |
| K71A2 | 138.94 | 1.10 × 104 | 8.81 × 10−6 | 1.48 × 10−2 | 1.57 × 10−2 | 2.47 × 10−7 |
| K71A3 | 156.86 | 1.07 × 104 | 6.32 × 10−6 | 1.69 × 10−2 | 1.73 × 10−2 | 2.86 × 10−7 |
| Mean | Rmax (RU) | ka1 (1/Ms) | kd1 (1/s) | ka2 (1/s) | kd2 (1/s) | KD (M) |
| WT | 176.73 | 2.99 × 103 | 1.97 × 10−4 | 2.71 × 10−3 | 4.29 × 10−3 | 1.74 × 10−6 |
| K71A | 148.83 | 1.08 × 104 | 5.94 × 10−6 | 1.80 × 10−2 | 1.81 × 10−2 | 2.75 × 10−7 |
| SD | Rmax (RU) | ka1 (1/Ms) | kd1 (1/s) | ka2 (1/s) | kd2 (1/s) | KD (M) |
| WT | 10.13 | 2.84 × 102 | 9.38 × 10−5 | 6.88 × 10−4 | 1.35 × 10−3 | 6.04 × 10−7 |
| K71A | 9.1 | 1.97 × 102 | 3.08 × 10−6 | 3.84 × 10−4 | 2.95 × 10−3 | 2.51 × 10−8 |
| t-test (WT vs. K71A) | ||||||
| p value | 0.0239 | <0.0001 | 0.0222 | 0.0024 | 0.0018 | 0.0135 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smulders, L.; Daniels, A.J.; Plescia, C.B.; Berger, D.; Stahelin, R.V.; Nikolaidis, N. Characterization of the Relationship between the Chaperone and Lipid-Binding Functions of the 70-kDa Heat-Shock Protein, HspA1A. Int. J. Mol. Sci. 2020, 21, 5995. https://doi.org/10.3390/ijms21175995
Smulders L, Daniels AJ, Plescia CB, Berger D, Stahelin RV, Nikolaidis N. Characterization of the Relationship between the Chaperone and Lipid-Binding Functions of the 70-kDa Heat-Shock Protein, HspA1A. International Journal of Molecular Sciences. 2020; 21(17):5995. https://doi.org/10.3390/ijms21175995
Chicago/Turabian StyleSmulders, Larissa, Amanda J. Daniels, Caroline B. Plescia, Devon Berger, Robert V. Stahelin, and Nikolas Nikolaidis. 2020. "Characterization of the Relationship between the Chaperone and Lipid-Binding Functions of the 70-kDa Heat-Shock Protein, HspA1A" International Journal of Molecular Sciences 21, no. 17: 5995. https://doi.org/10.3390/ijms21175995
APA StyleSmulders, L., Daniels, A. J., Plescia, C. B., Berger, D., Stahelin, R. V., & Nikolaidis, N. (2020). Characterization of the Relationship between the Chaperone and Lipid-Binding Functions of the 70-kDa Heat-Shock Protein, HspA1A. International Journal of Molecular Sciences, 21(17), 5995. https://doi.org/10.3390/ijms21175995