The Molecular Basis and Biologic Significance of the β-Dystroglycan-Emerin Interaction

 , , , , ,
, , , , ,
Abstract

1. Introduction
2. Results
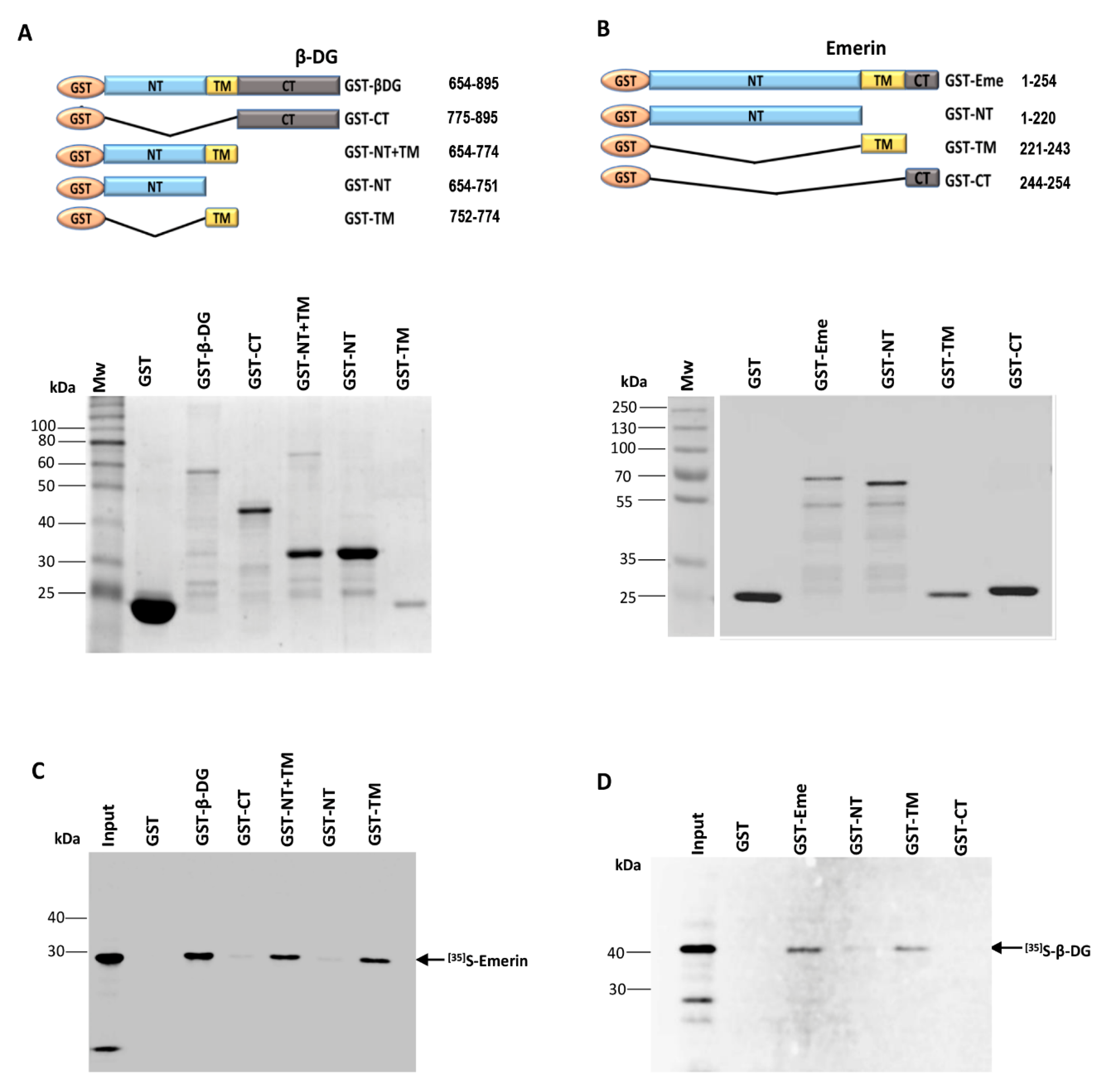
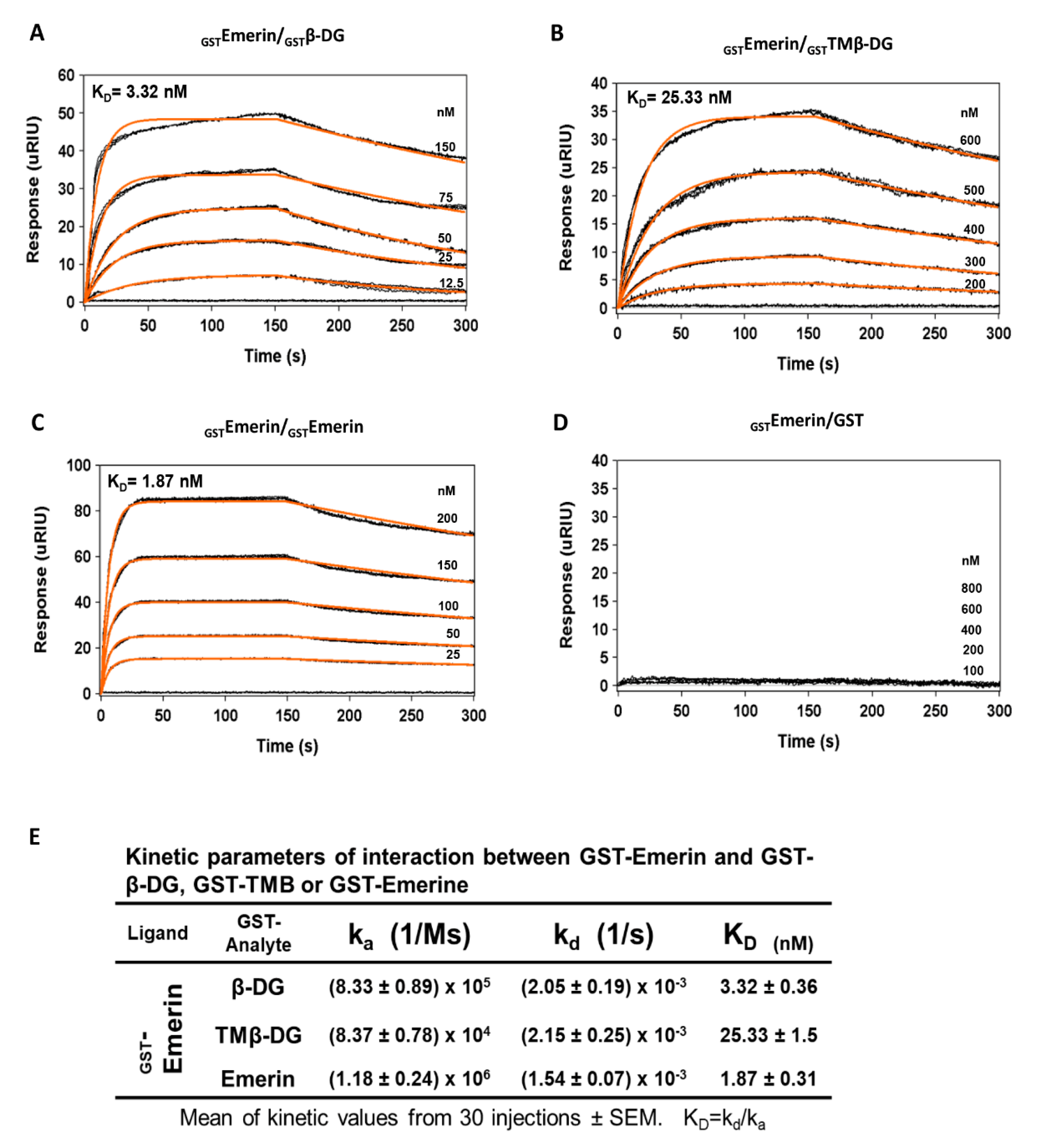
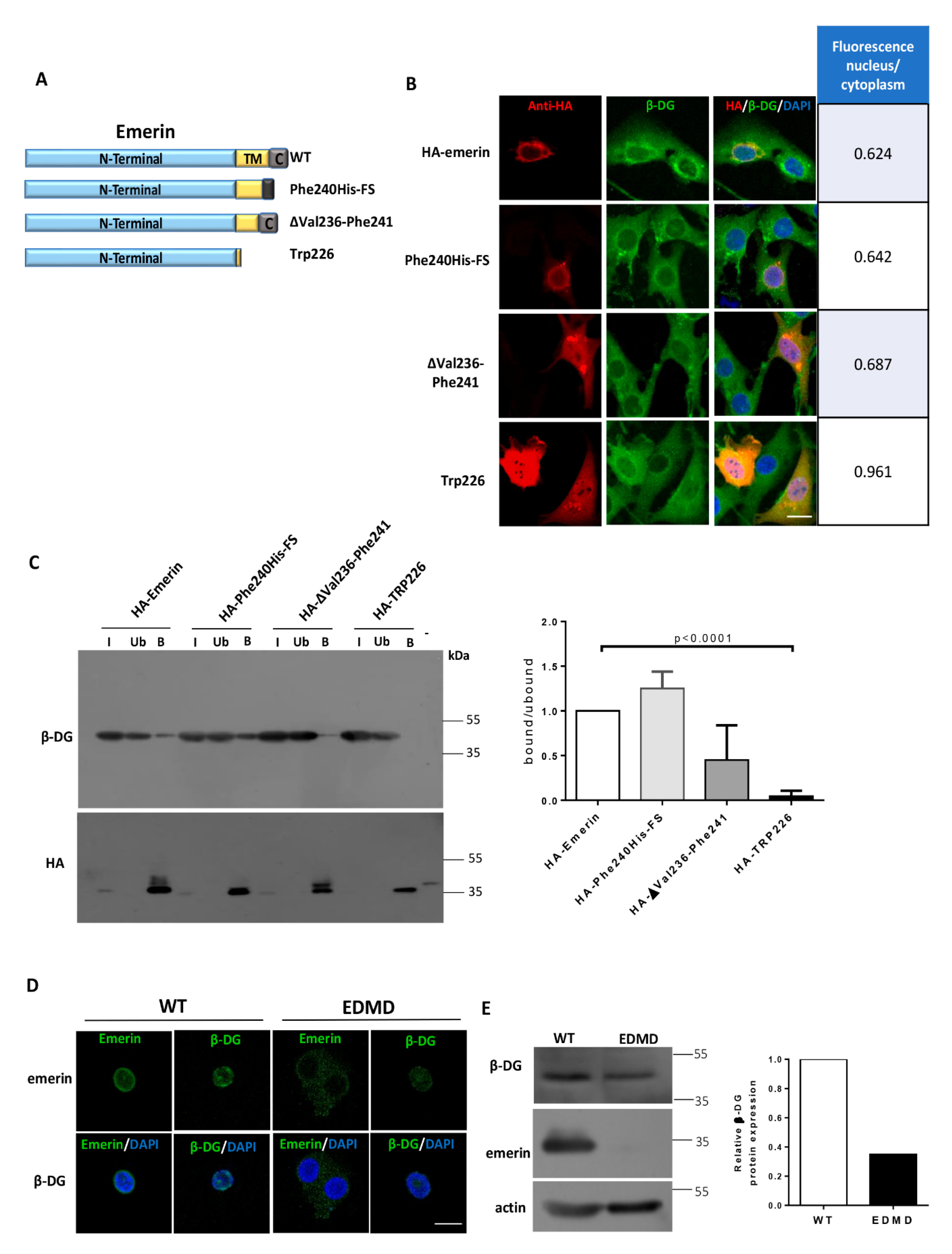
2.1. The Interaction between β-DG and Emerin Requires Their Respective Transmembrane Domains
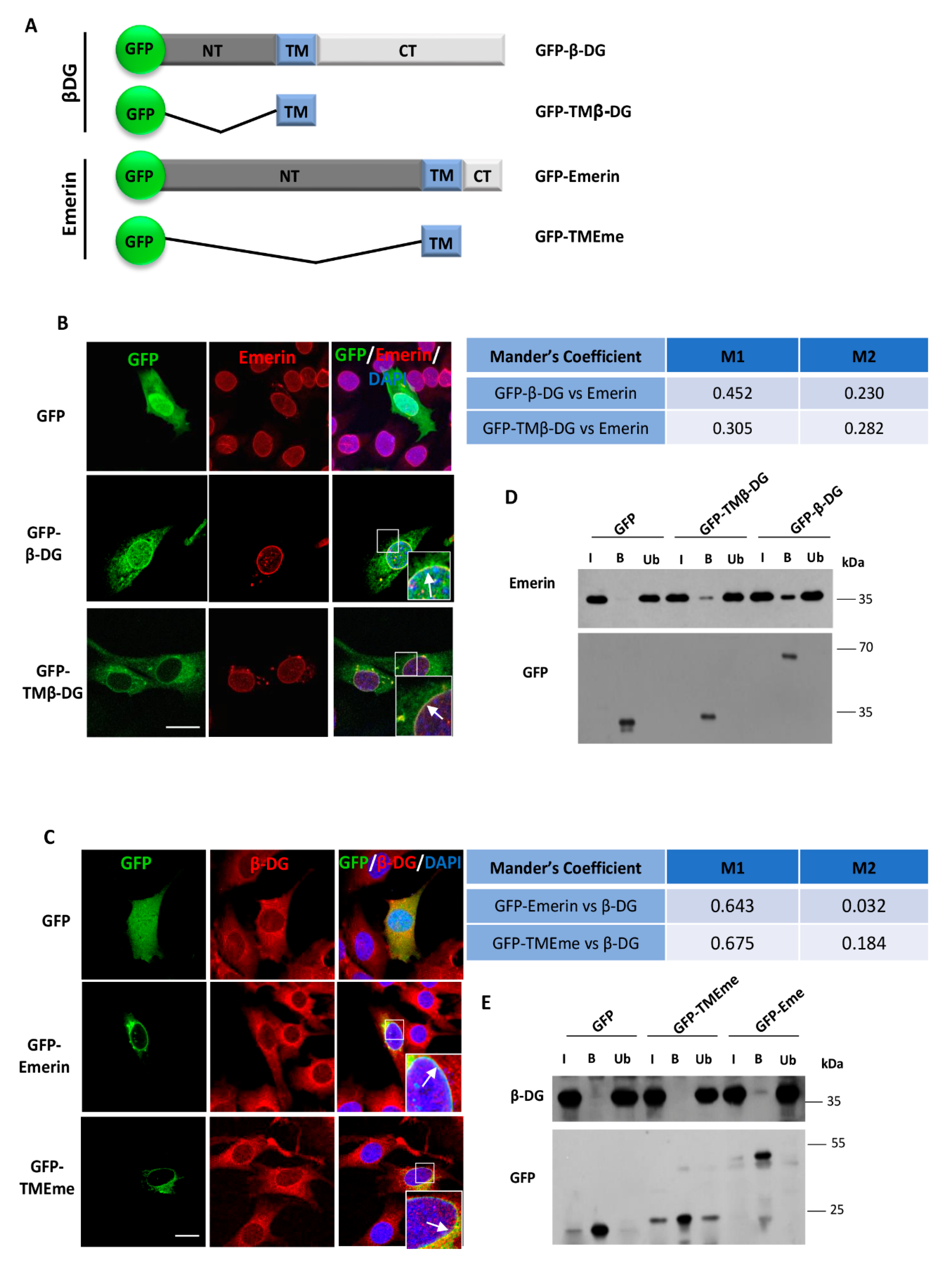
2.2. Localization of β-DG at the NE Depends on the Emerin TM Domain
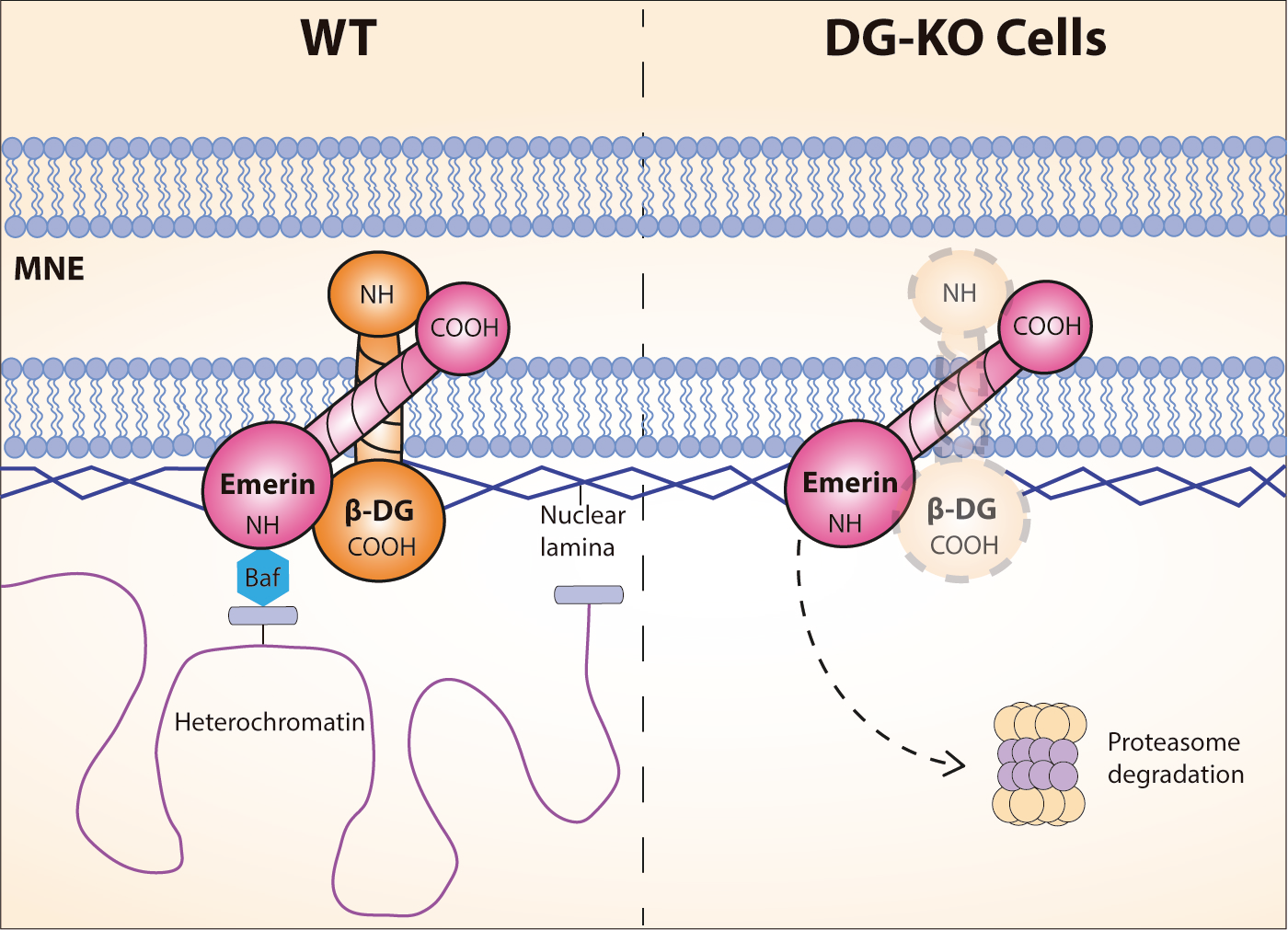
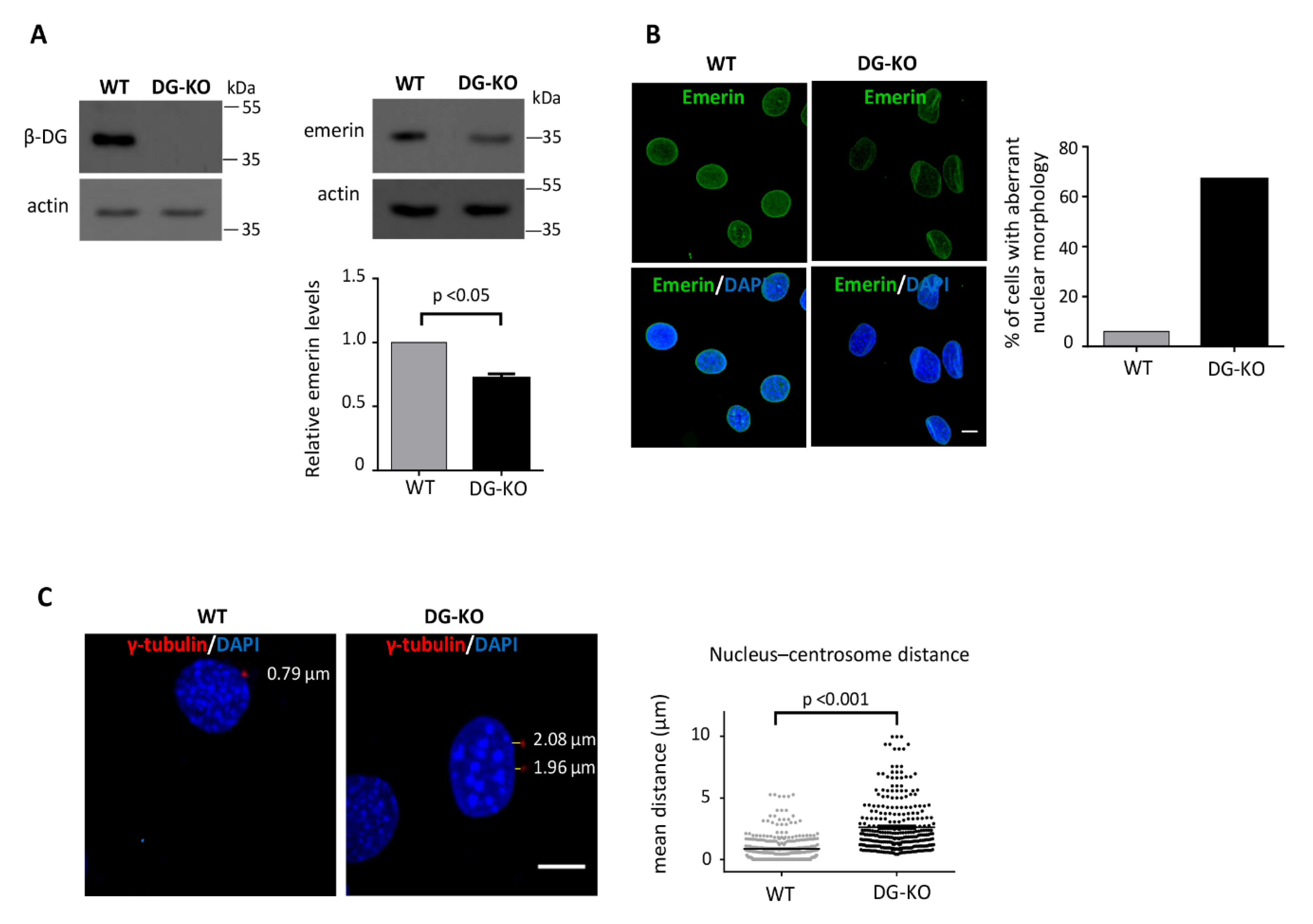
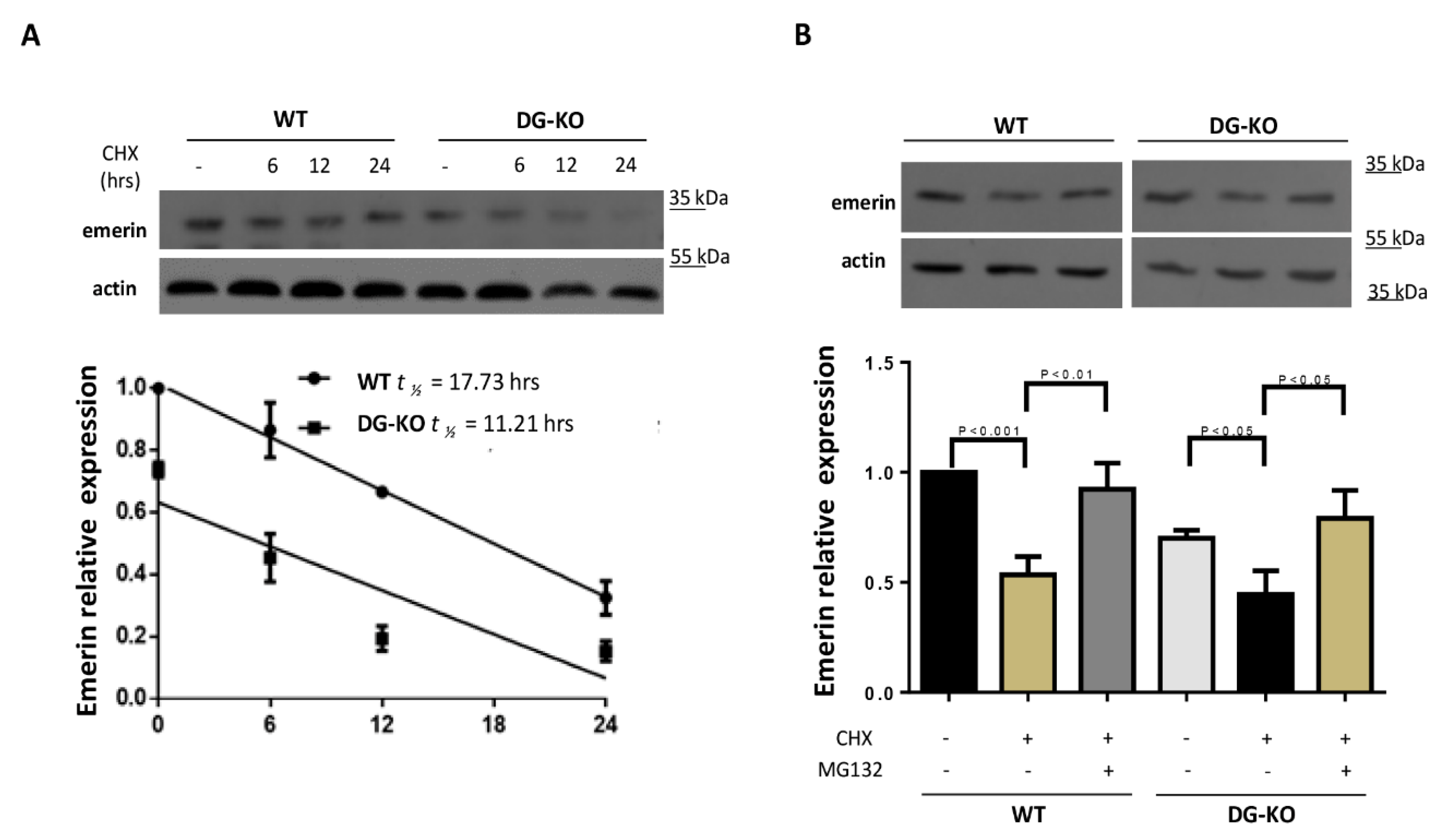
2.3. The Loss of Dystroglycan Impairs Emerin Stability and Function
3. Discussion
4. Materials and Methods
4.1. Cell Culturing and Transfection
4.2. Plasmid Constructs and Antibodies
4.3. In Vitro Transcription/Translation and GST Pull-Down Assay
4.4. Indirect Immunofluorescence and Confocal Microscopy Analysis
4.5. Western Blotting
4.6. Immunoprecipitation
4.7. Surface Plasmon Resonance
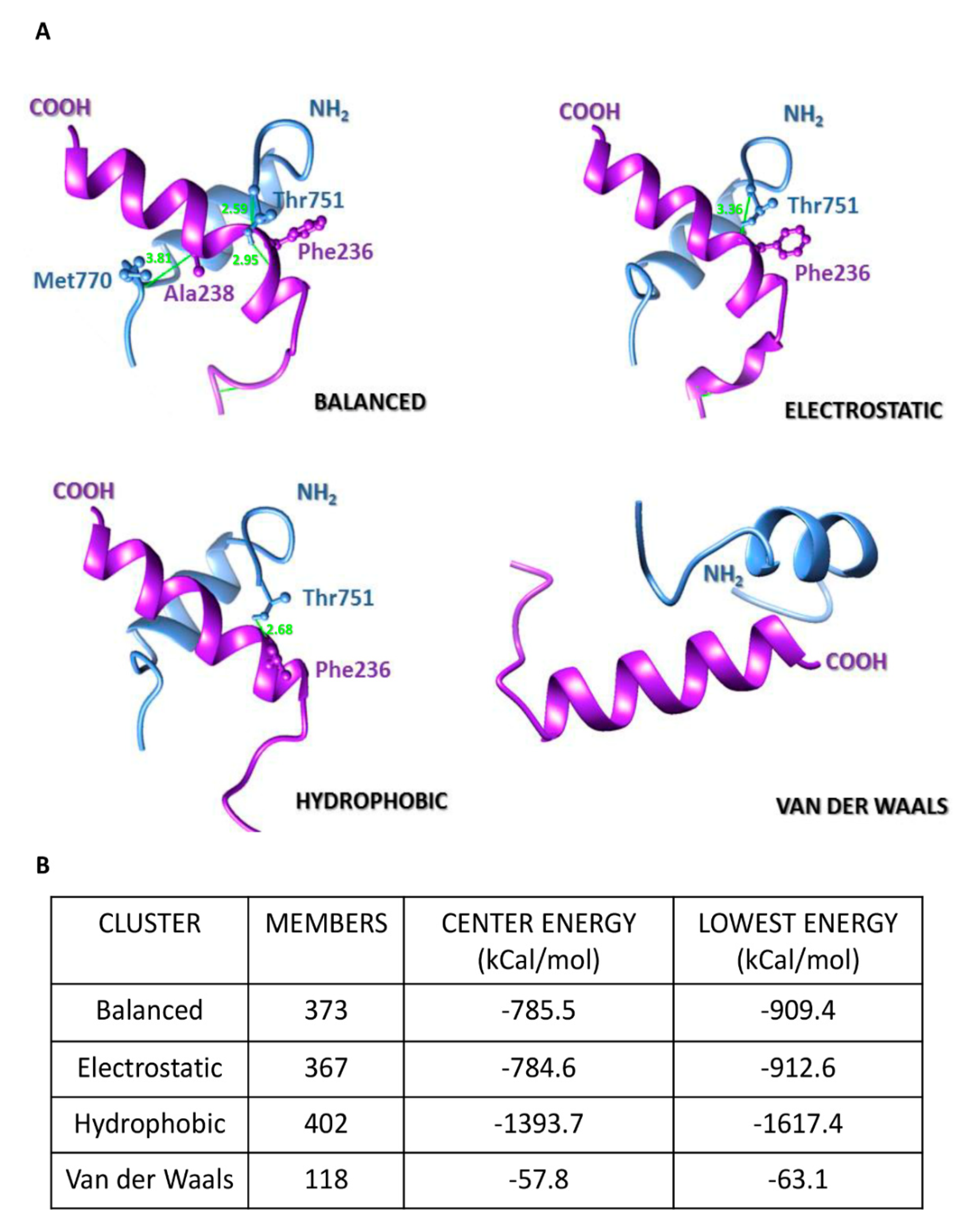
4.8. In Silico Analyses of Emerin-β-DG Interaction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tatli, M.; Medalia, O. Insight into the functional organization of nuclear lamins in health and disease. Curr. Opin. Cell Biol. 2018, 54, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Gesson, K.; Foisner, R. Lamina-independent lamins in the nuclear interior serve important functions. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Ungricht, R.; Kutay, U. Mechanisms and functions of nuclear envelope remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Vieyra, I.A.; Vásquez-Limeta, A.; González-Ramírez, R.; Morales-Lázaro, S.L.; Mondragón, M.; Mondragón, R.; Ortega, A.; Winder, S.J.; Cisneros, B. A role for β-dystroglycan in the organization and structure of the nucleus in myoblasts. Biochim. Et Biophys. Acta 2013, 1833, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.D.; Campbell, K.P. Dystroglycan inside and out. Curr. Opin. Cell Biol. 1999, 11, 602–607. [Google Scholar] [CrossRef]
- Chen, Y.J.; Spence, H.J.; Cameron, J.M.; Jess, T.; Ilsley, J.L.; Winder, S.J. Direct interaction of beta-dystroglycan with F-actin. Biochem. J. 2003, 375, 329–337. [Google Scholar] [CrossRef]
- Gracida-Jiménez, V.; Mondragón-González, R.; Vélez-Aguilera, G.; Vásquez-Limeta, A.; Laredo-Cisneros, M.S.; Gómez-López, J.D.; Vaca, L. Retrograde trafficking of β-dystroglycan from the plasma membrane to the nucleus. Sci. Rep. 2017, 7, 9906. [Google Scholar] [CrossRef]
- Lara-Chacón, B.; de León, M.B.; Leocadio, D.; Gómez, P.; Fuentes-Mera, L.; Martínez-Vieyra, I.; Ortega, A.; Jans, D.A.; Cisneros, B. Characterization of an Importin alpha/beta-recognized nuclear localization signal in beta-dystroglycan. J. Cell. Biochem. 2010, 110, 706–717. [Google Scholar] [CrossRef]
- Vélez-Aguilera, G.; de Dios Gómez-López, J.; Jiménez-Gutiérrez, G.E.; Vásquez-Limeta, A.; Laredo-Cisneros, M.S.; Gómez, P.; Winder, S.J.; Cisneros, B. Control of nuclear β-dystroglycan content is crucial for the maintenance of nuclear envelope integrity and function. Biochim. Et Biophys. Acta. Mol. Cell Res. 2018, 1865, 406–420. [Google Scholar] [CrossRef]
- Mathew, G.; Mitchell, A.; Down, J.M.; Jacobs, L.A.; Hamdy, F.C.; Eaton, C.; Rosario, D.J.; Cross, S.S.; Winder, S.J. Nuclear targeting of dystroglycan promotes the expression of androgen regulated transcription factors in prostate cancer. Sci. Rep. 2013, 3, 2792. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.J.; Holaska, J.M. Emerin in health and disease. Semin. Cell Dev. Biol. 2014, 29, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.A.; Shih, R.; Kalra, R.; Kang, P.B. Emery-Dreifuss muscular dystrophy. Muscle Nerve 2019. [Google Scholar] [CrossRef] [PubMed]
- Cerecedo, D.; Cisneros, B.; Suarez-Sanchez, R.; Hernandez-Gonzalez, E.; Galvan, I. beta-Dystroglycan modulates the interplay between actin and microtubules in human-adhered platelets. Br. J. Haematol. 2008, 141, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.A.; Davies, J.D.; Zhang, Q.; Emerson, L.J.; Hunt, J.; Shanahan, C.M.; Ellis, J.A. Distinct functional domains in nesprin-1alpha and nesprin-2beta bind directly to emerin and both interactions are disrupted in X-linked Emery-Dreifuss muscular dystrophy. Exp Cell Res 2007, 313, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Demmerle, J.; Koch, A.J.; Holaska, J.M. The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J. Biol. Chem. 2012, 287, 22080–22088. [Google Scholar] [CrossRef]
- Pfaff, J.; Rivera Monroy, J.; Jamieson, C.; Rajanala, K.; Vilardi, F.; Schwappach, B.; Kehlenbach, R.H. Emery-Dreifuss muscular dystrophy mutations impair TRC40-mediated targeting of emerin to the inner nuclear membrane. J. Cell Sci. 2016, 129, 502–516. [Google Scholar] [CrossRef]
- Berk, J.M.; Simon, D.N.; Jenkins-Houk, C.R.; Westerbeck, J.W.; Gronning-Wang, L.M.; Carlson, C.R.; Wilson, K.L. The molecular basis of emerin-emerin and emerin-BAF interactions. J. Cell Sci. 2014, 127, 3956–3969. [Google Scholar] [CrossRef]
- Bar, D.Z.; Davidovich, M.; Lamm, A.T.; Zer, H.; Wilson, K.L.; Gruenbaum, Y. BAF-1 mobility is regulated by environmental stresses. Mol. Biol. Cell 2014, 25, 1127–1136. [Google Scholar] [CrossRef]
- Lammerding, J.; Hsiao, J.; Schulze, P.C.; Kozlov, S.; Stewart, C.L.; Lee, R.T. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J. Cell Biol. 2005, 170, 781–791. [Google Scholar] [CrossRef]
- Salpingidou, G.; Smertenko, A.; Hausmanowa-Petrucewicz, I.; Hussey, P.J.; Hutchison, C.J. A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J. Cell Biol. 2007, 178, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994, 8, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.; Sal-Man, N.; Gerber, D.; Shai, Y. Transmembrane domains interactions within the membrane milieu: Principles, advances and challenges. Biochim. Et Biophys. Acta 2012, 1818, 974–983. [Google Scholar] [CrossRef]
- Holaska, J.M.; Lee, K.K.; Kowalski, A.K.; Wilson, K.L. Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. J. Biol. Chem. 2003, 278, 6969–6975. [Google Scholar] [CrossRef]
- Russ, W.P.; Engelman, D.M. TOXCAT: A measure of transmembrane helix association in a biological membrane. Proc. Natl. Acad. Sci. USA 1999, 96, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Holaska, J.M.; Rais-Bahrami, S.; Wilson, K.L. Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum. Mol. Genet. 2006, 15, 3459–3472. [Google Scholar] [CrossRef]
- Dedeic, Z.; Cetera, M.; Cohen, T.V.; Holaska, J.M. Emerin inhibits Lmo7 binding to the Pax3 and MyoD promoters and expression of myoblast proliferation genes. J. Cell Sci. 2011, 124, 1691–1702. [Google Scholar] [CrossRef]
- Markiewicz, E.; Tilgner, K.; Barker, N.; van de Wetering, M.; Clevers, H.; Dorobek, M.; Hausmanowa-Petrusewicz, I.; Ramaekers, F.C.; Broers, J.L.; Blankesteijn, W.M.; et al. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. Embo J. 2006, 25, 3275–3285. [Google Scholar] [CrossRef]
- Jamieson, C.; Lui, C.; Brocardo, M.G.; Martino-Echarri, E.; Henderson, B.R. Rac1 augments Wnt signaling by stimulating beta-catenin-lymphoid enhancer factor-1 complex assembly independent of beta-catenin nuclear import. J. Cell Sci. 2015, 128, 3933–3946. [Google Scholar] [CrossRef]
- Stubenvoll, A.; Rice, M.; Wietelmann, A.; Wheeler, M.; Braun, T. Attenuation of Wnt/beta-catenin activity reverses enhanced generation of cardiomyocytes and cardiac defects caused by the loss of emerin. Hum. Mol. Genet. 2015, 24, 802–813. [Google Scholar] [CrossRef]
- Sakaki, M.; Koike, H.; Takahashi, N.; Sasagawa, N.; Tomioka, S.; Arahata, K.; Ishiura, S. Interaction between emerin and nuclear lamins. J. Biochem. 2001, 129, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Margalit, A.; Brachner, A.; Gotzmann, J.; Foisner, R.; Gruenbaum, Y. Barrier-to-autointegration factor--a BAFfling little protein. Trends Cell Biol. 2007, 17, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Gutierrez, G.E.; Mondragon-Gonzalez, R.; Soto-Ponce, L.A.; Gómez-Monsiváis, W.L.; García-Aguirre, I.; Pacheco-Rivera, R.A.; Suárez-Sánchez, R.; Brancaccio, A. Loss of Dystroglycan Drives Cellular Senescence via Defective Mitosis-Mediated Genomic Instability. Int. J. Mol. Sci. 2020, 21, 4961. [Google Scholar] [CrossRef] [PubMed]
- Kituyi, S.N.; Edkins, A.L. Hop/STIP1 depletion alters nuclear structure via depletion of nuclear structural protein emerin. Biochem. Biophys. Res. Commun. 2018, 507, 503–509. [Google Scholar] [CrossRef]
- Meinke, P.; Kerr, A.R.W.; Czapiewski, R.; de Las Heras, J.I.; Dixon, C.R.; Harris, E.; Kolbel, H.; Muntoni, F.; Schara, U.; Straub, V.; et al. A multistage sequencing strategy pinpoints novel candidate alleles for Emery-Dreifuss muscular dystrophy and supports gene misregulation as its pathomechanism. EBioMedicine 2020, 51, 102587. [Google Scholar] [CrossRef]
- Vasquez-Limeta, A.; Wagstaff, K.M.; Ortega, A.; Crouch, D.H.; Jans, D.A.; Cisneros, B. Nuclear import of beta-dystroglycan is facilitated by ezrin-mediated cytoskeleton reorganization. PLoS ONE 2014, 9, e90629. [Google Scholar] [CrossRef]
- Aguilar, A.; Wagstaff, K.M.; Suarez-Sanchez, R.; Zinker, S.; Jans, D.A.; Cisneros, B. Nuclear localization of the dystrophin-associated protein alpha-dystrobrevin through importin alpha2/beta1 is critical for interaction with the nuclear lamina/maintenance of nuclear integrity. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 1842–1858. [Google Scholar] [CrossRef]
- Moscetti, I.; Cannistraro, S. Surface Plasmon Resonance Sensing of Biorecognition Interactions within the Tumor Suppressor p53 Network. Sensors 2017, 17, 2680. [Google Scholar] [CrossRef]
- Wang, H.; Karlsson, A.; Sjöström, I.; Wiman, B. The interaction between plasminogen and antiplasmin variants as studied by surface plasmon resonance. Biochim. Et Biophys. Acta 2006, 1764, 1730–1734. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmids | Oligonucleotide Sequences | Reference/Source |
|---|---|---|
| pSG5 plasmids | ||
| pSG5–emerin | Forward 5′-ACTGGATCCATGGACAACTACGCAGATCT-3′ | This study |
| Reverse 5′-ACTGGATCCCTAGAAGGGGTTGCCTT-3′, | ||
| pSG5–β-DG | Forward 5′-CCGAATTCATGTCCATCGTGGT-3′ | This study |
| Reverse 5′-CCGAATTCGATTAAGGTGGGACATA-3′ | ||
| GST-tagged β-DG plasmids | ||
| GST–β-DG | [14] | |
| GST-CT | Forward 5′-GCATGAATTCTACCGCAAGAAGCGGAAGG-3′ | This study |
| Reverse 5′-GCATGAATTCTTAAGGTGGGACATAGGGAG-3′ | ||
| GST–NT+TM | Forward 5′-GCATGAATTCTCCATCGTGGTGGAATGGAC-3′ | This study |
| Reverse 5′-GCATGAATTCTTAGCAGATCATGGCAATGATGC-3′ | ||
| GST–NT | Forward 5′-CTGAATTCTCCATCGTGGTGGAA-3′ | This study |
| Reverse 5′-CTGAATTCTTACAGGTAGACATCAT-3′ | ||
| GST–TM | Forward 5′-CTGAATTCCACACAGTCATTCC-3′ | This study |
| Reverse 5′-CTGAATTCTTAGCAGATCATGGCA-3′ | ||
| GST-tagged emerin plasmids | ||
| pGST–emerin | [15] | |
| pGST–NT | Forward 5′-ACTGGATCCATGGACAACTACGCAGATCT-3′ | This study |
| Reverse 5′-CTGGATCCCTAATCCTGGCCCA-3′ | ||
| pGST–TM | Forward 5′-CTGGATCCCGCCAGGTCCCG-3′ | This study |
| Reverse 5′-CTGGATCCCTAGTGGTAAATGAA-3′ | ||
| pGST–CT | [15] | |
| GFP-tagged plasmids | ||
| GFP–β-DG | [9] | |
| GFP–emerin | [16] | |
| GFP–TMβ-DG | Forward 5′-CTAGGATCCCACACAGTCATTCCG-3′ | This study |
| Reverse 5′-CTCGGATCCTTAAAGGGTAAGCTTGC-3′ | ||
| GFP–TM–Eme | Forward 5′-ATGGATCCCGTGCTCCTGGGGCT-3′ | This study |
| Reverse 5′-ATGGATCCCTAGTGGTAAATGAAGAAG-3′ | ||
| HA-tagged plasmids | ||
| pHA–emerin | [17] | |
| pTrp226 | [17] | |
| pΔVal236–Phe241 | [17] | |
| pPhe240His–FS | [17] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Monsivais, W.L.; Monterrubio-Ledezma, F.; Huerta-Cantillo, J.; Mondragon-Gonzalez, R.; Alamillo-Iniesta, A.; García-Aguirre, I.; Azuara-Medina, P.M.; Arguello-García, R.; Rivera-Monroy, J.E.; Holaska, J.M.; et al. The Molecular Basis and Biologic Significance of the β-Dystroglycan-Emerin Interaction. Int. J. Mol. Sci. 2020, 21, 5944. https://doi.org/10.3390/ijms21175944
Gómez-Monsivais WL, Monterrubio-Ledezma F, Huerta-Cantillo J, Mondragon-Gonzalez R, Alamillo-Iniesta A, García-Aguirre I, Azuara-Medina PM, Arguello-García R, Rivera-Monroy JE, Holaska JM, et al. The Molecular Basis and Biologic Significance of the β-Dystroglycan-Emerin Interaction. International Journal of Molecular Sciences. 2020; 21(17):5944. https://doi.org/10.3390/ijms21175944
Chicago/Turabian StyleGómez-Monsivais, Wendy Lilián, Feliciano Monterrubio-Ledezma, Jazmin Huerta-Cantillo, Ricardo Mondragon-Gonzalez, Alma Alamillo-Iniesta, Ian García-Aguirre, Paulina Margarita Azuara-Medina, Raúl Arguello-García, Jhon Erick Rivera-Monroy, James M. Holaska, and et al. 2020. "The Molecular Basis and Biologic Significance of the β-Dystroglycan-Emerin Interaction" International Journal of Molecular Sciences 21, no. 17: 5944. https://doi.org/10.3390/ijms21175944
APA StyleGómez-Monsivais, W. L., Monterrubio-Ledezma, F., Huerta-Cantillo, J., Mondragon-Gonzalez, R., Alamillo-Iniesta, A., García-Aguirre, I., Azuara-Medina, P. M., Arguello-García, R., Rivera-Monroy, J. E., Holaska, J. M., Hernández-Méndez, J. M. E., Garrido, E., Magaña, J. J., Winder, S. J., Brancaccio, A., Martínez-Vieyra, I., Navarro-Garcia, F., & Cisneros, B. (2020). The Molecular Basis and Biologic Significance of the β-Dystroglycan-Emerin Interaction. International Journal of Molecular Sciences, 21(17), 5944. https://doi.org/10.3390/ijms21175944