Histone H1 Post-Translational Modifications: Update and Future Perspectives


Abstract

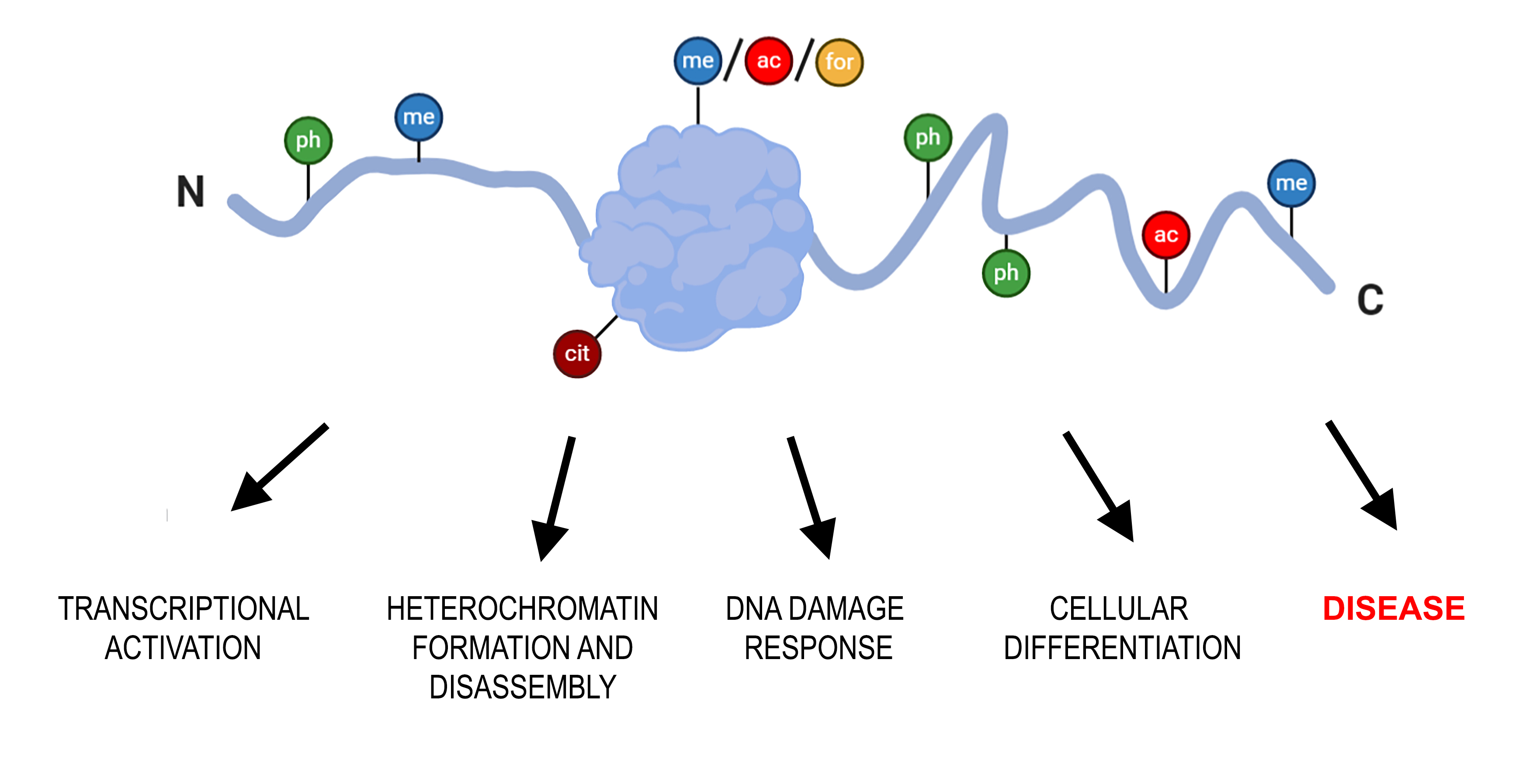{kind=link}
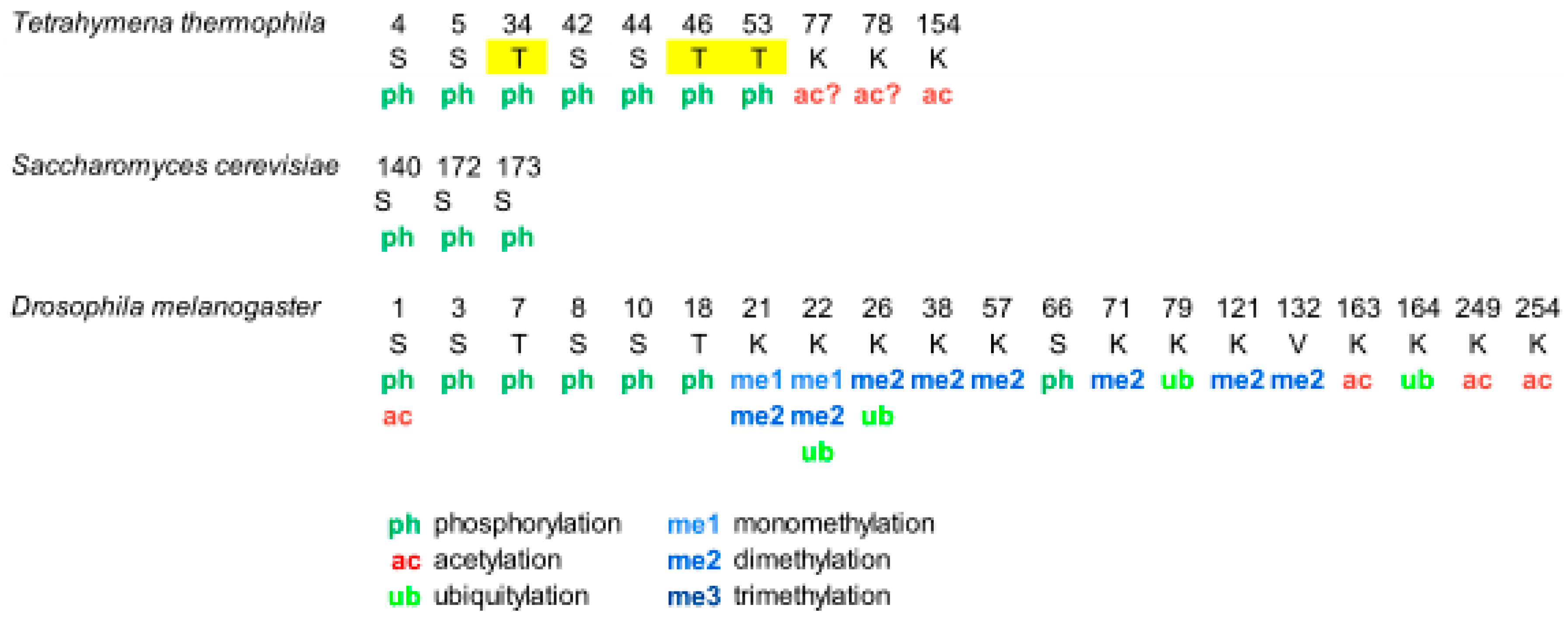{kind=link}
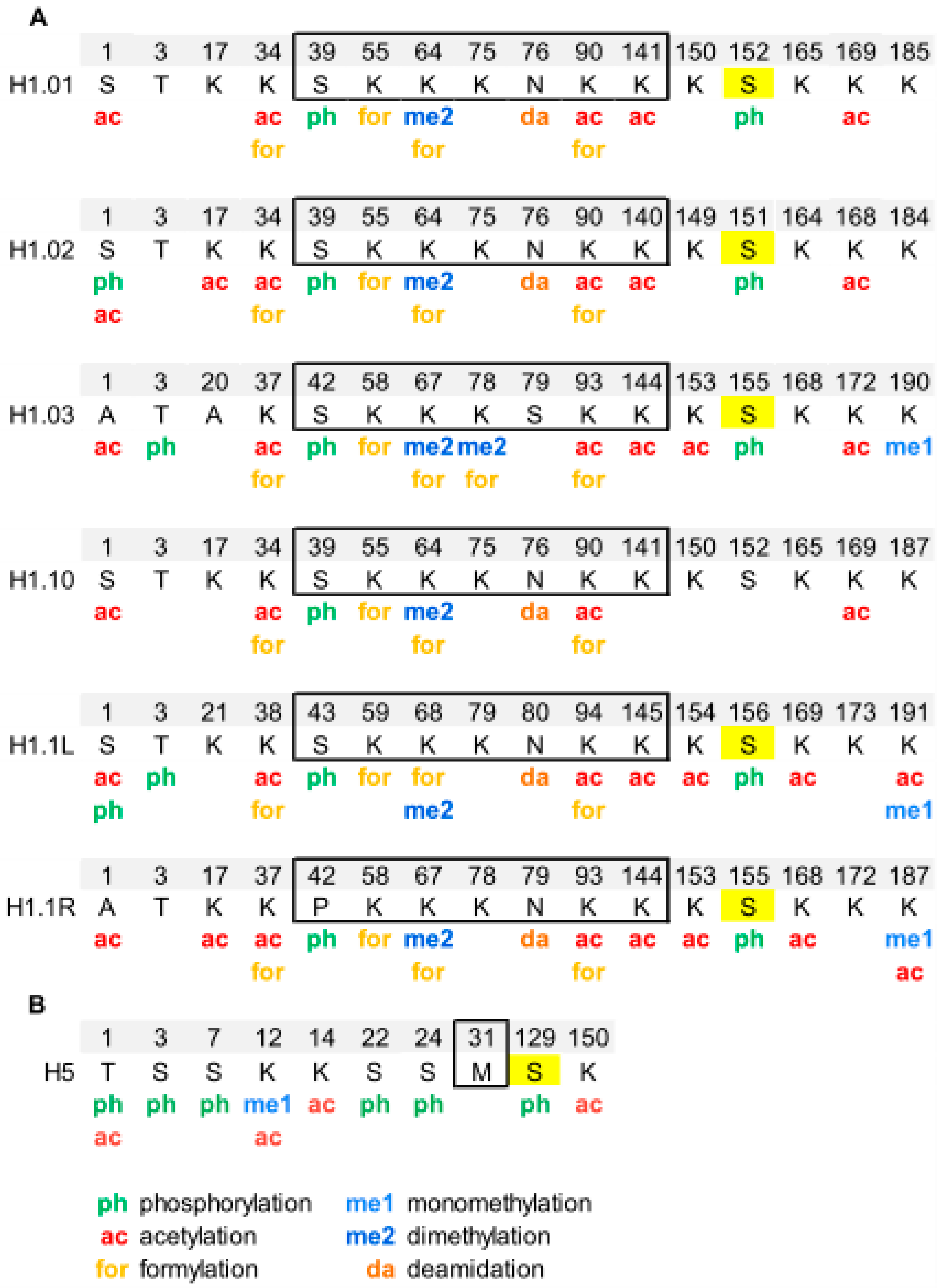{kind=link}
{kind=link}
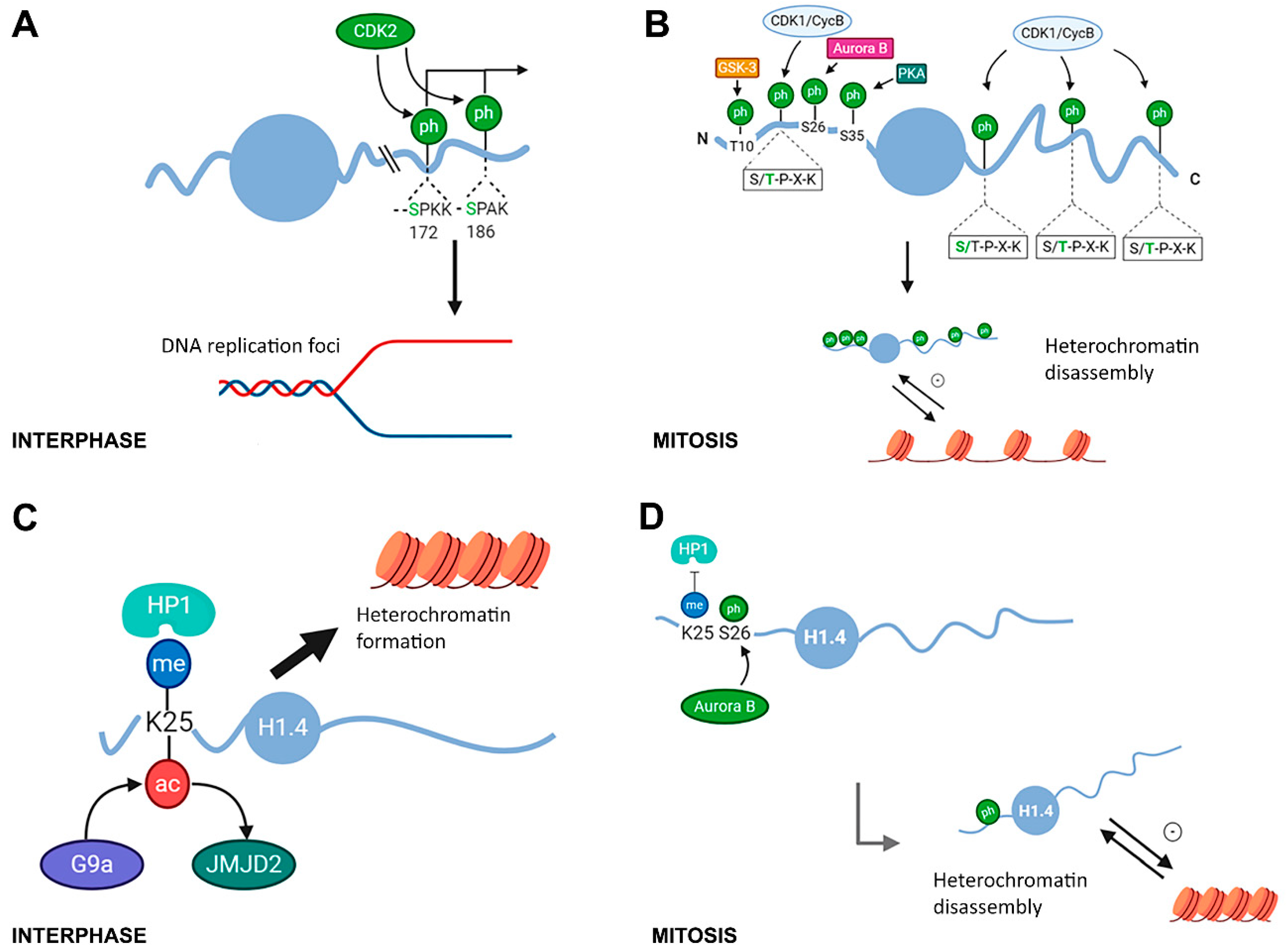{kind=link}
{kind=link}
{kind=link}
1. Introduction
Overview of Histone H1 PTMs from Lower Eukaryotes to Humans
2. Functional Role of H1 PTMs.
2.1. Modifications in Histone H1 Associated with Chromatin Compaction during the Cell Cycle
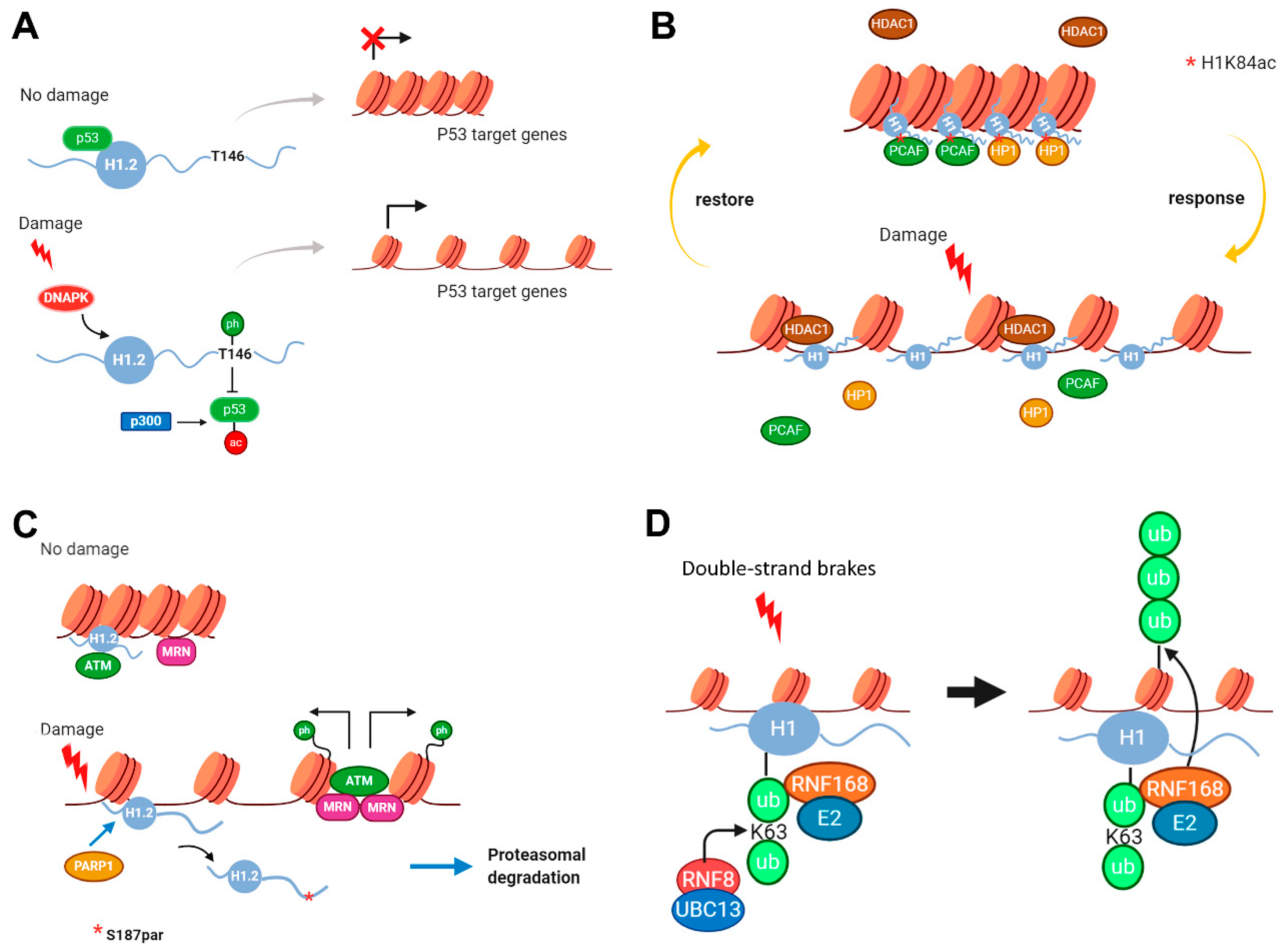
2.2. Modifications in Histone H1 Associated with DNA Damage Response
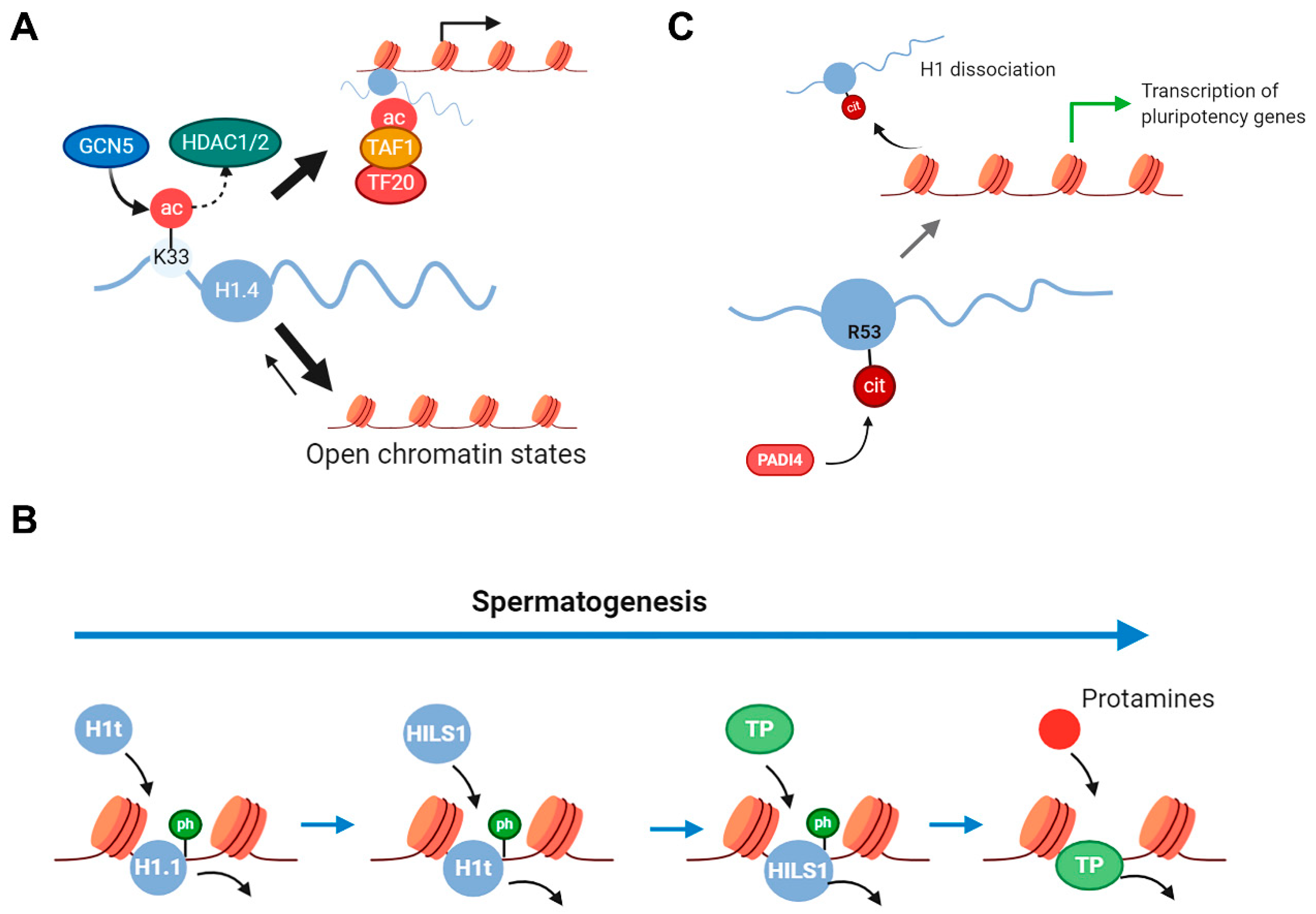
2.3. Histone H1 PTMs in Cell Differentiation and Aging
3. Histone H1 PTMs Associated with Disease
4. Future Perspectives and Challenges
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bradbury, E.M.; Cary, P.D.; Chapman, G.E.; Crane-Robinson, C.; Danby, S.E.; Rattle, H.W.; Boublik, M.; Palau, J.; Aviles, F.J. Studies on the role and mode of operation of the very-lysine-rich histone H1 (F1) in eukaryote chromatin. The conformation of histone H1. Eur. J. Biochem. 1975, 52, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Hansen, J.C. Identification of specific functional subdomains within the linker histone H10 C-terminal domain. J. Biol. Chem. 2004, 279, 8701–8707. [Google Scholar] [CrossRef]
- Th’ng, J.P.; Sung, R.; Ye, M.; Hendzel, M.J. H1 family histones in the nucleus. Control of binding and localization by the C-terminal domain. J. Biol Chem. 2005, 280, 27809–27814. [Google Scholar] [CrossRef] [PubMed]
- Roque, A.; Orrego, M.; Ponte, I.; Suau, P. The preferential binding of histone H1 to DNA scaffold-associated regions is determined by its C-terminal domain. Nucleic Acids Res. 2004, 32, 6111–6119. [Google Scholar] [CrossRef] [PubMed]
- Widlak, P.; Kalinowska, M.; Parseghian, M.H.; Lu, X.; Hansen, J.C.; Garrard, W.T. The histone H1 C-terminal domain binds to the apoptotic nuclease, DNA fragmentation factor (DFF40/CAD) and stimulates DNA cleavage. Biochemistry 2005, 44, 7871–7878. [Google Scholar] [PubMed]
- Roque, A.; Sortino, R.; Ventura, S.; Ponte, I.; Suau, P. Histone H1 Favors Folding and Parallel Fibrillar Aggregation of the 1–42 Amyloid-β Peptide. Langmuir 2015, 31, 6782–6790. [Google Scholar] [CrossRef]
- Kasinsky, H.E.; Lewis, J.D.; Dacks, J.B.; Ausió, J. Origin of H1 linker histones. FASEB J. 2001, 15, 34–42. [Google Scholar] [CrossRef]
- Ponte, I.; Romero, D.; Yero, D.; Suau, P.; Roque, A. Complex evolutionary history of the mammalian histone H1.1-H1.5 gene family. Mol. Biol. Evol. 2017, 34, 545–558. [Google Scholar] [CrossRef]
- Patterton, H.G.; Landel, C.C.; Landsman, D.; Peterson, C.L.; Simpson, R.T. The biochemical and phenotypic characterization of Hho1p, the putative linker histone H1 of Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 7268–7276. [Google Scholar] [CrossRef]
- Panday, A.; Grove, A. The high mobility group protein HMO1 functions as a linker histone in yeast. Epigenetics Chromatin 2016, 9, 13. [Google Scholar] [CrossRef]
- Talbert, P.B.; Ahmad, K.; Almouzni, G.; Ausió, J.; Berger, F.; Bhalla, P.L.; Bonner, W.M.; Cande, W.Z.; Chadwick, B.P.; Chan, S.W.L.; et al. A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin 2012, 5, 7. [Google Scholar] [CrossRef]
- Millán-Ariño, L.; Izquierdo-Bouldstridge, A.; Jordan, A. Specificities and genomic distribution of somatic mammalian histone H1 subtypes. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 510–519. [Google Scholar] [CrossRef]
- Ponte, I.; Vidal-Taboada, J.M.; Suau, P. Evolution of the vertebrate H1 histone class: Evidence for the functional differentiation of the subtypes. Mol. Biol. Evol. 1998, 15, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Orrego, M.; Ponte, I.; Roque, A.; Buschati, N.; Mora, X.; Suau, P. Differential affinity of mammalian histone H1 somatic subtypes for DNA and chromatin. BMC Biol. 2007, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Clausell, J.; Happel, N.; Hale, T.K.; Doenecke, D.; Beato, M. Histone H1 subtypes differentially modulate chromatin condensation without preventing ATP-dependent remodeling by SWI/SNF or NURF. PLoS ONE 2009, 4, e0007243. [Google Scholar] [CrossRef] [PubMed]
- Raghuram, N.; Carrero, G.; Th’ng, J.; Hendzel, M.J. Molecular dynamics of histone H1. Biochem. Cell Biol. 2009, 87, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Patterson, M.; Mikkola, H.K.A.; Lowry, W.E.; Kurdistani, S.K. Dynamic Distribution of Linker Histone H1.5 in Cellular Differentiation. PLoS Genet. 2012, 80, e1002879. [Google Scholar] [CrossRef]
- Izzo, A.; Kamieniarz-Gdula, K.; Ramírez, F.; Noureen, N.; Kind, J.; Manke, T.; van Steensel, B.; Schneider, R. The Genomic Landscape of the Somatic Linker Histone Subtypes H1.1 to H1.5 in Human Cells. Cell Rep. 2013, 3, 2142–2154. [Google Scholar] [CrossRef]
- Millán-Ariño, L.; Islam, A.B.M.M.K.; Izquierdo-Bouldstridge, A.; Mayor, R.; Terme, J.-M.; Luque, N.; Sancho, M.; López-Bigas, N.; Jordan, A. Mapping of six somatic linker histone H1 variants in human breast cancer cells uncovers specific features of H1.2. Nucleic Acids Res. 2014, 42, 4474–4493. [Google Scholar] [CrossRef]
- Mayor, R.; Izquierdo-Bouldstridge, A.; Millán-Ariño, L.; Bustillos, A.; Sampaio, C.; Luque, N.; Jordan, A. Genome distribution of replication-independent histone H1 variants shows H1.0 associated with nucleolar domains and H1X associated with RNA polymerase II-enriched regions. J. Biol. Chem. 2015, 290, 7474–7491. [Google Scholar] [CrossRef]
- Bradbury, E.M.; Inglis, R.J.; Matthews, H.R.; Sarner, N. Phosphorylation of Very-Lysine-Rich Histone in Physarum polycephalum Correlation with Chromosome Condensation. Eur. J. Biochem. 1973, 33, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Gorovsky, M.A.; Keevert, J.B.; Pleger, G.L. Histone F1 of Tetrahymena macronuclei: Unique electrophoretic properties and phosphorylation of F1 in an amitotic nucleus. J. Cell Biol. 1974, 1, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Gorovsky, M.A. Histone Phosphorylation in Macro and Micronuclei of Tetrahymena thermophila. Biochemistry 1981, 20, 3828–3833. [Google Scholar] [CrossRef] [PubMed]
- Blumenfeld, M. Phosphorylated H1 histone in Drosophila melanogaster. Biochem. Genet. 1979, 17, 163–166. [Google Scholar] [CrossRef]
- Sung, M.T.; Wagner, T.E.; Hartford, J.B.; Serra, M.; Vandegrift, V.; Sung, M.T. Phosphorylation and Dephosphorylation of Histone V (H5): Controlled Condensation of Avian Erythrocyte Chromatin. Appendix: Phosphorylation and Dephosphorylation of Histone H5. II. Circular Dichroic Studies. Biochemistry 1977, 16, 286–290. [Google Scholar] [CrossRef]
- Balhorn, R.; Chalkley, R.; Granner, D. Lysine-Rich Histone Phosphorylation. A Positive Correlation with Cell Replication. Biochemistry 1972, 11, 1094–1098. [Google Scholar] [CrossRef]
- Gurley, L.R.; Walters, R.A.; Tobey, R.A. Sequential phosphorylation of histone subfractions in the Chinese hamster cell cycle. J. Biol. Chem. 1975, 250, 3936–3944. [Google Scholar]
- Moradian, A.; Kalli, A.; Sweredoski, M.J.; Hess, S. The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics 2014, 14, 489–497. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Krüger, S.; Mann, M. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol. Cell. Proteom. 2007, 6, 72–87. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Mann, M. Nε-Formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 2008, 36, 570–577. [Google Scholar] [CrossRef]
- Garcia, B.A.; Mollah, S.; Ueberheide, B.M.; Busby, S.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, D.F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007, 2, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef]
- Sarg, B.; Lopez, R.; Lindner, H.; Ponte, I.; Suau, P.; Roque, A. Sequence conservation of linker histones between chicken and mammalian species. Data Brief. 2014, 1, 60–64. [Google Scholar] [CrossRef][Green Version]
- Sarg, B.; Faserl, K.; Kremser, L.; Halfinger, B.; Sebastiano, R.; Lindner, H.H. Comparing and combining capillary electrophoresis electrospray ionization mass spectrometry and nano-liquid chromatography electrospray ionization mass spectrometry for the characterization of post-translationally modified histones. Mol. Cell. Proteom. 2013, 12, 2640–2656. [Google Scholar] [CrossRef] [PubMed]
- Starkova, T.Y.; Artamonova, T.O.; Ermakova, V.V.; Chikhirzhina, E.V.; Khodorkovskii, M.A.; Tomilin, A.N. The Profile of Post-translational Modifications of Histone H1 in Chromatin of Mouse Embryonic Stem Cells. Acta Nat. 2019, 11, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Starkova, T.Y.; Polyanichko, A.M.; Artamonova, T.O.; Khodorkovskii, M.A.; Kostyleva, E.I.; Chikhirzhina, E.V.; Tomilin, A.N. Post-translational modifications of linker histone H1 variants in mammals. Phys. Biol. 2017, 14, 016005. [Google Scholar] [CrossRef]
- Perri, A.M.; Agosti, V.; Olivo, E.; Concolino, A.; De Angelis, M.T.; Tammè, L.; Fiumara, C.V.; Cuda, G.; Scumaci, D. Histone proteomics reveals novel post-translational modifications in breast cancer. Aging 2019, 11, 11722–11755. [Google Scholar] [CrossRef]
- Garcia, B.A.; Joshi, S.; Thomas, C.E.; Chitta, R.K.; Diaz, R.L.; Busby, S.A.; Andrews, P.C.; Ogorzalek Loo, R.R.; Shabanowitz, J.; Kelleher, N.L.; et al. Comprehensive phosphoprotein analysis of linker histone H1 from tetrahymena thermophila. Mol. Cell. Proteom. 2006, 5, 1593–1609. [Google Scholar] [CrossRef]
- Bonet-Costa, C.; Vilaseca, M.; Diema, C.; Vujatovic, O.; Vaquero, A.; Omeñaca, N.; Castejón, L.; Bernués, J.; Giralt, E.; Azorín, F. Combined bottom-up and top-down mass spectrometry analyses of the pattern of post-translational modifications of Drosophila melanogaster linker histone H1. J. Proteom. 2012, 75, 4124–4138. [Google Scholar] [CrossRef]
- Zheng, Y.; John, S.; Pesavento, J.J.; Schultz-Norton, J.R.; Schiltz, R.L.; Baek, S.; Nardulli, A.M.; Hager, G.L.; Kelleher, N.L.; Mizzen, C.A. Histone H1 phosphorylation is associated with transcription by RNA polymerases I and II. J. Cell Biol. 2010, 189, 407–415. [Google Scholar] [CrossRef]
- Chen, Y.; Hoover, M.E.; Dang, X.; Shomo, A.A.; Guan, X.; Marshall, A.G.; Freitas, M.A.; Young, N.L. Quantitative mass spectrometry reveals that intact histone H1 phosphorylations are variant specific and exhibit single molecule hierarchical dependence. Mol. Cell. Proteom. 2016, 15, 818–833. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A.; Busby, S.A.; Barber, C.M.; Shabanowitz, J.; Allis, C.D.; Hunt, D.F. Characterization of phosphorylation sites on histone H1 isoforms by tandem mass spectrometry. J. Proteome Res. 2004, 3, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Zougman, A.; Pudełko, M.; Bȩbenek, M.; Ziółkowski, P.; Mann, M.; Wiśniewski, J.R. Mapping of lysine monomethylation of linker histones in human breast and its cancer. J. Proteome Res. 2009, 8, 4207–4215. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Peng, C.; Montellier, E.; Lu, Z.; Chen, Y.; Ishii, H.; Debernardi, A.; Buchou, T.; Rousseaux, S.; Jin, F.; et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 2014, 10, 365–370. [Google Scholar] [CrossRef]
- Christophorou, M.A.; Castelo-Branco, G.; Halley-Stott, R.P.; Oliveira, C.S.; Loos, R.; Radzisheuskaya, A.; Mowen, K.A.; Bertone, P.; Silva, J.C.R.; Zernicka-Goetz, M.; et al. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 2014, 507, 104–108. [Google Scholar] [CrossRef]
- Roth, S.Y.; Schulman, I.G.; Richman, R.; Cook, R.G.; Allis, C.D. Characterization of phosphorylation sites in histone H1 in the amitotic macronucleus of Tetrahymena during different physiological states. J. Cell Biol. 1988, 107, 2473–2482. [Google Scholar] [CrossRef]
- Mizzen, C.A.; Dou, Y.; Liu, Y.; Cook, R.G.; Gorovsky, M.A.; Allis, C.D. Identification and mutation of phosphorylation sites in a linker histone. Phosphorylation of macronuclear H1 is not essential for viability in Tetrahymena. J. Biol. Chem. 1999, 274, 14533–14536. [Google Scholar] [CrossRef]
- Mukherjee, K.; English, N.; Meers, C.; Kim, H.; Jonke, A.; Storici, F.; Torres, M. Systematic analysis of linker histone PTM hotspots reveals phosphorylation sites that modulate homologous recombination and DSB repair. DNA Repair 2020, 86, 102763. [Google Scholar] [CrossRef]
- Bayona-Feliu, A.; Casas-Lamesa, A.; Carbonell, A.; Climent-Cantó, P.; Tatarski, M.; Pérez-Montero, S.; Azorín, F.; Bernués, J. Histone H1: Lessons from Drosophila. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 526–532. [Google Scholar] [CrossRef]
- Villar-Garea, A.; Imhof, A. Fine mapping of posttranslational modifications of the linker histone H1 from Drosophila melanogaster. PLoS ONE 2008, 3, e1553. [Google Scholar] [CrossRef]
- Thomas, J.O. The higher order structure of chromatin and histone H1. J. Cell Sci. 1984, 1, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Cogburn, L.A.; Porter, T.E.; Duclos, M.J.; Simon, J.; Burgess, S.C.; Zhu, J.J.; Cheng, H.H.; Dodgson, J.B.; Burnside, J. Functional genomics of the chicken-A model organism. Poult. Sci. 2007, 86, 2059–2094. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, A.; Pałyga, J. Chromatin compaction in terminally differentiated avian blood cells: The role of linker histone H5 and non-histone protein MENT. Chromosom. Res. 2011, 19, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Koutzamani, E.; Loborg, H.; Sarg, B.; Lindner, H.H.; Rundquist, I. Linker histone subtype composition and affinity for chromatin in situ in nucleated mature erythrocytes. J. Biol. Chem. 2002, 277, 44688–44694. [Google Scholar] [CrossRef] [PubMed]
- Sarg, B.; Lopez, R.; Lindner, H.; Ponte, I.; Suau, P.; Roque, A. Identification of novel post-translational modifications in linker histones from chicken erythrocytes. J. Proteom. 2015, 113, 162–177. [Google Scholar] [CrossRef]
- Snijders, A.P.L.; Pongdam, S.; Lambert, S.J.; Wood, C.M.; Baldwin, J.P.; Dickman, M.J. Characterization of post-translational modifications of the linker histones HI and H5 from chicken erythrocytes using mass spectrometry. J. Proteome Res. 2008, 7, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Luense, L.J.; Wang, X.; Schon, S.B.; Weller, A.H.; Lin Shiao, E.; Bryant, J.M.; Bartolomei, M.S.; Coutifaris, C.; Garcia, B.A.; Berger, S.L. Comprehensive analysis of histone post-translational modifications in mouse and human male germ cells. Epigenetics Chromatin 2016, 9, 24. [Google Scholar] [CrossRef]
- Mishra, L.N.; Gupta, N.; Rao, S.M.R. Mapping of post-translational modifications of spermatid-specific linker histone H1-like protein, HILS1. J. Proteom. 2015, 128, 218–230. [Google Scholar] [CrossRef]
- Pan, C.; Fan, Y. Role of H1 linker histones in mammalian development and stem cell differentiation. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 496–509. [Google Scholar] [CrossRef]
- Talasz, H.; Helliger, W.; Puschendorf, B.; Lindner, H. In vivo phosphorylation of histone H1 variants during the cell cycle. Biochemistry 1996, 35, 1761–1767. [Google Scholar] [CrossRef]
- Rose, K.L.; Li, A.; Zalenskaya, I.; Zhang, Y.; Unni, E.; Hodgson, K.C.; Yu, Y.; Shabanowitz, J.; Meistrich, M.L.; Hunt, D.F.; et al. C-terminal phosphorylation of murine testis-specific histone H1t in elongating spermatids. J. Proteome Res. 2008, 7, 4070–4078. [Google Scholar] [CrossRef]
- Sarg, B.; Chwatal, S.; Talasz, H.; Lindner, H.H. Testis-specific linker histone H1t is multiply phosphorylated during spermatogenesis. Identification of phosphorylation sites. J. Biol Chem. 2009, 284, 3610–3618. [Google Scholar] [CrossRef] [PubMed]
- Bernier, M.; Luo, Y.; Nwokelo, K.C.; Goodwin, M.; Dreher, S.J.; Zhang, P.; Parthun, M.R.; Fondufe-Mittendorf, Y.; Ottesen, J.J.; Poirier, M.G. Linker histone H1 and H3K56 acetylation are antagonistic regulators of nucleosome dynamics. Nat. Commun. 2015, 6, 10152. [Google Scholar] [CrossRef] [PubMed]
- Simithy, J.; Sidoli, S.; Yuan, Z.F.; Coradin, M.; Bhanu, N.V.; Marchione, D.M.; Klein, B.J.; Bazilevsky, G.A.; McCullough, C.E.; Magin, R.S.; et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 2017, 8, 1141. [Google Scholar] [CrossRef] [PubMed]
- Tweedie-Cullen, R.Y.; Reck, J.M.; Mansuy, I.M. Comprehensive mapping of post-translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. J. Proteome Res. 2009, 8, 4966–4982. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Hergeth, S.P.; Schneider, R. The H1 linker histones: Multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015, 16, 1439–1453. [Google Scholar] [CrossRef]
- Izzo, A.; Schneider, R. The role of linker histone H1 modifications in the regulation of gene expression and chromatin dynamics. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 486–495. [Google Scholar] [CrossRef]
- Daujat, S.; Zeissler, U.; Waldmann, T.; Happel, N.; Schneider, R. HP1 binds specifically to Lys26-methylated histone H1.4, whereas simultaneous Ser27 phosphorylation blocks HP1 binding. J. Biol. Chem. 2005, 280, 38090–380905. [Google Scholar] [CrossRef]
- Talasz, H.; Sarg, B.; Lindner, H.H. Site-specifically phosphorylated forms of H1.5 and H1.2 localized at distinct regions of the nucleus are related to different processes during the cell cycle. Chromosoma 2009, 118, 693–709. [Google Scholar] [CrossRef]
- Chu, C.S.; Hsu, P.H.; Lo, P.W.; Scheer, E.; Tora, L.; Tsai, H.J.; Tsai, M.D.; Juan, L.J. Protein kinase A-mediated serine 35 phosphorylation dissociates histone H1.4 from mitotic chromosome. J. Biol. Chem. 2011, 286, 35843–35851. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Z.; Dong, L.; Tang, M.; Zhang, P.; Zhang, C.; Cao, Z.; Zhu, Q.; Chen, Y.; Wang, H.; et al. Histone H1 acetylation at lysine 85 regulates chromatin condensation and genome stability upon DNA damage. Nucleic Acids Res. 2018, 46, 7716–7730. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Jeong, K.W.; Kim, H.; Choi, J.; Lu, W.; Stallcup, M.R.; An, W. Functional interplay between p53 acetylation and H1.2 phosphorylation in p53-regulated transcription. Oncogene 2012, 31, 4290–4301. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Tang, M.; Peng, B.; Lu, X.; Yang, Q.; Zhu, Q.; Hou, T.; Li, M.; Liu, C.; et al. Destabilization of linker histone H1.2 is essential for ATM activation and DNA damage repair. Cell Res. 2018, 28, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef]
- Kamieniarz, K.; Izzo, A.; Dundr, M.; Tropberger, P.; Ozretić, L.; Kirfel, J.; Scheer, E.; Tropel, P.; Wiśniewski, J.R.; Tora, L.; et al. A dual role of linker histone H1.4 Lys 34 acetylation in transcriptional activation. Genes Dev. 2012, 26, 797–802. [Google Scholar] [CrossRef]
- Happel, N.; Doenecke, D.; Sekeri-Pataryas, K.E.; Sourlingas, T.G. H1 histone subtype constitution and phosphorylation state of the ageing cell system of human peripheral blood lymphocytes. Exp. Gerontol. 2008, 43, 184–199. [Google Scholar] [CrossRef]
- Gréen, A.; Sarg, B.; Gréen, H.; Lönn, A.; Lindner, H.H.; Rundquist, I. Histone H1 interphase phosphorylation becomes largely established in G 1or early S phase and differs in G1between T-lymphoblastoid cells and normal T cells. Epigenetics Chromatin 2011, 4, 15. [Google Scholar] [CrossRef]
- Lopez, R.; Sarg, B.; Lindner, H.; Bartolomé, S.; Ponte, I.; Suau, P.; Roque, A. Linker histone partial phosphorylation: Effects on secondary structure and chromatin condensation. Nucleic Acids Res. 2015, 43, 4463–4476. [Google Scholar] [CrossRef]
- Alexandrow, M.G.; Hamlin, J.L. Chromatin decondensation in S-phase involves recruitment or Cdk2 by Cdc45 and histone H1 phosphorylation. J. Cell Biol. 2005, 168, 875–886. [Google Scholar] [CrossRef]
- Koop, R.; Di Croce, L.; Beato, M. Histone H1 enhances synergistic activation of the MMTV promoter in chromatin. EMBO J. 2003, 22, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Vicent, G.P.; Nacht, A.S.; Font-Mateu, J.; Castellano, G.; Gaveglia, L.; Ballaré, C.; Beato, M. Four enzymes cooperate to displace histone H1 during the first minute of hormonal gene activation. Genes Dev. 2011, 25, 845–862. [Google Scholar] [CrossRef]
- Happel, N.; Stoldt, S.; Schmidt, B.; Doenecke, D. M Phase-Specific Phosphorylation of Histone H1.5 at Threonine 10 by GSK-3. J. Mol. Biol. 2009, 386, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Hergeth, S.P.; Dundr, M.; Tropberger, P.; Zee, B.M.; Garcia, B.A.; Daujat, S.; Schneider, R. Isoform-specific phosphorylation of human linker histone H1.4 in mitosis by the kinase Aurora B. J. Cell Sci. 2011, 124, 1623–1628. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Th’ng, J.P.H.; Guo, X.W.; Swank, R.A.; Crissman, H.A.; Bradbury, E.M. Inhibition of histone phosphorylation by staurosporine leads to chromosome decondensation. J. Biol. Chem. 1994, 269, 9568–9573. [Google Scholar] [PubMed]
- Gibson, B.A.; Doolittle, L.K.; Schneider, M.W.G.; Jensen, L.E.; Gamarra, N.; Henry, L.; Gerlich, D.W.; Redding, S.; Rosen, M.K. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 2019, 179, 470–484. [Google Scholar] [CrossRef]
- Turner, A.L.; Watson, M.; Wilkins, O.G.; Cato, L.; Travers, A.; Thomas, J.O.; Stott, K. Highly disordered histone H1-DNA model complexes and their condensates. Proc. Natl. Acad. Sci. USA 2018, 115, 11964–11969. [Google Scholar] [CrossRef]
- Trojer, P.; Zhang, J.; Yonezawa, M.; Schmidt, A.; Zheng, H.; Jenuwein, T.; Reinberg, D. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the jumonji domain-containing JMJD2/KDM4 proteins. J. Biol. Chem. 2009, 284, 8395–8405. [Google Scholar] [CrossRef]
- Weiss, T.; Hergeth, S.; Zeissler, U.; Izzo, A.; Tropberger, P.; Zee, B.M.; Dundr, M.; Garcia, B.A.; Daujat, S.; Schneider, R. Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenetics Chromatin 2010, 3, 7. [Google Scholar] [CrossRef]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef]
- Terme, J.M.; Millán-Ariño, L.; Mayor, R.; Luque, N.; Izquierdo-Bouldstridge, A.; Bustillos, A.; Sampaio, C.; Canes, J.; Font, I.; Sima, N.; et al. Dynamics and dispensability of variant-specific histone H1 Lys-26/Ser-27 and Thr-165 post-translational modifications. FEBS Lett. 2014, 588, 2353–2362. [Google Scholar] [CrossRef] [PubMed]
- Pradeepa, M.M.; Rao, M.R. Chromatin remodeling during mammalian spermatogenesis: Role of testis specific histone variants and transition proteins. Soc. Reprod. Fertil. Suppl. 2007, 63, 1–10. [Google Scholar] [PubMed]
- Sekeri-Pataryas, K.E.; Sourlingas, T.G. The differentiation-associated linker histone, H1.0, during the in vitro aging and senescence of human diploid fibroblasts. Ann. N. Y. Acad. Sci. 2007, 1100, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Lindner, H.; Sarg, B.; Hoertnagl, B.; Helliger, W. The Microheterogeneity of the Mammalian H1 0 Histone. J. Biol. Chem. 1998, 273, 13324–13330. [Google Scholar] [CrossRef] [PubMed]
- Telu, K.H.; Abbaoui, B.; Thomas-Ahner, J.M.; Zynger, D.L.; Clinton, S.K.; Freitas, M.A.; Mortazavi, A. Alterations of histone H1 phosphorylation during bladder carcinogenesis. J. Proteome Res. 2013, 12, 3317–3326. [Google Scholar] [CrossRef]
- Shi, S.; Zhang, J.; Liu, M.; Dong, H.; Li, N. Ras-ERK signalling represses H1.4 phosphorylation at serine 36 to promote non-small-cell lung carcinoma cells growth and migration. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2343–2351. [Google Scholar] [CrossRef]
- Li, Y.H.; Zhong, M.; Zang, H.L.; Tian, X.F. MTA1 Promotes Hepatocellular Carcinoma Progression by Downregulation of DNA-PK-Mediated H1.2T146 Phosphorylation. Front. Oncol. 2020, 10, 256. [Google Scholar] [CrossRef]
- Saloura, V.; Vougiouklakis, T.; Bao, R.; Kim, S.; Baek, S.; Zewde, M.; Bernard, B.; Burkitt, K.; Nigam, N.; Izumchenko, E.; et al. WHSC1 monomethylates histone H1 and induces stem-cell like features in squamous cell carcinoma of the head and neck. Neoplasia 2020, 22, 283–293. [Google Scholar] [CrossRef]
- Dwivedi, N.; Neeli, I.; Schall, N.; Wan, H.; Desiderio, D.M.; Csernok, E.; Thompson, P.R.; Dali, H.; Briand, J.P.; Muller, S.; et al. Deimination of linker histones links neutrophil extracellular trap release with autoantibodies in systemic autoimmunity. FASEB J. 2014, 28, 2840–2851. [Google Scholar] [CrossRef]
- Lesner, A.; Kartvelishvili, A.; Lesniak, J.; Nikolov, D.; Kartvelishvili, M.; Trillo-Pazos, G.; Zablotna, E.; Simm, M. Monoubiquitinated histone H1B is required for antiviral protection in CD4+T cells resistant to HIV-1. Biochemistry 2004, 43, 16203–16211. [Google Scholar] [CrossRef]
- Bauden, M.; Kristl, T.; Sasor, A.; Andersson, B.; Marko-Varga, G.; Andersson, R.; Ansari, D. Histone profiling reveals the H1.3 histone variant as a prognostic biomarker for pancreatic ductal adenocarcinoma. BMC Cancer 2017, 17, 810. [Google Scholar] [CrossRef]
- Kulej, K.; Avgousti, D.C.; Sidoli, S.; Herrmann, C.; Della Fera, A.N.; Kim, E.T.; Garcia, B.A.; Weitzman, M.D. Time-resolved global and chromatin proteomics during herpes simplex virus type 1 (HSV-1) infection. Mol. Cell. Proteom. 2017, 16, S92–S107. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Tian, F.; Chen, X.; Liu, Z.; Wu, C.; Zhao, Z. Ras-ERK1/2 signaling participates in the progression of gastric cancer through repressing Aurora B-mediated H1.4 phosphorylation at Ser27. J. Cell. Physiol. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef]
- Noberini, R.; Bonaldi, T. A Super-SILAC Strategy for the Accurate and Multiplexed Profiling of Histone Posttranslational Modifications. Methods Enzymol. 2017, 586, 311–333. [Google Scholar]
- Lin, S.; Wein, S.; Gonzales-Cope, M.; Otte, G.L.; Yuan, Z.F.; Afjehi-Sadat, L.; Maile, T.; Berger, S.L.; Rush, J.; Lill, J.R.; et al. Stable-isotope-labeled histone peptide library for histone post-translational modification and variant quantification by mass spectrometry. Mol. Cell. Proteom. 2014, 13, 2450–2466. [Google Scholar] [CrossRef]
- Savaryn, J.P.; Catherman, A.D.; Thomas, P.M.; Abecassis, M.M.; Kelleher, N.L. The emergence of top-down proteomics in clinical research. Genome Med. 2013, 5, 53. [Google Scholar] [CrossRef]
- Karch, K.R.; Sidoli, S.; Garcia, B.A. Identification and Quantification of Histone PTMs Using High-Resolution Mass Spectrometry. Methods Enzymol. 2016, 574, 3–29. [Google Scholar]
- Noberini, R.; Osti, D.; Miccolo, C.; Richichi, C.; Lupia, M.; Corleone, G.; Hong, S.P.; Colombo, P.; Pollo, B.; Fornasari, L.; et al. Extensive and systematic rewiring of histone post-translational modifications in cancer model systems. Nucleic Acids Res. 2018, 46, 3817–3832. [Google Scholar] [CrossRef]
- Sidoli, S.; Kori, Y.; Lopes, M.; Yuan, Z.F.; Kim, H.J.; Kulej, K.; Janssen, K.A.; Agosto, L.M.; Da Cunha, J.P.C.; Andrews, A.J.; et al. One minute analysis of 200 histone posttranslational modifications by direct injection mass spectrometry. Genome Res. 2019, 29, 978–987. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrés, M.; García-Gomis, D.; Ponte, I.; Suau, P.; Roque, A. Histone H1 Post-Translational Modifications: Update and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 5941. https://doi.org/10.3390/ijms21165941
Andrés M, García-Gomis D, Ponte I, Suau P, Roque A. Histone H1 Post-Translational Modifications: Update and Future Perspectives. International Journal of Molecular Sciences. 2020; 21(16):5941. https://doi.org/10.3390/ijms21165941
Chicago/Turabian StyleAndrés, Marta, Daniel García-Gomis, Inma Ponte, Pedro Suau, and Alicia Roque. 2020. "Histone H1 Post-Translational Modifications: Update and Future Perspectives" International Journal of Molecular Sciences 21, no. 16: 5941. https://doi.org/10.3390/ijms21165941
APA StyleAndrés, M., García-Gomis, D., Ponte, I., Suau, P., & Roque, A. (2020). Histone H1 Post-Translational Modifications: Update and Future Perspectives. International Journal of Molecular Sciences, 21(16), 5941. https://doi.org/10.3390/ijms21165941