Linking LOXL2 to Cardiac Interstitial Fibrosis

 , ,
, ,  , ,
, ,  , , and
, , and 
Abstract
1. Introduction
2. The ECM is Compromised during the Development of Fibrosis
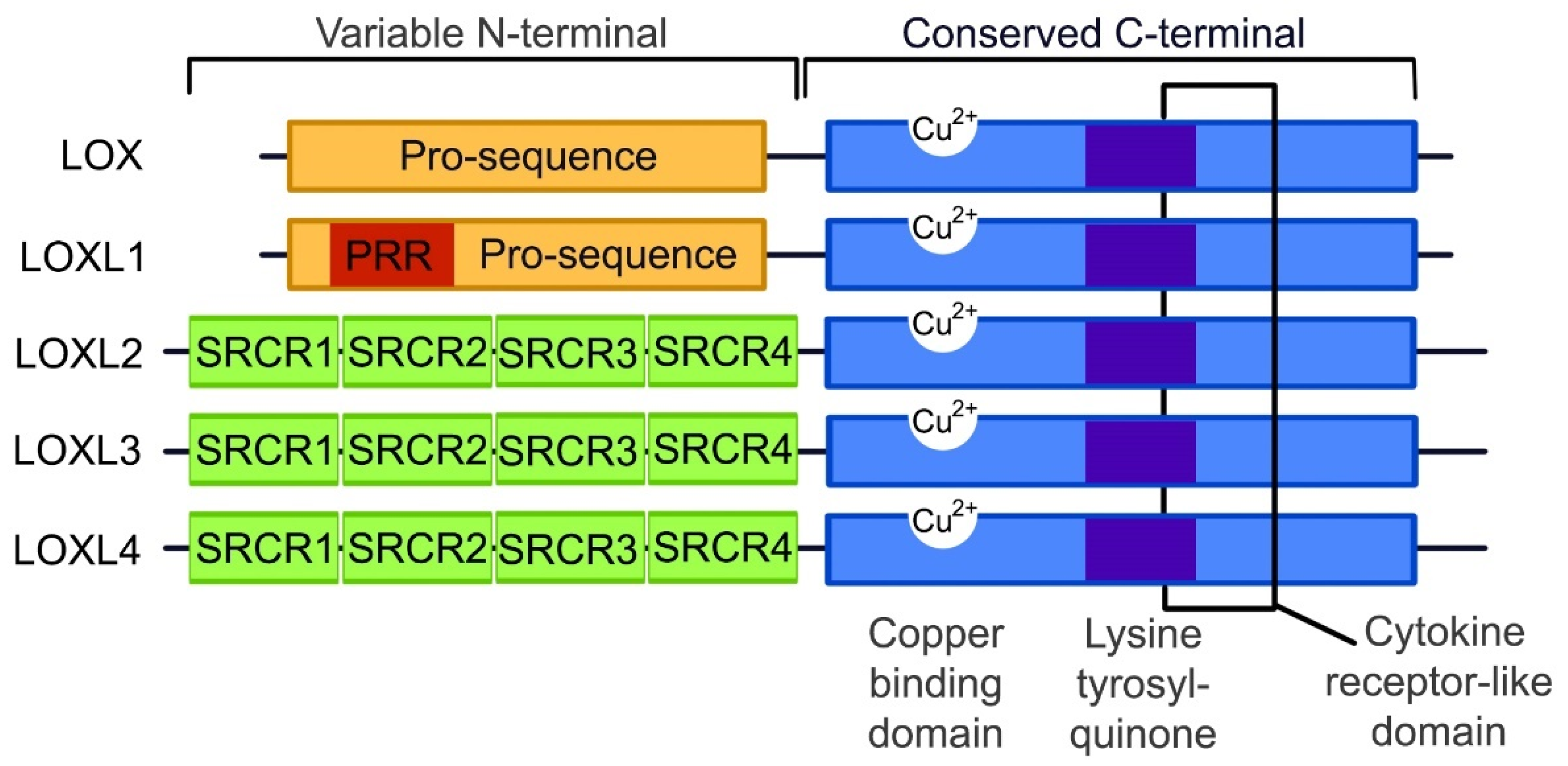
3. The Lysyl Oxidase Gene Family
4. LOXL2 in Disease
The Role of LOXL2 in the Development of Cardiovascular Disease
5. LOXL2 Activity and Its Gene Regulatory Network
6. Epigenetic Control of LOXL2 Expression
6.1. LOXL2 and DNA Methylation
6.2. LOXL2 and Histone Modification
7. Future Considerations
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| α-SMA | Alpha smooth muscle actin |
| AGEs | Advanced glycation end-products |
| CTGF | Connective tissue growth factor |
| CVD | Cardiovascular disease |
| DNMTs | DNA methyltransferases |
| ECM | Extracellular matrix |
| GAL3 | Galectin 3 |
| HF | Heart failure |
| HIF-1 | Hypoxia-inducible factor 1 |
| LOX | Lysyl oxidase |
| LOXL1-4 | Lysyl Oxidase-Like 1-4 |
| mTOR | Mechanistic target of rapamycin |
| NFκB | Nuclear factor kappa beta |
| PI3K | Phosphoinositide 3-kinase |
| Sp-1 | Specificity protein 1 |
| TGF-α | Transforming growth factor alpha |
| TGF-β | Transforming growth factor beta |
| WHO | World Health Organization |
References
- World Health Statistics (WHO). Monitoring Health for the SDGs; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- World Health Statistics (WHO). New Initiative Launched to Tackle Cardiovascular Disease: The World’s Number One Killer; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- World Health Statistics (WHO). Global Hearts Initiative: Working Together to Promote Cardiovascular Health; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- World Health Statistics (WHO). Obesity and Overweight Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 12 November 2018).
- International Diabetes Federation (IDF). IDF Diabetes Atlas, 9th ed.; IDF: Brussels, Belgium, 2019. [Google Scholar]
- World Health Statistics (WHO). Cardiovascular Diseases (CVDs) Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 12 September 2019).
- CDC. Heart Failure Fact Sheet. Available online: https://www.cdc.gov/dhdsp/data_statistics/fact_sheets/fs_heart_failure.htm (accessed on 13 July 2019).
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular inflammation and oxidative stress: Major triggers for cardiovascular disease. Oxid. Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [PubMed]
- López, B.; Querejeta, R.; González, A.; Beaumont, J.; Larman, M.; Díez, J. Impact of treatment on myocardial lysyl oxidase expression and collagen cross-linking in patients with heart failure. Hypertension 2009, 53, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and heart failure. Heart Fail. Rev. 2014, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Miner, E.C.; Miller, W.L. A look between the cardiomyocytes: The extracellular matrix in heart failure. Mayo Clin. Proc. 2006, 81, 71–76. [Google Scholar] [CrossRef]
- Schaper, J.; Froede, R.; Hein, S.; Buck, A.; Hashizume, H.; Speiser, B.; Friedl, A.; Bleese, N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 1991, 83, 504–514. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The extracellular matrix in ischemic and nonischemic heart failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Kasner, M.; Westermann, D.; Lopez, B.; Gaub, R.; Escher, F.; Kühl, U.; Schultheiss, H.-P.; Tschöpe, C. Diastolic tissue doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J. Am. Coll. Cardiol. 2011, 57, 977–985. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Collagen: The fibrous proteins of the matrix. In Molecular Cell Biology, 4th ed.; W. H. Freeman and Company: New York, NY, USA, 2000. [Google Scholar]
- Henriksen, K.; Karsdal, M.A. Type I collagen. In Biochemistry of Collagens, Laminins and Elastin: Structure, Function and Biomarkers; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 1–11. ISBN 9780128098998. [Google Scholar]
- Gao, L.; Orth, P.; Cucchiarini, M.; Madry, H. Effects of solid acellular type I/III collagen biomaterials on in vitro and in vivo chondrogenesis of mesenchymal stem cells. Expert Rev. Med. Devices 2017, 14, 717–732. [Google Scholar] [CrossRef]
- Karsenty, G.; Park, R.W. Regulation of type I collagen genes expression. Int. Rev. Immunol. 1995, 12, 177–185. [Google Scholar] [CrossRef]
- Yamauchi, M.; Sricholpech, M. Lysine post-translational modifications of collagen. Essays Biochem. 2012, 52, 113–133. [Google Scholar] [CrossRef]
- Nishioka, T.; Eustace, A.; West, C. Lysyl oxidase: From basic science to future cancer treatment. Cell Struct. Funct. 2012, 37, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Santamaría, P.G.; Moreno-Bueno, G. LOXL2 in epithelial cell plasticity and tumor progression. Futur. Oncol. 2012, 8, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.; Martínez-González, J. The role of lysyl oxidase enzymes in cardiac function and remodeling. Cells 2019, 8, 1483. [Google Scholar] [CrossRef] [PubMed]
- Al-u’datt, D.; Allen, B.G.; Nattel, S. Role of the lysyl oxidase enzyme family in cardiac function and disease. Cardiovasc. Res. 2019, 115, 1820–1837. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Savvatis, K.; Kang, J.S.; Fan, P.; Zhong, H.; Schwartz, K.; Barry, V.; Mikels-Vigdal, A.; Karpinski, S.; Kornyeyev, D.; et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat. Commun. 2016, 7, 13710. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tang, K.; Tianbao, X.; Wang, J.; Yang, J.; Li, D. Increased serum lysyl oxidase-like 2 levels correlate with the degree of left atrial fibrosis in patients with atrial fibrillation. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- Trackman, P.C. Enzymatic and non-enzymatic functions of the lysyl oxidase family in bone. Matrix Biol. 2016, 52–54, 7–18. [Google Scholar] [CrossRef]
- González, A.; López, B.; Ravassa, S.; San José, G.; Díez, J. The complex dynamics of myocardial interstitial fibrosis in heart failure. Focus on collagen cross-linking. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1421–1432. [Google Scholar] [CrossRef]
- Fong, S.F.T.; Dietzsch, E.; Fong, K.S.K.; Hollosi, P.; Asuncion, L.; He, Q.; Parker, M.I.; Csiszar, K. Lysyl oxidase-like 2 expression is increased in colon and esophageal tumors and associated with less differentiated colon tumors. Genes Chromosom. Cancer 2007, 46, 644–655. [Google Scholar] [CrossRef]
- Saxton, A.; Bordoni, B. Anatomy, Thorax, Cardiac Muscle; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. Physiol. 1983, 245, C1–C14. [Google Scholar] [CrossRef]
- Cheng, H.; Lederer, W.; Cannell, M. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Marks, A.R. Calcium and the heart: A question of life and death. J. Clin. Invest. 2003, 111, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.F.; Factor, S.M.; Sonnenblick, E.H. The heart as a suction pump. Sci. Am. 1986, 254, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Jackson, C.J. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef]
- Deshmukh, S.; Dive, A.; Moharil, R.; Munde, P. Enigmatic insight into collagen. J. Oral Maxillofac. Pathol. 2016, 20, 276. [Google Scholar] [CrossRef]
- Eckes, B.; Nischt, R.; Krieg, T. Cell-matrix interactions in dermal repair and scarring. Fibrogenesis Tissue Repair 2010, 3, 4. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Campana, L.; Iredale, J.P. Extracellular matrix metabolism and fibrotic disease. Curr. Pathobiol. Rep. 2014, 2, 217–224. [Google Scholar] [CrossRef]
- Querejeta, R.; López, B.; González, A.; Sánchez, E.; Larman, M.; Martínez Ubago, J.L.; Díez, J. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: Relation to myocardial fibrosis. Circulation 2004, 110, 1263–1268. [Google Scholar] [CrossRef]
- Martos, R.; Baugh, J.; Ledwidge, M.; O’Loughlin, C.; Conlon, C.; Patle, A.; Donnelly, S.C.; McDonald, K. Diastolic heart failure: Evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 2007, 115, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.A.; Trafford, A.W. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J. Mol. Cell. Cardiol. 2016, 93, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Agha, G.; Baccarelli, A.A. The role of DNA methylation in cardiovascular risk and disease. Circ. Res. 2016, 118, 119–131. [Google Scholar] [CrossRef]
- Zibadi, S.; Vazquez, R.; Larson, D.F.; Watson, R.R. T Lymphocyte regulation of lysyl oxidase in diet-induced cardiac fibrosis. Cardiovasc. Toxicol. 2010, 10, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Lucero, H.A.; Kagan, H.M. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 2006, 63, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.-J.; Finney, J.; Ronnebaum, T.; Mure, M. Human lysyl oxidase-like. Bioorg. Chem. 2014, 57, 231–241. [Google Scholar] [CrossRef]
- Xu, X.-H.; Jia, Y.; Zhou, X.; Xie, D.; Huang, X.; Jia, L.; Zhou, Q.; Zheng, Q.; Zhou, X.; Wang, K.; et al. Downregulation of lysyl oxidase and lysyl oxidase-like protein 2 suppressed the migration and invasion of trophoblasts by activating the TGF-β/collagen pathway in preeclampsia. Exp. Mol. Med. 2019, 51, 20. [Google Scholar] [CrossRef]
- Davidson, J.M.; Zoia, O.; Liu, J.-M. Modulation of transforming growth factor-beta 1 stimulated elastin and collagen production and proliferation in porcine vascular smooth muscle cells and skin fibroblasts by basic fibroblast growth factor, transforming growth factor-α, and insulin-like growth factor-I. J. Cell. Physiol. 1993, 155, 149–156. [Google Scholar] [CrossRef]
- Rodriguez, C.; Martinez-Gonzalez, J.; Raposo, B.; Alcudia, J.F.; Guadall, A.; Badimon, L. Regulation of lysyl oxidase in vascular cells: Lysyl oxidase as a new player in cardiovascular diseases. Cardiovasc. Res. 2008, 79, 7–13. [Google Scholar] [CrossRef]
- Wu, L.; Zhu, Y. The function and mechanisms of action of LOXL2 in cancer: A review. Int. J. Mol. Med. 2015, 36, 1200–1204. [Google Scholar] [CrossRef]
- Molnar, J.; Fong, K.S.K.; He, Q.P.; Hayashi, K.; Kim, Y.; Fong, S.F.T.; Fogelgren, B.; Szauter, K.M.; Mink, M.; Csiszar, K. Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim. Biophys. Acta 2003, 1647, 220–224. [Google Scholar] [CrossRef]
- Trackman, P.C. Functional importance of lysyl oxidase family propeptide regions. J. Cell Commun. Signal. 2018, 12, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, R.; Sodek, K.L.; Xu, W.-P.; Bais, M.V.; Saxena, D.; Faibish, M.; Trackman, P.C. A novel function for lysyl oxidase in pluripotent mesenchymal cell proliferation and relevance to inflammation-associated osteopenia. PLoS ONE 2014, 9, e100669. [Google Scholar] [CrossRef] [PubMed]
- Liguori, T.T.A.; Liguori, G.R.; Moreira, L.F.P.; Harmsen, M.C. Fibroblast growth factor-2, but not the adipose tissue-derived stromal cells secretome, inhibits TGF-β1-induced differentiation of human cardiac fibroblasts into myofibroblasts. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Ohta, H. Pathophysiological roles of FGF signaling in the heart. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, Y.; Gao, J.; Pawlyk, B.; Starcher, B.; Spencer, J.A.; Yanagisawa, H.; Zuo, J.; Li, T. Elastic fiber homeostasis requires lysyl oxidase–like 1 protein. Nat. Genet. 2004, 36, 178–182. [Google Scholar] [CrossRef]
- Ohmura, H.; Yasukawa, H.; Minami, T.; Sugi, Y.; Oba, T.; Nagata, T.; Kyogoku, S.; Ohshima, H.; Aoki, H.; Imaizumi, T. Cardiomyocyte-specific transgenic expression of lysyl oxidase-like protein-1 induces cardiac hypertrophy in mice. Hypertens. Res. 2012, 35, 1063–1068. [Google Scholar] [CrossRef]
- Busnadiego, O.; Gonzalez-Santamaria, J.; Lagares, D.; Guinea-Viniegra, J.; Pichol-Thievend, C.; Muller, L.; Rodriguez-Pascual, F. LOXL4 is induced by transforming growth factor 1 through smad and JunB/Fra2 and contributes to vascular matrix remodeling. Mol. Cell. Biol. 2013, 33, 2388–2401. [Google Scholar] [CrossRef]
- Alzahrani, F.; Al Hazzaa, S.A.; Tayeb, H.; Alkuraya, F.S. LOXL3 encoding lysyl oxidase-like 3, is mutated in a family with autosomal recessive Stickler syndrome. Hum. Genet. 2015, 134, 451–453. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, R.; Liu, Z.; Hou, C.; Zong, W.; Zhang, A.; Sun, X.; Gao, J. Loss of lysyl oxidase-like 3 causes cleft palate and spinal deformity in mice. Hum. Mol. Genet. 2015, 24, 6174–6185. [Google Scholar] [CrossRef]
- Bignon, M.; Pichol-Thievend, C.; Hardouin, J.; Malbouyres, M.; Brechot, N.; Nasciutti, L.; Barret, A.; Teillon, J.; Guillon, E.; Etienne, E.; et al. Lysyl oxidase-like protein-2 regulates sprouting angiogenesis and type IV collagen assembly in the endothelial basement membrane. Blood 2011, 118, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Salvador, F.; Moreno-Bueno, G.; Floristán, A.; Ruiz-Herguido, C.; Cuevas, E.P.; Morales, S.; Santos, V.; Csiszar, K.; Dubus, P.; et al. Lysyl oxidase-like 2 represses Notch1 expression in the skin to promote squamous cell carcinoma progression. EMBO J. 2015, 34, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Barry-Hamilton, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.M.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C.; et al. Allosteric inhibition of lysyl oxidase–like-2 impedes the development of a pathologic microenvironment. Nat. Med. 2010, 16, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 373, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Nxele, X.; Cour, M.; Sangweni, N.; Jooste, T.; Hadebe, N.; Samodien, E.; Benjeddou, M.; Mazino, M.; Louw, J.; et al. Identification of potential biomarkers for predicting the early onset of diabetic cardiomyopathy in a mouse model. Sci. Rep. 2020, 10, 12352. [Google Scholar] [CrossRef]
- Puente, A.; Fortea, J.I.; Cabezas, J.; Arias Loste, M.T.; Iruzubieta, P.; Llerena, S.; Huelin, P.; Fábrega, E.; Crespo, J. LOXL2—A new target in antifibrogenic therapy? Int. J. Mol. Sci. 2019, 20, 1634. [Google Scholar] [CrossRef]
- Lytle, K.A.; Wong, C.P.; Jump, D.B. Docosahexaenoic acid blocks progression of western diet-induced nonalcoholic steatohepatitis in obese Ldlr-/- mice. PLoS ONE 2017, 12, e0173376. [Google Scholar] [CrossRef]
- Wright, E.; Scism-Bacon, J.L.; Glass, L.C.; Glass, L. Oxidative stress in type 2 diabetes: The role of fasting and postprandial glycaemia. Int. J. Clin. Pract. 2006, 60, 308–314. [Google Scholar] [CrossRef]
- Michael Brownlee, M.D. Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med. 1995, 46, 223–234. [Google Scholar] [CrossRef]
- Kislinger, T.; Tanji, N.; Wendt, T.; Qu, W.; Lu, Y.; Ferran, L.J.; Taguchi, A.; Olson, K.; Bucciarelli, L.; Goova, M.; et al. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 905–910. [Google Scholar] [CrossRef]
- Candido, R.; Forbes, J.M.; Thomas, M.C.; Thallas, V.; Dean, R.G.; Burns, W.C.; Tikellis, C.; Ritchie, R.H.; Twigg, S.M.; Cooper, M.E.; et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ. Res. 2003, 92, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J. Hypertens. 2003, 21, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Brown, K.K.; Collard, H.R.; Cottin, V.; Gibson, K.F.; Kaner, R.J.; Lederer, D.J.; Martinez, F.J.; Noble, P.W.; Song, J.W.; et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: A randomised, double-blind, controlled, phase 2 trial. Lancet Respir. Med. 2017, 5, 22–32. [Google Scholar] [CrossRef]
- Mižíková, I.; Palumbo, F.; Tábi, T.; Herold, S.; Vadász, I.; Mayer, K.; Seeger, W.; Morty, R.E. Perturbations to lysyl oxidase expression broadly influence the transcriptome of lung fibroblasts. Physiol. Genom. 2017, 49, 416–429. [Google Scholar] [CrossRef]
- Craighead, D.H.; Wang, H.; Santhanam, L.; Alexander, L.M. Acute lysyl oxidase inhibition alters microvascular function in normotensive but not hypertensive men and women. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H424–H433. [Google Scholar] [CrossRef]
- Steppan, J.; Wang, H.; Bergman, Y.; Rauer, M.J.; Tan, S.; Jandu, S.; Nandakumar, K.; Barreto-Ortiz, S.; Cole, R.N.; Boronina, T.N.; et al. Lysyl oxidase-like 2 depletion is protective in age-associated vascular stiffening. Am. J. Physiol. Circ. Physiol. 2019, 317, H49–H59. [Google Scholar] [CrossRef]
- Torregrosa-Carrión, R.; Luna-Zurita, L.; García-Marqués, F.; D’Amato, G.; Piñeiro-Sabarís, R.; Bonzón-Kulichenko, E.; Vázquez, J.; de la Pompa, J.L. NOTCH activation promotes valve formation by regulating the endocardial secretome. Mol. Cell. Proteom. 2019, 18, 1782–1795. [Google Scholar] [CrossRef]
- Schilter, H.; Findlay, A.D.; Perryman, L.; Yow, T.T.; Moses, J.; Zahoor, A.; Turner, C.I.; Deodhar, M.; Foot, J.S.; Zhou, W.; et al. The lysyl oxidase like 2/3 enzymatic inhibitor, PXS-5153A, reduces crosslinks and ameliorates fibrosis. J. Cell. Mol. Med. 2018, 23, jcmm.14074. [Google Scholar] [CrossRef]
- Dragomir, A.-C.D.; Sun, R.; Choi, H.; Laskin, J.D.; Laskin, D.L. Role of Galectin-3 in classical and alternative macrophage activation in the liver following acetaminophen intoxication. J. Immunol. 2012, 189, 5934–5941. [Google Scholar] [CrossRef]
- Li, Y.; Komai-Koma, M.; Gilchrist, D.S.; Hsu, D.K.; Liu, F.-T.; Springall, T.; Xu, D. Galectin-3 is a negative regulator of lipopolysaccharide-mediated inflammation. J. Immunol. 2008, 181, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A.; Olobatoke, A.; Vanhecke, T.E. Galectin-3: A novel blood test for the evaluation and management of patients with heart failure. Rev. Cardiovasc. Med. 2011, 12, 200–210. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.A.; Voors, A.A.; Muntendam, P.; van Gilst, W.H.; van Veldhuisen, D.J. Galectin-3: A novel mediator of heart failure development and progression. Eur. J. Heart Fail. 2009, 11, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Pez, F.; Dayan, F.; Durivault, J.; Kaniewski, B.; Aimond, G.; Le Provost, G.S.; Deux, B.; Clézardin, P.; Sommer, P.; Pouysségur, J.; et al. The HIF-1–Inducible lysyl oxidase activates HIF-1 via the akt pathway in a positive regulation loop and synergizes with HIF-1 in promoting tumor cell growth. Cancer Res. 2011, 71, 1647–1657. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 2013, 123, 3664–3671. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Molecular pathways: Connecting fibrosis and solid tumor metastasis. Clin. Cancer Res. 2014, 20, 3637–3643. [Google Scholar] [CrossRef]
- Chaudhury, A.; Howe, P.H. The tale of transforming growth factor-beta (TGFβ) signaling: A soigné enigma. IUBMB Life 2009, 61, 929–939. [Google Scholar] [CrossRef]
- Lu, M.; Qin, Q.; Yao, J.; Sun, L.; Qin, X. Induction of LOX by TGF-β1/Smad/AP-1 signaling aggravates rat myocardial fibrosis and heart failure. IUBMB Life 2019, 71, 1729–1739. [Google Scholar] [CrossRef]
- Eulertaimor, G.; Heger, J. The complex pattern of SMAD signaling in the cardiovascular system. Cardiovasc. Res. 2006, 69, 15–25. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Tsutsui, H.; Shiomi, T.; Matsusaka, H.; Matushima, S.; Wen, J.; Kubota, T.; Takeshita, A. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc. Res. 2004, 64, 526–535. [Google Scholar] [CrossRef]
- Hao, J.; Wang, B.; Jones, S.C.; Jassal, D.S.; Dixon, I.M.C. Interaction between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitro. Am. J. Physiol. Circ. Physiol. 2000, 279, H3020–H3030. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.C.; Jones, P.A. Epigenetics and microRNAs. Pediatr. Res. 2007, 61, 24R–29R. [Google Scholar] [CrossRef] [PubMed]
- Morales, S.; Monzo, M.; Navarro, A. Epigenetic regulation mechanisms of microRNA expression. Biomol. Concepts 2017, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- NCBI. LOXL2 lysyl Oxidase Like 2 [Homo sapiens (Human)] Gene. Available online: https://www.ncbi.nlm.nih.gov/gene?cmd=Retrieve&dopt=full_report&list_uids=4017 (accessed on 16 May 2019).
- Ensembl Genome Browser. LOXL2 (ENSG00000134013) Gene (Homo sapiens). Available online: http://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000134013,r=8:23297189-23425328 (accessed on 16 May 2015).
- Hollosi, P.; Yakushiji, J.K.; Fong, K.S.K.; Csiszar, K.; Fong, S.F.T. Lysyl oxidase-like 2 promotes migration in noninvasive breast cancer cells but not in normal breast epithelial cells. Int. J. Cancer 2009, 125, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, R.; Chiyomaru, T.; Enokida, H.; Inoguchi, S.; Ishihara, T.; Matsushita, R.; Goto, Y.; Fukumoto, I.; Nakagawa, M.; Seki, N. Tumour-suppressive microRNA-29s directly regulate LOXL2 expression and inhibit cancer cell migration and invasion in renal cell carcinoma. FEBS Lett. 2015, 589, 2136–2145. [Google Scholar] [CrossRef]
- Martin, E.M.; Fry, R.C. Environmental influences on the epigenome: Exposure—Associated DNA methylation in human populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef]
- Łuczak, M.W.; Jagodziński, P.P. The role of DNA methylation in cancer development. Folia Histochem. Cytobiol. 2006, 44, 143–154. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Alshenibr, W.; Tashkandi, M.M.; Alsaqer, S.F.; Alkheriji, Y.; Wise, A.; Fulzele, S.; Mehra, P.; Goldring, M.B.; Gerstenfeld, L.C.; Bais, M. V Anabolic role of lysyl oxidase like-2 in cartilage of knee and temporomandibular joints with osteoarthritis. Arthritis Res. Ther. 2017, 19, 179. [Google Scholar] [CrossRef]
- Dong, Y.; Huang, Y.; Gutin, B.; Raed, A.; Dong, Y.; Zhu, H. Associations between global DNA methylation and telomere length in healthy adolescents. Sci. Rep. 2017, 7, 4210. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009, 9, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-κB in the heart: To be or not to NF-κB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, N.; Elliott, H.R.; Burrows, K.; Tillin, T.; Hughes, A.D.; Chaturvedi, N.; Gaunt, T.R.; Relton, C.L. Associations between high blood pressure and DNA methylation. J. Clin. Epigenetics 2018, 4, 16. [Google Scholar]
- Makar, K.W.; Wilson, C.B. DNA methylation is a nonredundant repressor of the Th2 effector program. J. Immunol. 2004, 173, 4402–4406. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Yu, L.-M.; Xu, Y. Epigenetic regulation in cardiac fibrosis. World J. Cardiol. 2015, 7, 784–791. [Google Scholar] [CrossRef]
- Bargagli, E.; Piccioli, C.; Rosi, E.; Torricelli, E.; Turi, L.; Piccioli, E.; Pistolesi, M.; Ferrari, K.; Voltolini, L. Pirfenidone and Nintedanib in idiopathic pulmonary fibrosis: Real-life experience in an Italian referral centre. Pulmonology 2019, 25, 149–153. [Google Scholar] [CrossRef]
- Baer, C.; Claus, R.; Plass, C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res. 2013, 73, 473–477. [Google Scholar] [CrossRef]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef]
- Alegría-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Martínez-González, M.A.; Gea, A.; Ruiz-Canela, M. The Mediterranean diet and cardiovascular health: A critical review. Circ. Res. 2019, 124, 779–798. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Study Design | Findings | References |
|---|---|---|---|
| Loxl2+/− knockout mice | Mice: Underwent transaortic constriction followed by LOXL2 expression analysis and histology. | Transgenic mice: cardiac stress results in↑ LOXL2 → myocardial fibrosis & dysfunction. Inhibition of LOXL2 activity: ↓ cardiac fibrosis and ↑ cardiac function. | Yang et al. (2016) [24] |
| Human | Human: Patients presenting with HFpEF and diastolic dysfunction without symptoms underwent right-ventricular biopsies for evaluation of cardiomyopathy. | LOXL2 acts via the PI3K/AKT pathway to activate TGF-β2. Diseased human hearts: LOXL2 ↑ in the interstitial space and serum ↑ LOXL2 expression correlated with ↑ fibrosis and myocardial dysfunction. | |
| Human | Patients (aged 45–85) with idiopathic pulmonary fibrosis were treated with simtuzumab or a placebo once a week and its effects studied. | Simtuzumab, did not improve survival rates in patients with idiopathic pulmonary fibrosis. | Raghu et al. (2017) [74] |
| Human | Patients with atrial fibrillation were assessed in terms of serum LOXL2 levels, left atrial size and left ventricular function. | Atrial fibrillation patients: ↑ serum LOXL2 Positively associated with increased left atrial size. | Zhao et al. (2017) [25] |
| Primary cells isolated from C57Bl/6J mice, macrophages and endothelial cells, and mouse pups | Primary cells: cultured in the presence of a LOX inhibitor or LOX, LOXL1 and LOXL2 knocked down with siRNA. Gene expression, amine oxidase activity and microarray analyses were performed Mice: a bronchopulmonary dysplasia model was established, and lungs harvested for expression analysis. | Lox, Loxl1, and Loxl2 are highly expressed in primary mouse lung fibroblasts. Knockdown of Lox, Loxl1, and Loxl2: associated with change in gene expression (primary mouse lung fibroblasts). BAPN: no impact on mRNA levels of LOX target-genes, in lung fibroblasts or in BAPN-treated mice. | Mižíková et al. (2017) [75] |
| Human | Intradermal microdialysis fibers were placed in the forearm of young, normotensive and hypertensive individuals. Fibers treated with β-aminopropionitrile, a LOX inhibitor, or acted as a control. Norepinephrine was used to examine the vasoconstrictor function and sodium nitroprusside to study smooth muscle vasodilation. | LOX inhibition augmented vasoconstrictor sensitivity in young and normotensive but not hypertensive patients. ECM-bound LOX expression: ↑ in hypertensive subjects vs. younger patients. Vascular stiffness & microvascular dysfunction in hypertension could be due to ↑ LOX expression. | Craighead et al. (2018) [76] |
| Human aortic smooth muscle cells and LOXL2+/− mice | Human aortic smooth muscle cells were cultured and the secretome analyzed. Mice: nitric oxide production was assessed in the aortic rings. | Proteomic analysis: LOXL2: important mediator of age-associated vascular stiffening in smooth muscle cells. Nitric oxide assessment: it ↓ LOXL2 abundance and activity in the ECM of isolated smooth muscle cells. Knock out mice: protected from age-associated vascular stiffening. Isolated aortic rings: LOXL2 mediates vascular stiffening in aging by promoting smooth muscle cell stiffness, contractility, and matrix deposition. | Steppan et al. (2018) [77] |
| Mouse embryonic endocardial cells, human aortic smooth muscle cells and LOXL2+/− mice | Mouse embryonic endocardinal cells were stimulated with DLL4 and JAG1, with or without NOTCH inhibitors. Proteomics analysis of the media was conducted to identified proteins that are secreted in response to NOTCH signaling manipulation. | Secretome analysis identified 129 factors that showed a change in expression when NOTCH was activated or repressed. NOTCH activation correlated with ↑ expression of TGF-β2 and collagen. | Torregrosa-Carrión et al. (2019) [78] |
| Wistar rats, Sprague Dawley rats, C57/BL6 mice | A LOXL2/LOXL3 inhibitor, PXS-5153A, was developed and its effect on LOXL2/3 in relation to collagen cross-linking and fibrosis was assessed. | PXS-5153A ↓ collagen cross-linking in vitro. PXS-5153A ↓ collagen expression and cross-linking, thereby ↑ liver function. In a model of myocardial infarction, addition PXS-5153A, ↑ cardiac output. This shows that inhibition of LOXL2/LOXL3 activity could be a viable treatment option for liver fibrosis. | Schilter et al. (2019) [79] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erasmus, M.; Samodien, E.; Lecour, S.; Cour, M.; Lorenzo, O.; Dludla, P.; Pheiffer, C.; Johnson, R. Linking LOXL2 to Cardiac Interstitial Fibrosis. Int. J. Mol. Sci. 2020, 21, 5913. https://doi.org/10.3390/ijms21165913
Erasmus M, Samodien E, Lecour S, Cour M, Lorenzo O, Dludla P, Pheiffer C, Johnson R. Linking LOXL2 to Cardiac Interstitial Fibrosis. International Journal of Molecular Sciences. 2020; 21(16):5913. https://doi.org/10.3390/ijms21165913
Chicago/Turabian StyleErasmus, Melisse, Ebrahim Samodien, Sandrine Lecour, Martin Cour, Oscar Lorenzo, Phiwayinkosi Dludla, Carmen Pheiffer, and Rabia Johnson. 2020. "Linking LOXL2 to Cardiac Interstitial Fibrosis" International Journal of Molecular Sciences 21, no. 16: 5913. https://doi.org/10.3390/ijms21165913
APA StyleErasmus, M., Samodien, E., Lecour, S., Cour, M., Lorenzo, O., Dludla, P., Pheiffer, C., & Johnson, R. (2020). Linking LOXL2 to Cardiac Interstitial Fibrosis. International Journal of Molecular Sciences, 21(16), 5913. https://doi.org/10.3390/ijms21165913