Critical Analysis of Genome-Wide Association Studies: Triple Negative Breast Cancer Quae Exempli Causa


 , , , and
, , , and 
Abstract
1. Introduction
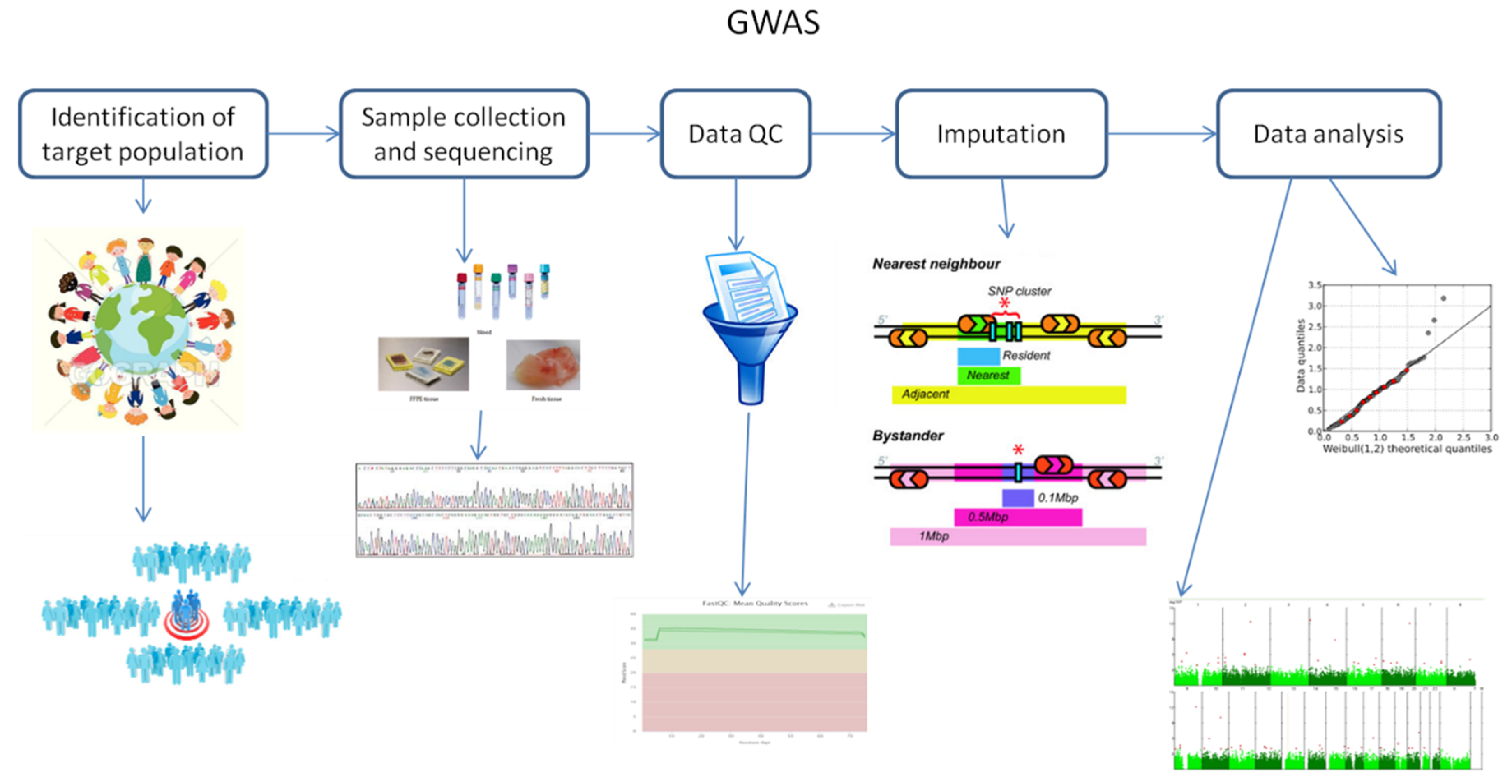
2. Fundamental Principles of Genome-Wide Association Studies
3. Challenges of Genome-Wide Association Studies
4. Genome-Wide Association Studies on Triple Negative Breast Cancer
4.1. Triple Negative Breast Cancer
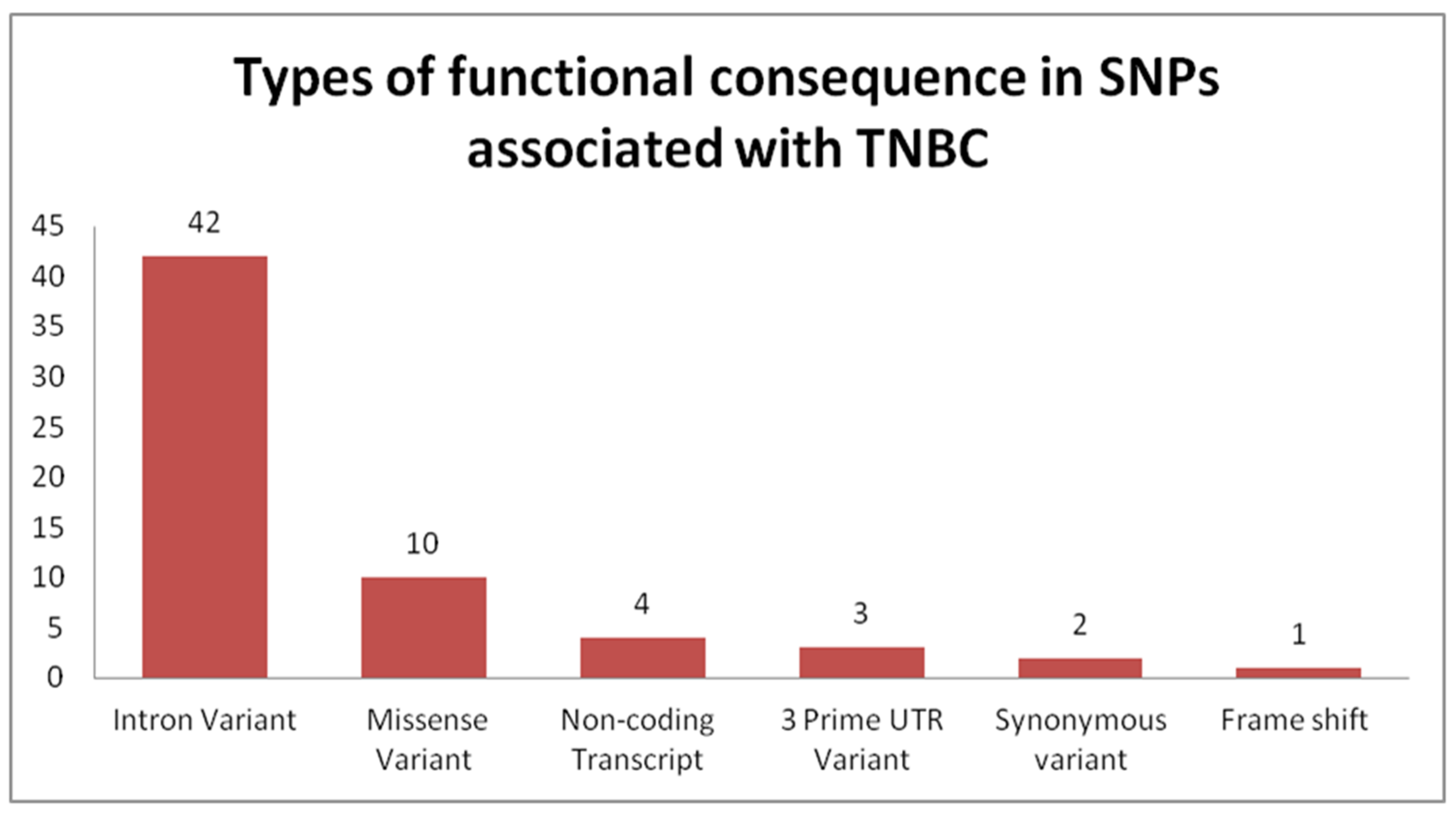
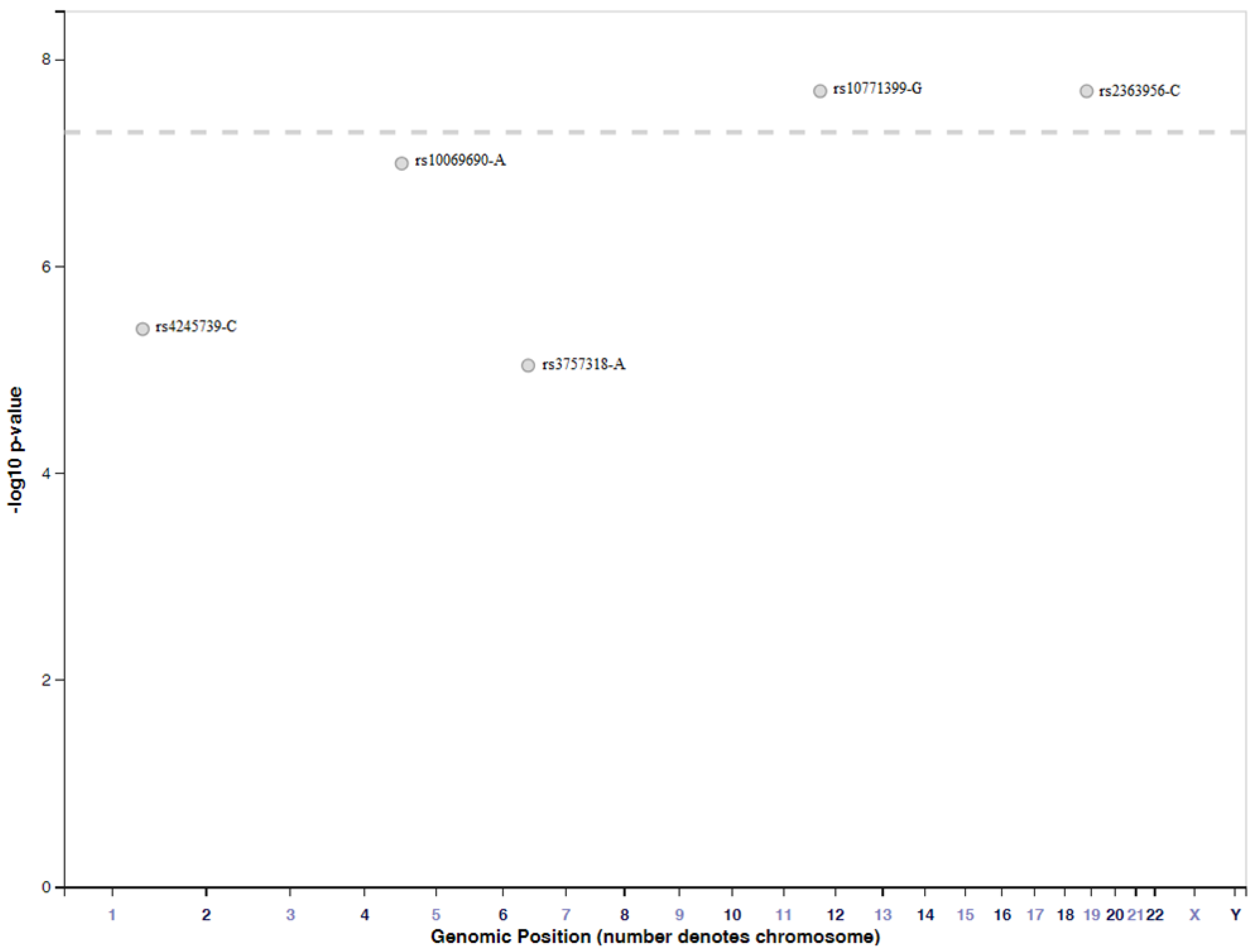
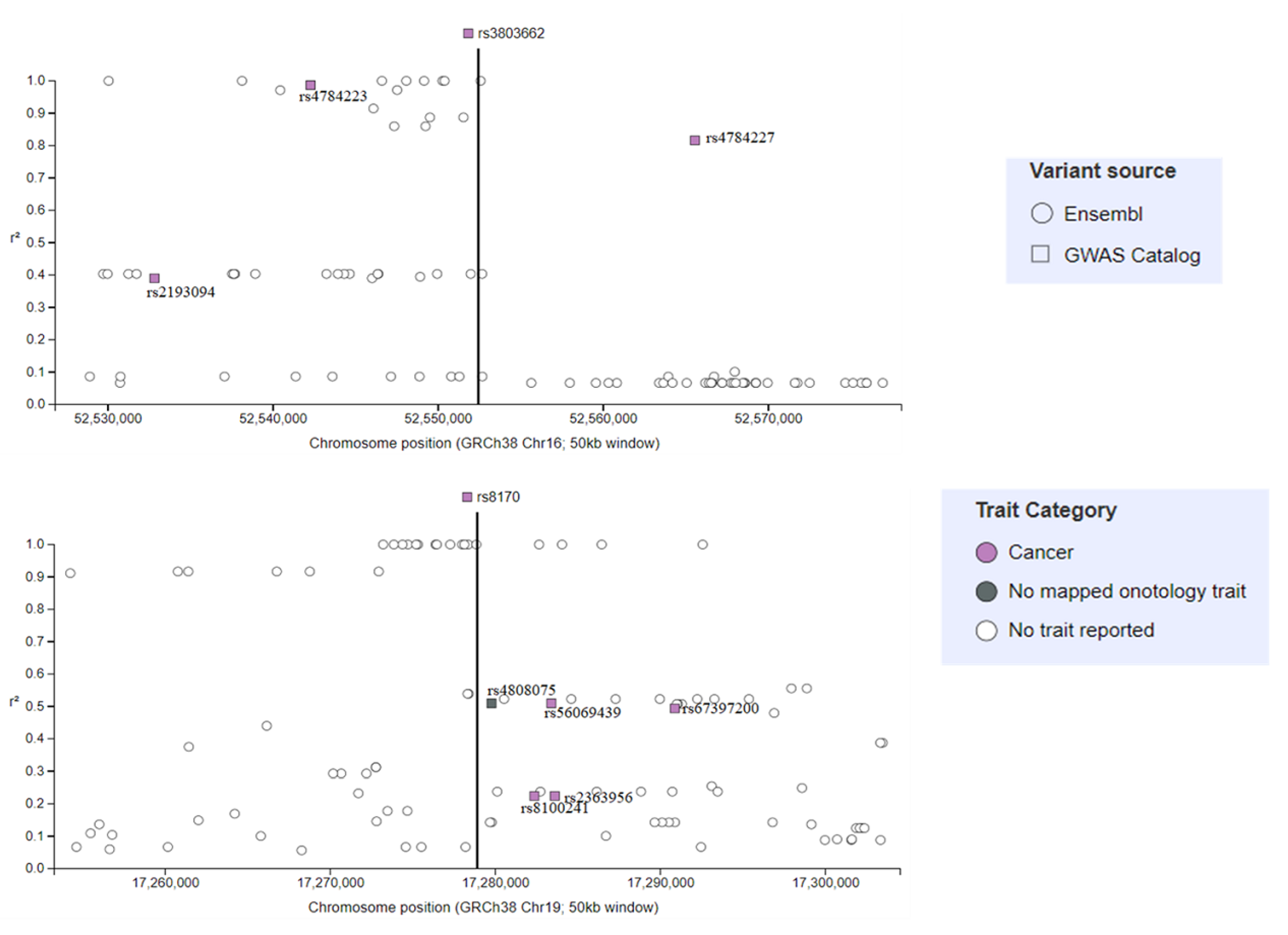
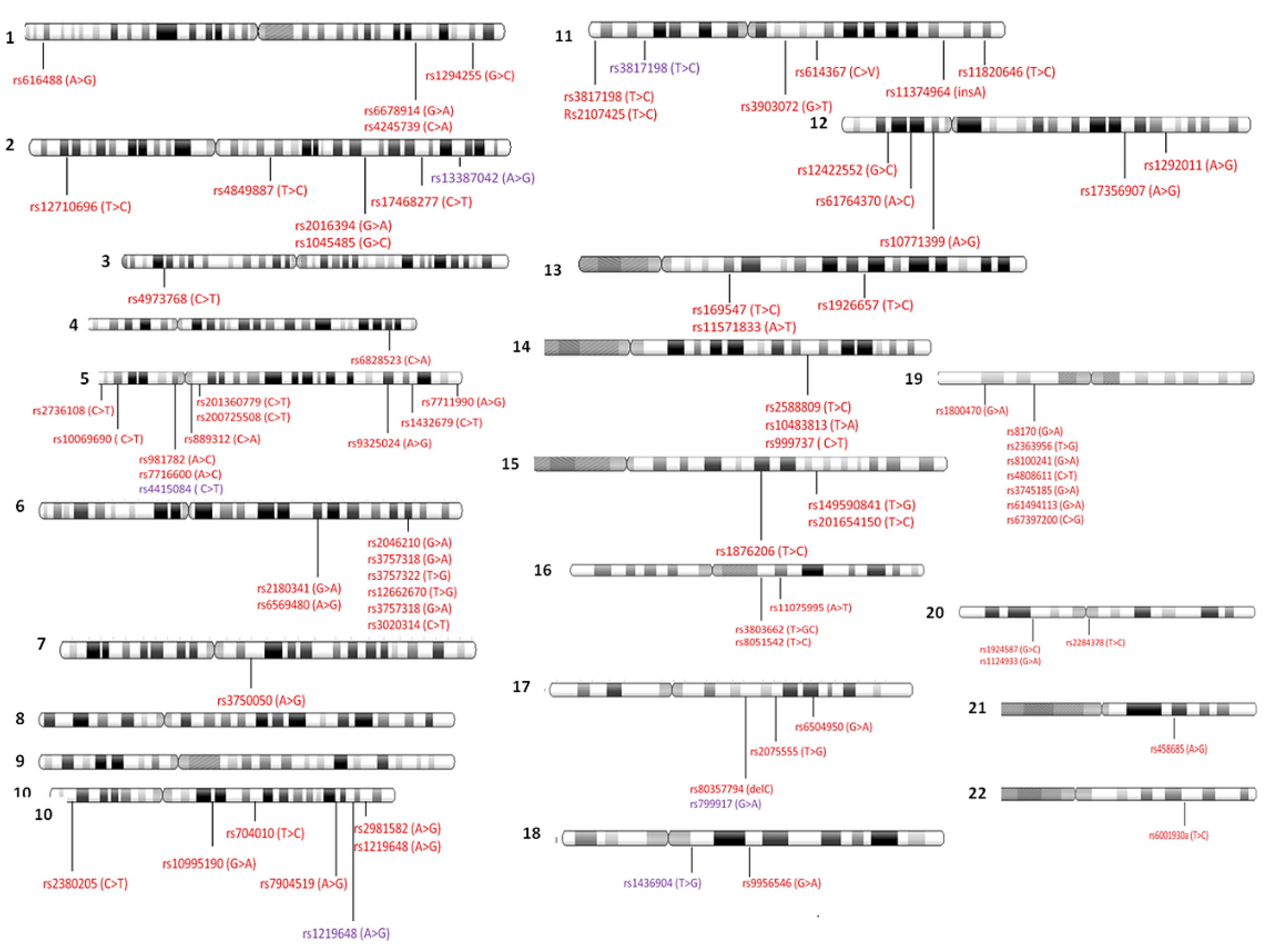
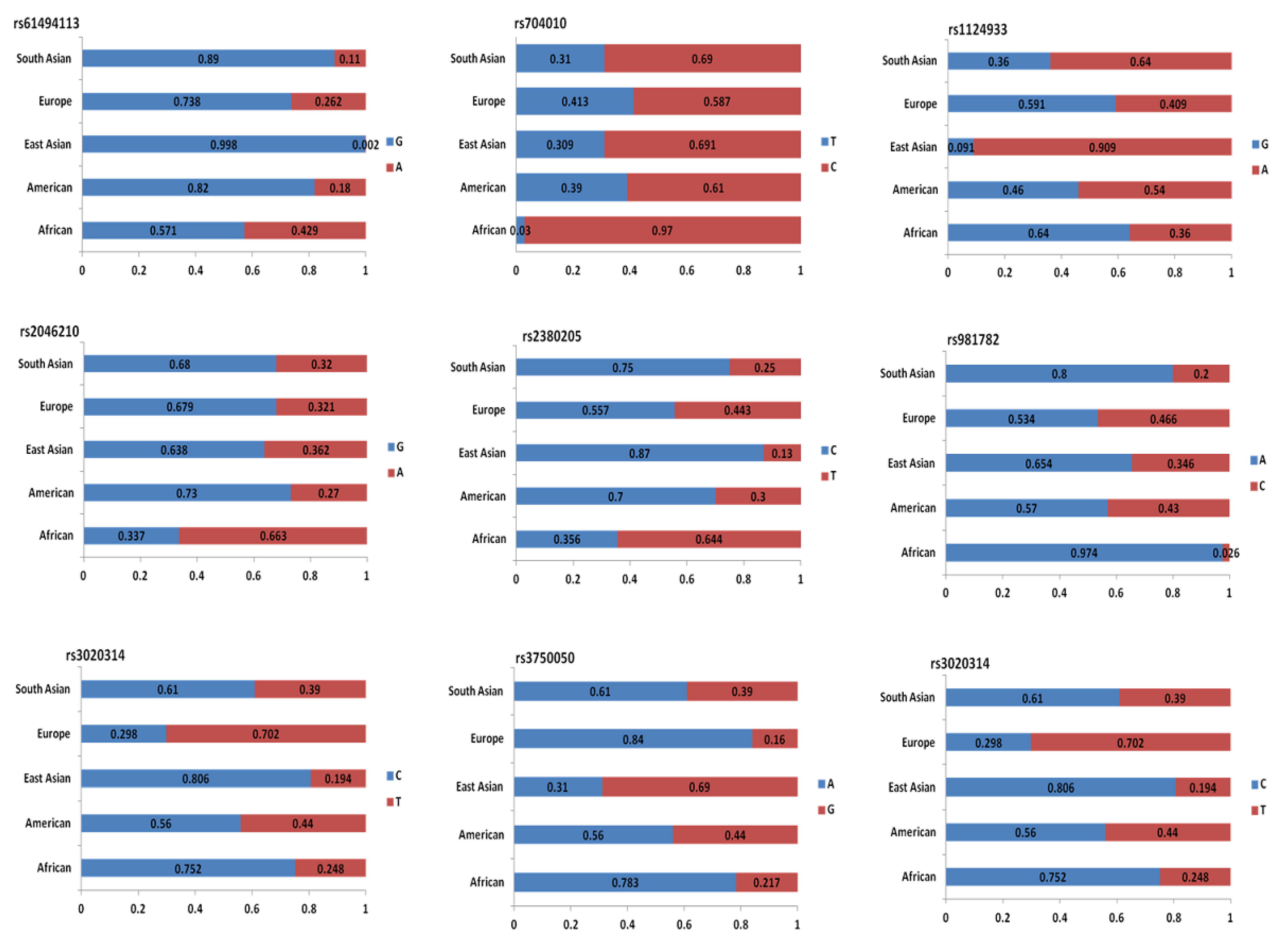
4.2. Genome-Wide Association Studies Identifying SNPs for TNBC
4.3. Importance of the Relationship between SNPs and miRNAs in BC
5. Risk Factor Scores for Prediction of Breast Cancer
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Pharoah, P.D.; Michailidou, K.; Tyrer, J.; Brook, M.N.; Bolla, M.K.; Wang, Q.; Dennis, J.; Dunning, A.M.; Shah, M.; et al. Prediction of breast cancer risk based on profiling with common genetic variants. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Banks, T.W.; Cloutier, S. SNP discovery through next-generation sequencing and its applications. Int. J. Plant Genom. 2012, 2012, 831460. [Google Scholar] [CrossRef]
- Slatkin, M. Linkage disequilibrium—Understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 2008, 9, 477–485. [Google Scholar] [CrossRef]
- Sachs, T. Epigenetic selection: An alternative mechanism of pattern formation. J. Theor. Biol. 1988, 134, 547–559. [Google Scholar] [CrossRef]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1093. [Google Scholar] [CrossRef]
- Reich, D.E.; Lander, E.S. On the allelic spectrum of human disease. Trends Genet. TIG 2001, 17, 502–510. [Google Scholar] [CrossRef]
- McClellan, J.; King, M.C. Genetic heterogeneity in human disease. Cell 2010, 141, 210–217. [Google Scholar] [CrossRef]
- Wang, K.; Bucan, M.; Grant, S.F.; Schellenberg, G.; Hakonarson, H. Strategies for genetic studies of complex diseases. Cell 2010, 142, 351–353; author reply 353–355. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klein, R.J.; Xu, X.; Mukherjee, S.; Willis, J.; Hayes, J. Successes of genome-wide association studies. Cell 2010, 142, 350–351; author reply 353–355. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Ioannidis, J.P. Meta-analysis in genome-wide association studies. Pharmacogenomics 2009, 10, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Panagiotou, O.A.; Willer, C.J.; Hirschhorn, J.N.; Ioannidis, J.P. The power of meta-analysis in genome-wide association studies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 441–465. [Google Scholar] [CrossRef] [PubMed]
- Dimou, N.L.; Tsirigos, K.D.; Elofsson, A.; Bagos, P.G. GWAR: Robust analysis and meta-analysis of genome-wide association studies. Bioinformatics (Oxford, England) 2017, 33, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, E.; Ioannidis, J.P. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 2013, 14, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Closas, M.; Couch, F.J.; Lindstrom, S.; Michailidou, K.; Schmidt, M.K.; Brook, M.N.; Orr, N.; Rhie, S.K.; Riboli, E.; Feigelson, H.S.; et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat. Genet. 2013, 45, 392–398, 398e1–398e2. [Google Scholar] [CrossRef]
- Eeles, R.A.; Olama, A.A.; Benlloch, S.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Ghoussaini, M.; Luccarini, C.; Dennis, J.; Jugurnauth-Little, S.; et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat. Genet. 2013, 45, 385–391, 391e1–391e2. [Google Scholar] [CrossRef]
- Pharoah, P.D.; Tsai, Y.Y.; Ramus, S.J.; Phelan, C.M.; Goode, E.L.; Lawrenson, K.; Buckley, M.; Fridley, B.L.; Tyrer, J.P.; Shen, H.; et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat. Genet. 2013, 45, 362–370, 370e1–370e2. [Google Scholar] [CrossRef]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361, 361e1–361e2. [Google Scholar] [CrossRef]
- Consortium, T.B.C.A. BCAC. University of Cambrige: Cambrige, UK. Available online: http://bcac.ccge.medschl.cam.ac.uk/ (accessed on 10 July 2020).
- Brigham, M.G. ISC. Mass General Brigham: Boston, MA, USA. Available online: https://www.massgeneral.org/ (accessed on 10 July 2020).
- MAGIC Consortium. Sanger Institute: Cambrige, UK. Available online: https://www.magicinvestigators.org/ (accessed on 10 July 2020).
- Sud, A.; Kinnersley, B.; Houlston, R.S. Genome-wide association studies of cancer: Current insights and future perspectives. Nat. Rev. Cancer 2017, 17, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Globocan Breast Worldwide. Available online: https://gco.iarc.fr/ (accessed on 10 July 2020).
- Globocan Breast Romania. Available online: https://gco.iarc.fr/ (accessed on 10 July 2020).
- Li, C.I.; Uribe, D.J.; Daling, J.R. Clinical characteristics of different histologic types of breast cancer. Br. J. Cancer 2005, 93, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.K.R.; LiVolsi, V.; Page, D.; Patchefsky, A.; Silverberg, S. Recommendations for the reporting of breast carcinoma. Association of Directors of Anatomic and Surgical Pathology. Am. J. Clin. Pathol. 1995, 104, 614–619. [Google Scholar]
- Lester, S.C.; Bose, S.; Chen, Y.Y.; Connolly, J.L.; de Baca, M.E.; Fitzgibbons, P.L.; Hayes, D.F.; Kleer, C.; O’Malley, F.P.; Page, D.L.; et al. Protocol for the examination of specimens from patients with invasive carcinoma of the breast. Arch. Pathol. Lab. Med. 2009, 133, 1515–1538. [Google Scholar] [CrossRef]
- Bustos, M.A.; Salomon, M.P.; Nelson, N.; Hsu, S.C.; DiNome, M.L.; Hoon, D.S.; Marzese, D.M. Genome-wide chromatin accessibility, DNA methylation and gene expression analysis of histone deacetylase inhibition in triple-negative breast cancer. Genom. Data 2017, 12, 14–16. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Gluz, O.; Liedtke, C.; Gottschalk, N.; Pusztai, L.; Nitz, U.; Harbeck, N. Triple-negative breast cancer—Current status and future directions. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2009, 20, 1913–1927. [Google Scholar] [CrossRef]
- Braicu, C.; Chiorean, R.; Irimie, A.; Chira, S.; Tomuleasa, C.; Neagoe, E.; Paradiso, A.; Achimas-Cadariu, P.; Lazar, V.; Berindan-Neagoe, I. Novel insight into triple-negative breast cancers, the emerging role of angiogenesis, and antiangiogenic therapy. Expert Rev. Mol. Med. 2016, 18, e18. [Google Scholar] [CrossRef]
- Pop, L.A.; Cojocneanu-Petric, R.M.; Pileczki, V.; Morar-Bolba, G.; Irimie, A.; Lazar, V.; Lombardo, C.; Paradiso, A.; Berindan-Neagoe, I. Genetic alterations in sporadic triple negative breast cancer. Breast 2018, 38, 30–38. [Google Scholar] [CrossRef]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Balacescu, O.; Balacescu, L.; Virtic, O.; Visan, S.; Gherman, C.; Drigla, F.; Pop, L.; Bolba-Morar, G.; Lisencu, C.; Fetica, B.; et al. Blood genome-wide transcriptional profiles of HER2 negative breast cancers patients. Mediat. Inflamm. 2016, 2016, 3239167. [Google Scholar] [CrossRef]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet (London, England) 2017, 389, 2430–2442. [Google Scholar] [CrossRef]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Hoffman, J.; Fejerman, L.; Hu, D.; Huntsman, S.; Li, M.; John, E.M.; Torres-Mejia, G.; Kushi, L.; Ding, Y.C.; Weitzel, J.; et al. Identification of novel common breast cancer risk variants at the 6q25 locus among Latinas. Breast Cancer Res. BCR 2019, 21, 3. [Google Scholar] [CrossRef]
- Mhatre, S.; Wang, Z.; Nagrani, R.; Badwe, R.; Chiplunkar, S.; Mittal, B.; Yadav, S.; Zhang, H.; Chung, C.C.; Patil, P.; et al. Common genetic variation and risk of gallbladder cancer in India: A case-control genome-wide association study. Lancet Oncol. 2017, 18, 535–544. [Google Scholar] [CrossRef]
- Nagrani, R.; Mhatre, S.; Rajaraman, P.; Chatterjee, N.; Akbari, M.R.; Boffetta, P.; Brennan, P.; Badwe, R.; Gupta, S.; Dikshit, R. Association of Genome-Wide Association Study (GWAS) identified SNPs and risk of breast cancer in an indian population. Sci. Rep. 2017, 7, 40963. [Google Scholar] [CrossRef]
- Swierniak, M.; Wojcicka, A.; Czetwertynska, M.; Dlugosinska, J.; Stachlewska, E.; Gierlikowski, W.; Kot, A.; Gornicka, B.; Koperski, L.; Bogdanska, M.; et al. Association between GWAS-derived rs966423 genetic variant and overall mortality in patients with differentiated thyroid cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1111–1119. [Google Scholar] [CrossRef]
- Kang, B.W.; Jeon, H.S.; Chae, Y.S.; Lee, S.J.; Park, J.Y.; Choi, J.E.; Park, J.S.; Choi, G.S.; Kim, J.G. Association between GWAS-identified genetic variations and disease prognosis for patients with colorectal cancer. PLoS ONE 2015, 10, e0119649. [Google Scholar] [CrossRef]
- Barrdahl, M.; Canzian, F.; Lindstrom, S.; Shui, I.; Black, A.; Hoover, R.N.; Ziegler, R.G.; Buring, J.E.; Chanock, S.J.; Diver, W.R.; et al. Association of breast cancer risk loci with breast cancer survival. Int. J. Cancer 2015, 137, 2837–2845. [Google Scholar] [CrossRef]
- Bayraktar, S.; Thompson, P.A.; Yoo, S.Y.; Do, K.A.; Sahin, A.A.; Arun, B.K.; Bondy, M.L.; Brewster, A.M. The relationship between eight GWAS-identified single-nucleotide polymorphisms and primary breast cancer outcomes. Oncologist 2013, 18, 493–500. [Google Scholar] [CrossRef]
- Pagani, F.; Baralle, F.E. Genomic variants in exons and introns: Identifying the splicing spoilers. Nat. Rev. Genet. 2004, 5, 389–396. [Google Scholar] [CrossRef]
- Giral, H.; Landmesser, U.; Kratzer, A. Into the wild: GWAS exploration of non-coding RNAs. Front. Cardiovasc. Med. 2018, 5, 181. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Akhurst, R.J. Transforming growth factor-beta in breast cancer: Too much, too late. Breast Cancer Res. BCR 2009, 11, 202. [Google Scholar] [CrossRef]
- De Blasio, A.; Di Fiore, R.; Morreale, M.; Carlisi, D.; Drago-Ferrante, R.; Montalbano, M.; Scerri, C.; Tesoriere, G.; Vento, R. Unusual roles of caspase-8 in triple-negative breast cancer cell line MDA-MB-231. Int. J. Oncol. 2016, 48, 2339–2348. [Google Scholar] [CrossRef]
- Cox, A.; Dunning, A.M.; Garcia-Closas, M.; Balasubramanian, S.; Reed, M.W.; Pooley, K.A.; Scollen, S.; Baynes, C.; Ponder, B.A.; Chanock, S.; et al. A common coding variant in CASP8 is associated with breast cancer risk. Nat. Genet. 2007, 39, 352–358. [Google Scholar] [CrossRef]
- Skeeles, L.E.; Fleming, J.L.; Mahler, K.L.; Toland, A.E. The impact of 3’UTR variants on differential expression of candidate cancer susceptibility genes. PLoS ONE 2013, 8, e58609. [Google Scholar] [CrossRef]
- Paranjape, T.; Heneghan, H.; Lindner, R.; Keane, F.K.; Hoffman, A.; Hollestelle, A.; Dorairaj, J.; Geyda, K.; Pelletier, C.; Nallur, S.; et al. A 3′-untranslated region KRAS variant and triple-negative breast cancer: A case-control and genetic analysis. Lancet Oncol. 2011, 12, 377–386. [Google Scholar] [CrossRef]
- Hunt, R.C.; Simhadri, V.L.; Iandoli, M.; Sauna, Z.E.; Kimchi-Sarfaty, C. Exposing synonymous mutations. Trends Genet. TIG 2014, 30, 308–321. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Sinilnikova, O.M.; McGuffog, L.; Healey, S.; Nevanlinna, H.; Heikkinen, T.; Simard, J.; Spurdle, A.B.; Beesley, J.; Chen, X.; et al. Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum. Mol. Genet. 2009, 18, 4442–4456. [Google Scholar] [CrossRef]
- Huo, D.; Zheng, Y.; Ogundiran, T.O.; Adebamowo, C.; Nathanson, K.L.; Domchek, S.M.; Rebbeck, T.R.; Simon, M.S.; John, E.M.; Hennis, A.; et al. Evaluation of 19 susceptibility loci of breast cancer in women of African ancestry. Carcinogenesis 2012, 33, 835–840. [Google Scholar] [CrossRef]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef]
- Ripperger, T.; Gadzicki, D.; Meindl, A.; Schlegelberger, B. Breast cancer susceptibility: Current knowledge and implications for genetic counselling. Eur. J. Hum. Genet. 2009, 17, 722–731. [Google Scholar] [CrossRef]
- Stratton, M.R.; Rahman, N. The emerging landscape of breast cancer susceptibility. Nat. Genet. 2008, 40, 17–22. [Google Scholar] [CrossRef]
- Ellsworth, D.L.; Turner, C.E.; Ellsworth, R.E. A review of the hereditary component of triple negative breast cancer: High- and moderate-penetrance breast cancer genes, low-penetrance loci, and the role of nontraditional genetic elements. J. Oncol. 2019, 2019, 4382606. [Google Scholar] [CrossRef]
- Zhang, F.; Lupski, J.R. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015, 24, R102–R110. [Google Scholar] [CrossRef] [PubMed]
- Nishizaki, S.S.; Boyle, A.P. Mining the unknown: Assigning function to noncoding single nucleotide polymorphisms. Trends Genet. TIG 2017, 33, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Moszynska, A.; Gebert, M.; Collawn, J.F.; Bartoszewski, R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. 2017, 7. [Google Scholar] [CrossRef]
- Gong, J.; Tong, Y.; Zhang, H.M.; Wang, K.; Hu, T.; Shan, G.; Sun, J.; Guo, A.Y. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum. Mutat. 2012, 33, 254–263. [Google Scholar] [CrossRef]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef]
- Brendle, A.; Lei, H.; Brandt, A.; Johansson, R.; Enquist, K.; Henriksson, R.; Hemminki, K.; Lenner, P.; Forsti, A. Polymorphisms in predicted microRNA-binding sites in integrin genes and breast cancer: ITGB4 as prognostic marker. Carcinogenesis 2008, 29, 1394–1399. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Upadhyaya, A.; Smith, R.A.; Chacon-Cortes, D.; Revechon, G.; Bellis, C.; Lea, R.A.; Haupt, L.M.; Chambers, S.K.; Youl, P.H.; Griffiths, L.R. Association of the microRNA-Single Nucleotide Polymorphism rs2910164 in miR146a with sporadic breast cancer susceptibility: A case control study. Gene 2016, 576, 256–260. [Google Scholar] [CrossRef]
- Qian, F.; Feng, Y.; Zheng, Y.; Ogundiran, T.O.; Ojengbede, O.; Zheng, W.; Blot, W.; Ambrosone, C.B.; John, E.M.; Bernstein, L.; et al. Genetic variants in microRNA and microRNA biogenesis pathway genes and breast cancer risk among women of African ancestry. Hum. Genet. 2016, 135, 1145–1159. [Google Scholar] [CrossRef]
- Naccarati, A.; Pardini, B.; Stefano, L.; Landi, D.; Slyskova, J.; Novotny, J.; Levy, M.; Polakova, V.; Lipska, L.; Vodicka, P. Polymorphisms in miRNA-binding sites of nucleotide excision repair genes and colorectal cancer risk. Carcinogenesis 2012, 33, 1346–1351. [Google Scholar] [CrossRef]
- Mullany, L.E.; Wolff, R.K.; Herrick, J.S.; Buas, M.F.; Slattery, M.L. SNP Regulation of microRNA Expression and Subsequent Colon Cancer Risk. PLoS ONE 2015, 10, e0143894. [Google Scholar] [CrossRef]
- Nicoloso, M.S.; Sun, H.; Spizzo, R.; Kim, H.; Wickramasinghe, P.; Shimizu, M.; Wojcik, S.E.; Ferdin, J.; Kunej, T.; Xiao, L.; et al. Single-nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer Res. 2010, 70, 2789–2798. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Greco, D.; Michailidou, K.; Milne, R.L.; Muranen, T.A.; Heikkinen, T.; Aaltonen, K.; Dennis, J.; Bolla, M.K.; Liu, J.; et al. MicroRNA related polymorphisms and breast cancer risk. PLoS ONE 2014, 9, e109973. [Google Scholar] [CrossRef]
- Yang, H.; Dinney, C.P.; Ye, Y.; Zhu, Y.; Grossman, H.B.; Wu, X. Evaluation of genetic variants in microRNA-related genes and risk of bladder cancer. Cancer Res. 2008, 68, 2530–2537. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Song, F.; Calin, G.A.; Wei, Q.; Hao, X.; Zhang, W. Polymorphisms in microRNA targets: A gold mine for molecular epidemiology. Carcinogenesis 2008, 29, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Salzman, D.W.; Weidhaas, J.B. SNPing cancer in the bud: microRNA and microRNA-target site polymorphisms as diagnostic and prognostic biomarkers in cancer. Pharmacol. Ther. 2013, 137, 55–63. [Google Scholar] [CrossRef]
- Wilk, G.; Braun, R. regQTLs: Single nucleotide polymorphisms that modulate microRNA regulation of gene expression in tumors. PLoS Genet. 2018, 14, e1007837. [Google Scholar] [CrossRef]
- Kalapanida, D.; Zagouri, F.; Gazouli, M.; Zografos, E.; Dimitrakakis, C.; Marinopoulos, S.; Giannos, A.; Sergentanis, T.N.; Kastritis, E.; Terpos, E.; et al. Evaluation of pre-mir-34a rs72631823 single nucleotide polymorphism in triple negative breast cancer: A case-control study. Oncotarget 2018, 9, 36906–36913. [Google Scholar] [CrossRef]
- Haiman, C.A.; Chen, G.K.; Vachon, C.M.; Canzian, F.; Dunning, A.; Millikan, R.C.; Wang, X.; Ademuyiwa, F.; Ahmed, S.; Ambrosone, C.B.; et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nat. Genet. 2011, 43, 1210–1214. [Google Scholar] [CrossRef]
- Stevens, K.N.; Fredericksen, Z.; Vachon, C.M.; Wang, X.; Margolin, S.; Lindblom, A.; Nevanlinna, H.; Greco, D.; Aittomaki, K.; Blomqvist, C.; et al. 19p13.1 is a triple-negative-specific breast cancer susceptibility locus. Cancer Res. 2012, 72, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Wynendaele, J.; Bohnke, A.; Leucci, E.; Nielsen, S.J.; Lambertz, I.; Hammer, S.; Sbrzesny, N.; Kubitza, D.; Wolf, A.; Gradhand, E.; et al. An illegitimate microRNA target site within the 3′ UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010, 70, 9641–9649. [Google Scholar] [CrossRef] [PubMed]
- Purrington, K.S.; Slager, S.; Eccles, D.; Yannoukakos, D.; Fasching, P.A.; Miron, P.; Carpenter, J.; Chang-Claude, J.; Martin, N.G.; Montgomery, G.W.; et al. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis 2014, 35, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, F.; Li, T.; Lu, M.; Wang, L.; Yue, W.; Zhang, D. MirSNP, a database of polymorphisms altering miRNA target sites, identifies miRNA-related SNPs in GWAS SNPs and eQTLs. BMC Genom. 2012, 13, 661. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, K.; Liao, Z.; Li, Y.; Yang, H.; Chen, C.; Zhou, Y.A.; Tao, Y.; Guo, M.; Ren, T.; et al. Promoter mutation of tumor suppressor microRNA-7 is associated with poor prognosis of lung cancer. Mol. Clin. Oncol. 2015, 3, 1329–1336. [Google Scholar] [CrossRef]
- Milne, R.L.; Kuchenbaecker, K.B.; Michailidou, K.; Beesley, J.; Kar, S.; Lindstrom, S.; Hui, S.; Lemacon, A.; Soucy, P.; Dennis, J.; et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat. Genet. 2017, 49, 1767–1778. [Google Scholar] [CrossRef]
- Wade, M.; Wang, Y.V.; Wahl, G.M. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell. Biol. 2010, 20, 299–309. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Griffith, O.L.; Montgomery, S.B.; Bernier, B.; Chu, B.; Kasaian, K.; Aerts, S.; Mahony, S.; Sleumer, M.C.; Bilenky, M.; Haeussler, M.; et al. ORegAnno: An open-access community-driven resource for regulatory annotation. Nucleic Acids Res. 2008, 36, D107–D113. [Google Scholar] [CrossRef]
- Hiard, S.; Charlier, C.; Coppieters, W.; Georges, M.; Baurain, D. Patrocles: A database of polymorphic miRNA-mediated gene regulation in vertebrates. Nucleic Acids Res. 2010, 38, D640–D651. [Google Scholar] [CrossRef]
- Hariharan, M.; Scaria, V.; Brahmachari, S.K. dbSMR: A novel resource of genome-wide SNPs affecting microRNA mediated regulation. BMC Bioinform. 2009, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. PolymiRTS Database 3.0: Linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014, 42, D86–D91. [Google Scholar] [CrossRef] [PubMed]
- Barnholtz-Sloan, J.S.; Shetty, P.B.; Guan, X.; Nyante, S.J.; Luo, J.; Brennan, D.J.; Millikan, R.C. FGFR2 and other loci identified in genome-wide association studies are associated with breast cancer in African-American and younger women. Carcinogenesis 2010, 31, 1417–1423. [Google Scholar] [CrossRef]
- Fletcher, O.; Johnson, N.; Orr, N.; Hosking, F.J.; Gibson, L.J.; Walker, K.; Zelenika, D.; Gut, I.; Heath, S.; Palles, C.; et al. Novel breast cancer susceptibility locus at 9q31.2: Results of a genome-wide association study. J. Natl. Cancer Inst. 2011, 103, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Gold, B.; Kirchhoff, T.; Stefanov, S.; Lautenberger, J.; Viale, A.; Garber, J.; Friedman, E.; Narod, S.; Olshen, A.B.; Gregersen, P.; et al. Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc. Natl. Acad. Sci. USA 2008, 105, 4340–4345. [Google Scholar] [CrossRef] [PubMed]
- Hein, R.; Maranian, M.; Hopper, J.L.; Kapuscinski, M.K.; Southey, M.C.; Park, D.J.; Schmidt, M.K.; Broeks, A.; Hogervorst, F.B.; Bueno-de-Mesquita, H.B.; et al. Comparison of 6q25 breast cancer hits from Asian and European Genome Wide Association Studies in the Breast Cancer Association Consortium (BCAC). PLoS ONE 2012, 7, e42380. [Google Scholar] [CrossRef]
- Kim, H.C.; Lee, J.Y.; Sung, H.; Choi, J.Y.; Park, S.K.; Lee, K.M.; Kim, Y.J.; Go, M.J.; Li, L.; Cho, Y.S.; et al. A genome-wide association study identifies a breast cancer risk variant in ERBB4 at 2q34: Results from the Seoul Breast Cancer Study. Breast Cancer Res. BCR 2012, 14, R56. [Google Scholar] [CrossRef]
- Thomas, G.; Jacobs, K.B.; Kraft, P.; Yeager, M.; Wacholder, S.; Cox, D.G.; Hankinson, S.E.; Hutchinson, A.; Wang, Z.; Yu, K.; et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1). Nat. Genet. 2009, 41, 579–584. [Google Scholar] [CrossRef]
- Turnbull, C.; Ahmed, S.; Morrison, J.; Pernet, D.; Renwick, A.; Maranian, M.; Seal, S.; Ghoussaini, M.; Hines, S.; Healey, C.S.; et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 2010, 42, 504–507. [Google Scholar] [CrossRef]
- Zheng, W.; Long, J.; Gao, Y.T.; Li, C.; Zheng, Y.; Xiang, Y.B.; Wen, W.; Levy, S.; Deming, S.L.; Haines, J.L.; et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat. Genet. 2009, 41, 324–328. [Google Scholar] [CrossRef]
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.H.; Wang, Q.; Bolla, M.K.; et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, A.; Song, M.; Brook, M.N.; Milne, R.L.; Mavaddat, N.; Michailidou, K.; Bolla, M.K.; Wang, Q.; Dennis, J.; Wilcox, A.N.; et al. Joint associations of a polygenic risk score and environmental risk factors for breast cancer in the Breast Cancer Association Consortium. Int. J. Epidemiol. 2018, 47, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Lindstrom, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemacon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Maas, P.; Barrdahl, M.; Joshi, A.D.; Auer, P.L.; Gaudet, M.M.; Milne, R.L.; Schumacher, F.R.; Anderson, W.F.; Check, D.; Chattopadhyay, S.; et al. Breast cancer risk from modifiable and nonmodifiable risk factors among white women in the United States. JAMA Oncol. 2016, 2, 1295–1302. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP | miRNA | Targeted Gene | Effect |
|---|---|---|---|
| rs4245739 | hsa-miR-191-5p | MDM4 | Create |
| hsa-miR-3545-3p | Break | ||
| hsa-miR-3669 | Create | ||
| hsa-miR-4427 | Break | ||
| hsa-miR-887 | Create | ||
| rs72993667 | hsa-let-7a-3p | ESR1 | Break |
| hsa-let-7b-3p | Break | ||
| hsa-let-7f-1-3p | Break | ||
| hsa-miR-3613-3p | Break | ||
| hsa-miR-548n | Decrease | ||
| rs4973768 | hsa-miR-302a-5p | SLC4A7 | Create |
| rs4808616 | hsa-miR-3121-3p | ABHD8 | Break |
| hsa-miR-3189-3p | Decrease | ||
| hsa-miR-635 | Decrease | ||
| rs61764370 | hsa-miR-1262 | KRAS | Create |
| hsa-miR-34b-3p | Create | ||
| hsa-miR-4701-3p | Create | ||
| hsa-miR-4701-3p | Decrease |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurj, M.-A.; Buse, M.; Zimta, A.-A.; Paradiso, A.; Korban, S.S.; Pop, L.-A.; Berindan-Neagoe, I. Critical Analysis of Genome-Wide Association Studies: Triple Negative Breast Cancer Quae Exempli Causa. Int. J. Mol. Sci. 2020, 21, 5835. https://doi.org/10.3390/ijms21165835
Jurj M-A, Buse M, Zimta A-A, Paradiso A, Korban SS, Pop L-A, Berindan-Neagoe I. Critical Analysis of Genome-Wide Association Studies: Triple Negative Breast Cancer Quae Exempli Causa. International Journal of Molecular Sciences. 2020; 21(16):5835. https://doi.org/10.3390/ijms21165835
Chicago/Turabian StyleJurj, Maria-Ancuta, Mihail Buse, Alina-Andreea Zimta, Angelo Paradiso, Schuyler S. Korban, Laura-Ancuta Pop, and Ioana Berindan-Neagoe. 2020. "Critical Analysis of Genome-Wide Association Studies: Triple Negative Breast Cancer Quae Exempli Causa" International Journal of Molecular Sciences 21, no. 16: 5835. https://doi.org/10.3390/ijms21165835
APA StyleJurj, M.-A., Buse, M., Zimta, A.-A., Paradiso, A., Korban, S. S., Pop, L.-A., & Berindan-Neagoe, I. (2020). Critical Analysis of Genome-Wide Association Studies: Triple Negative Breast Cancer Quae Exempli Causa. International Journal of Molecular Sciences, 21(16), 5835. https://doi.org/10.3390/ijms21165835